

Abstract
Several lines of evidence suggest that follicular cell-derived thyroid cancers represent a continuum of disease that progresses from the highly curable well-differentiated thyroid cancers to the universally fatal anaplastic cancers. However, the genetic mechanisms underlying thyroid cancer progression remain ill defined. We compared the molecular-cytogenetic profiles derived from comparative genomic hybridization (CGH) analysis of major histological variants of thyroid cancer to define genetic variables associated with progression. Overall, a sequential increase in chromosomal complexity was observed from well-differentiated papillary thyroid cancer to poorly differentiated and anaplastic carcinomas, both in terms of the presence of CGH detectable abnormalities (P = 0.003) and the median number of abnormalities per case (P < 0.001). The presence of multiple abnormalities common to all thyroid cancer variants, including gains of 5p15, 5q11–13, 19p, and 19q and loss of 8p, suggests that these tumors are derived from a common genetic pathway. Gains of 1p34–36, 6p21, 9q34, 17q25, and 20q and losses of 1p11-p31, 2q32–33, 4q11–13, 6q21, and 13q21–31 may represent secondary events in progression, as they were only detected in poorly differentiated and anaplastic carcinomas. Finally, recurrent gains at 3p13–14 and 11q13, and loss of 5q11–31 were unique to anaplastic carcinomas, suggesting they may be markers for anaplastic transformation. Our data suggests that the development of chromosomal instability underlies the progression to more aggressive phenotypes of thyroid cancer and sheds light on the possible genomic aberrations that may be selected for during this process.
Cancer is a progressive genetic disorder that may advance through well-defined clinicopathological stages. Accumulating evidence indicates that follicular cell derived thyroid cancer constitutes a biological continuum progressing from the highly curable well-differentiated carcinomas (WDTC) to the universally fatal anaplastic carcinomas (ATC). 1,2 Poorly differentiated carcinomas (PDTC) occupy an intermediate position in this progression concept. 3,4 Morphological support for this model includes the gradual loss of papillary and follicular growth patterns and simultaneous increase in the presence of solid growth, mitoses, necrosis, and nuclear pleomorphism that is observed from WDTC to ATC. 5 Moreover, the majority of ATC exhibit “residual” foci of differentiated thyroid carcinoma. 6 Although this admixture can be seen in the initial pathological specimen, it is also observed that poorly differentiated or undifferentiated thyroid carcinomas develop as a recurrence months or years after the removal of a well-differentiated neoplasm. 6-8 Although these clinicopathological features strongly support a progression concept, little is known regarding the possible mechanisms underlying this process. It is important to decipher these mechanisms, since poorly differentiated and anaplastic carcinomas account for the majority of thyroid cancer-related deaths.
As a result of a prevailing genomic instability, cancer development and progression are characterized by a random accumulation of genetic abnormalities. 9 This trait allows for a Darwinian selection of cells that have acquired the crucial genetic changes capable of driving development and progression of malignant disease. 10 If WDTC, PDTC, and ATC are linked in a progressive relationship, a sequential increase in genomic complexity is anticipated from WDTC to ATC. Specific abnormalities that may have occurred at different stages of the progression may be present at different frequency in WDTC, PDTC, and ATC. Accordingly, one approach for deciphering molecular factors associated with thyroid tumor progression is the direct genetic comparison of WDTC, PDTC, and ATC.
Although several types of genomic instability have been investigated, recent studies indicate that papillary, follicular, and anaplastic thyroid cancers are predominantly characterized by chromosomal instability. 11-17 However, since none of these studies have meticulously categorized the morphological phenotypes of the investigated specimens, a comparison of the chromosomal constitution of WDTC, PDTC, and ATC is currently unavailable. Comparative genomic hybridization (CGH), known for its genome-wide detection of chromosomal gains, losses, and amplifications, is a particularly attractive method to study chromosomal abnormalities. 18 Our previous work and that of others indicates that the independent utilization of CGH is sufficient for a chromosomal instability assessment. 19-21 Using CGH, we show here that the chromosomal complexity sequentially increases in a well-characterized panel from WDTC, PDTC, and ATC.
Materials and Methods
Tissue Samples
All patients treated at the Memorial Sloan-Kettering Cancer Center for PDTC and ATC between 1940 and 1999 were identified from a search of the Department of Pathology database. Cases of WDTC were randomly selected from the institutional tissue bank. To assemble a uniform group, only papillary carcinomas of follicular or classical variant were included.
Histological Classification
Critical histopathological review of all specimens was performed by at least two pathologists based on the criteria described in the Armed Forces Institute of Pathology (AFIP) fascicle and all of the pathological findings were subsequently confirmed by an independent reviewer. 6 Briefly, WDTC included papillary thyroid cancers displaying “ground glass” nuclei (“Orphan Annie eyed nuclei”) without a significant degree of mitotic activity, necrosis, or nuclear pleomorphism. PDTC were identified by the presence of marked mitosis, necrosis and/or nuclear pleomorphism and displaying a definite degree of thyroid follicular cell differentiation at the morphological level. ATC were defined as thyroid-based highly undifferentiated neoplasms with extensive mitotic rate, necrosis, and nuclear pleomorphism and some degree of epithelial differentiation. ATC did not show any follicular thyroid cell differentiation at the morphological level. In more than half of the ATC, residual areas of WDTC and sometimes PDTC were identified. PDTC and ATC were only included if a minimum of 70% of the specimen contained a poorly differentiated or anaplastic phenotype. Based on these criteria, we obtained tissue from 12 PDTC (paraffin-embedded, n = 5) and 15 ATC (paraffin-embedded, n = 12). Fifteen cases of WDTC with available frozen tissue were randomly chosen for inclusion in this study. All 42 cases were represented by tissue from primary tumors. Table 1 ▶ summarizes the clinicopathological features of these cases.
Table 1.
Comparison of Clinical Characteristics of Well-differentiated-(WDTC), Poorly Differentiated-(PDTC), and Anaplastic Thyroid Carcinomas (ATC) at a Median Follow-up of 43 Months
| Clinical factor | WDTC (n = 15) | PDTC (n = 12) | ATC (n = 15) | p-value |
|---|---|---|---|---|
| Female sex | 6 (40%) | 9 (75%) | 9 (60%) | 0.1 |
| Age less than 45 years | 4 (27%) | 0 (0%) | 1 (7%) | 0.05 |
| Extrathyroidal extension | 5 (33%) | 8 (73%) | 15 (100%) | <0.0001 |
| Tumor size > 4.0 cm | 2 (13%) | 8 (73%) | 9 (69%) | 0.0001 |
| Lymph node metastasis | 7 (47%) | 7 (64%) | 15 (100%) | 0.0007 |
| Distant metastasis | 0 (0%) | 6 (50%) | 13 (87%) | <0.0001 |
| 5-years disease-free survival | 91% | 51% | 0% | <0.0001 |
| 5-years cause-specific survival | 100% | 70% | 0% | <0.0001 |
| Overall survival | 100% | 70% | 0% | <0.0001 |
Comparative Genomic Hybridization Analysis
DNA extraction of frozen and formalin-fixed, paraffin-embedded samples was performed as described previously. 22 Tumor DNA was labeled by nick translation (Life Technologies, Inc., Rockville, MD) with fluorescein-12-dUTP (FITC) (New England Nuclear-DuPont, Boston, MA). Reference DNA was extracted from normal placenta and labeled with Texas red-5-dUTP (New England Nuclear-DuPont). CGH was performed using previously published methods. 18,19 For analysis, 7 to 10 separate metaphases were captured and processed using the Quantitative Image Processing System (Quips Pathvysion System; Applied Imaging, Santa Clara, CA). Red, green, and blue fluorescence intensities were analyzed for all metaphase spreads, normalized to a standard length, and statistically combined to show the red/green signal ratio and 95% confidence intervals for the entire chromosome. Copy number changes were detected based on the variance of the red/green ratio profile from the standard of 1. Ratio values of 1.2 and 2.0 were defined as thresholds for gains and amplifications, respectively, and losses were defined as a ratio value of 0.8 or less.
Fluorescent in Situ Hybridization
Dual color probe CEP 17 (green) and HER-2 (red) was obtained from Vysis (Downer’s Grove, IL) and used to asses 17q copy number in two well-differentiated thyroid carcinoma samples with normal 17q copy number by CGH and four cases of poorly differentiated or anaplastic thyroid cancer known to harbor 17q copy number changes (PD5, PD12, ATC11, and ATC6). The BAC clone from chromosome 13q12 (RP11–556N21) was labeled with spectrum red dUTP by nick translation and used to assess chromosome 13 copy number in two well-differentiated thyroid carcinoma samples with normal chromosome 13 copy number by CGH and 4 poorly differentiated or anaplastic thyroid cancer cases known to harbor chromosome 13 copy number changes (PD2, PD6, ATC11, and ATC12). In these experiments CEP 10 (Vysis) was used as a control. FISH was performed on paraffin sections as described previously. 23
Statistical Analysis
Statistical analyses were performed using the JMP4 statistical software package (SAS Institute Inc, Cary, NC.). Statistical significance was defined as a two-tailed P ≤ 0.05. Qualitative and quantitative differences between the histopathological variants of thyroid carcinomas were assessed using the χ2 test and Kruskal-Wallis analysis of variance, respectively.
Results
Validation of CGH Findings
Our own experience and that of others indicates that CGH is a reliable technique for the detection of DNA copy number changes not only in fresh frozen tissue specimens but also on formalin-fixed, paraffin-embedded tissue specimens. 22,24 To validate the use of paraffin-embedded cases we performed several control experiments. Firstly, we hybridized FITC-labeled DNA extracted from fresh frozen normal placenta against Texas-red labeled DNAs extracted from several formalin-fixed, paraffin-embedded normal lymph nodes that were pathologically confirmed. As expected, these experiments generated clean hybridizations of which the ratio profiles were within the normal limits. Second, 10 tumor cases were selected and CGH profiles from corresponding frozen and paraffin-embedded tissues were compared. In these experiments, only one case featured a single abnormality discrepancy in the ratio profiles from the frozen sample relative to that of the paraffin-embedded tissue. In the frozen sample of this case, a gain of 17q and losses of 16q and 17p were detected. However, in the corresponding paraffin-embedded sample only the chromosome 17 abnormalities were detected. We believe that this discrepancy may have been a result of intratumoral heterogeneity that is common in neoplasia. In addition, we used FISH to corroborate some of the CGH findings in cases from the present study of which only paraffin-embedded tissue was available. We analyzed gain of chromosome 17q in 2 well-differentiated thyroid carcinoma samples and some of our poorly differentiated and anaplastic thyroid cancer cases (Cases PD5, PD12, ATC11, and ATC6). FISH confirmed CGH results with normal copy numbers detected in paraffin-embedded well-differentiated carcinomas and increased copy numbers in all investigated poorly differentiated and anaplastic tumor samples (PD5, PD12, ATC11, and ATC6), ranging from 3 to 6 copies (Figure 1) ▶ . Similarly, FISH confirmed normal 13q copy number in well-differentiated thyroid cancer samples and 13q deletion in poorly differentiated and anaplastic thyroid cancer samples (PD2, PD6, ATC11, and ATC12) as detected by CGH.
Figure 1.

Fluorescent in situ hybridization analysis of poorly differentiated (A, C) and anaplastic thyroid cancer (B, D). Spectrum red-labeled 13q12 BAC clone and spectrum green-labeled CEP 10 probes hybridized to the 5-μm section confirming decreased copy numbers in chromosome 13 identified by comparative genomic hybridization in these cases, as depicted by arrows identifying the signals (A, B). Spectrum red-labeled HER-2 probe and spectrum green-labeled CEP 17 probes hybridized to the 5-μm section confirming increased numbers in chromosome 17 identified by comparative genomic hybridization in these cases (C, D).
Comparison of Chromosomal Instability in WDTC, PDTC, and ATC
To determine whether the sequential deterioration in clinical behavior and loss of pathological differentiation from WDTC to ATC is accompanied by a progressive accumulation of genetic abnormalities, we compared WDTC, PDTC, and ATC for the presence of CGH detectable abnormalities and the median number of abnormalities per case (Figure 2) ▶ . Both PDTC (83%) and ATC (80%) displayed a higher proportion of cases with CGH-detectable chromosomal abnormalities as compared to WDTC (30%) (P = 0.003). Moreover, the median number of chromosomal abnormalities sequentially increased from WDTC 1 to PDTC (5.5) and ATC 10 (P < 0.001).
Figure 2.

Comparison of chromosomal instability of WDTC, PDTC, and ATC. Both PDTC and ATC displayed a significantly higher percentage of cases with CGH detectable abnormalities as compared to WDTC (A). The median number of chromosomal abnormalities in cases with abnormalities increased sequentially and significantly from WDTC to PDTC and finally ATC (B).
Comparison of Molecular-Cytogenetic Profile of WDTC, PDTC, and ATC
Figure 3 ▶ details a compilation of all chromosomal abnormalities detected in WDTC, PDTC, and ATC. To identify possible chromosomal alterations associated with progression, we compared the frequency distribution of individual abnormalities among WDTC, PDTC, and ATC. This comparison showed a significant overlap in the chromosomal profiles among the three subtypes of thyroid cancer. Of the 11 different copy number changes detected in WDTC, 8 showed overlap with recurrent abnormalities in PDTC and 7 showed overlap with abnormalities detected in ATC. Of the 34 different abnormalities detected in PDTC, 28 showed overlap with ATC. However, several genetic differences among WDTC, PDTC, and ATC were present. Figure 4 ▶ details the frequency distribution of the most common abnormalities among WDTC, PDTC, and ATC. The most common abnormalities detectable in all three subtypes included gains at 5p, 5q, 19p, and 19q, and losses of 8p. Also, the two recurrent abnormalities detected in WDTC, loss of 22 and gain of 8q were detected in PDTC and ATC, respectively. Common abnormalities restricted to PDTC and ATC included gains of 1p, 3q, 6p, 7p, 7q, 9q, 12q, 16q, 17p, 17q, and 20q and losses of 1p11–31, 2q, 4p, 4q, 6q, 9p, 11q, 12q, 13q, and 18. Abnormalities exclusively detectable in ATC included gains of 3p, 11p, and 11q and losses of 5q.
Figure 3.

Ideogram showing DNA copy number changes identified by CGH in WDTC (A), PDTC (B), and ATC (C). Thin vertical lines on either side of the ideogram indicate losses (left) and gains (right) of the chromosomal region. The chromosomal regions of the high-level amplification are shown by thick lines (right).
Figure 4.
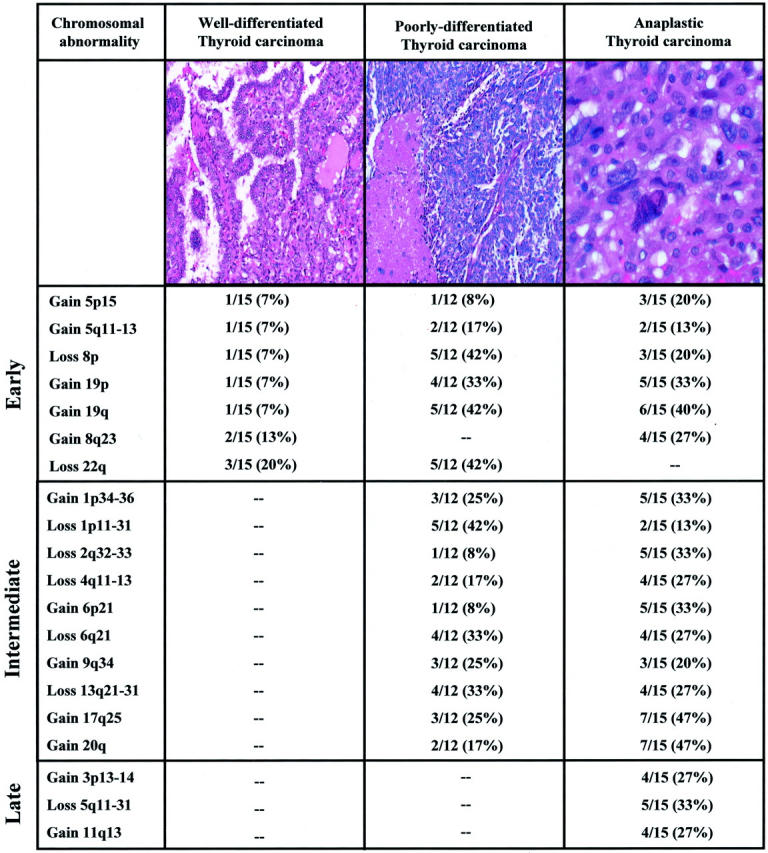
Representative histological sections of well differentiated, poorly differentiated, and anaplastic carcinomas analyzed in this study and frequency distribution of most commonly detected chromosomal abnormalities among the three groups. The well-differentiated papillary carcinoma (left) is of the classical type. In addition to the presence of well-defined papillae seen at this power, the tumor nuclei are clear (“ground glass”) with nuclear grooves and pseudoinclusions (H&E; magnification, ×100). In contrast, the poorly differentiated tumor (center) features a solid growth pattern and has many areas of tumor necrosis (lower left corner) with increased mitotic activity (H&E; magnification, ×100). The highly undifferentiated anaplastic carcinoma (right) exhibits marked nuclear enlargement, pleomorphism with hyperchromasia and very coarse chromatin. The degree of nuclear atypia is substantially higher than that seen in poorly differentiated thyroid carcinomas (H&E; magnification, ×200). Chromosomal abnormalities detectable in all three thyroid cancer subtypes may be early events in thyroid tumorigenesis. Chromosomal abnormalities solely present in poorly differentiated and anaplastic carcinomas may be intermediate events in the progression from well-differentiated to poorly differentiated subtypes. Finally, chromosomal abnormalities exclusively detectable in anaplastic carcinomas may be late events in anaplastic transformation.
Discussion
Well-differentiated papillary carcinoma accounts for 80% of thyroid cancers and is characterized by indolent clinical behavior and an excellent overall prognosis. 25 However, strong circumstantial evidence indicates that individual cases may be capable of progressing to the highly malignant ATC, a transition associated with devastating consequences. 6 The identification of poorly differentiated thyroid carcinomas has reinforced the progression hypothesis by providing a “clinicopathological bridge” between WDTC and ATC. 3
As a first step to decipher possible underlying mechanisms of thyroid tumor progression, we have compared the molecular-cytogenetic profile of WDTC, PDTC, and ATC using CGH. This analysis has shown a sequential accumulation of chromosomal abnormalities from WDTC to ATC. Previous studies have addressed papillary, follicular, and anaplastic thyroid carcinomas individually for the presence of genome-wide abnormalities and confirm that thyroid cancers are commonly characterized by chromosomal instability. 11-17 However, these reports show a wide variation in the detection rate of abnormalities and suggest that rates of chromosomal complexity are similar between papillary, follicular, and anaplastic carcinomas. Since none of these studies performed a detailed histopathological appraisal, the inclusion of morphologically heterogeneous study populations likely confounds the data presented. Our data clearly demonstrate the need for strict morphological characterization of thyroid cancer specimens in molecular genetic studies. In our study, the increase in the presence of DNA copy number abnormalities was most pronounced from WDTC to PDTC. This was evident from both the percentage of cases with abnormalities as well as the median number of accumulated abnormalities per case (Figure 2) ▶ . These data suggest that the development of chromosomal instability underlies the progression to more aggressive phenotypes of thyroid cancer. Interestingly, recent studies report that deregulation of the p53 and Wnt pathways follow a similar trend, rarely present in WDTC, but common in poorly differentiated and ATC. 26,27 It remains to be elucidated whether abnormalities in p53 and Wnt pathways, both of which may promote genetic instability, 28,29 are a cause rather than a result of the observed chromosomal instability in thyroid cancer.
Although chromosomal instability is most likely a nonspecific process, WDTC, PDTC, and ATC are characterized by a non-random pattern of chromosomal abnormalities. This suggests that tumor cells that harbor the presently reported abnormalities are selected during the progression because these abnormalities may impart survival advantages. Analogous to previous studies of tumor progression in other tumor systems, 30,31 we can speculate the relative oncogenic time frame where chromosomal abnormalities occur by assessing their prevalence among the different histopathological subtypes (Figure 4) ▶ . For example, gains of 5p15, 5q11–13, 19p, 19q, and losses of 8p were detectable in all three subtypes, suggesting that they may be early events in the development of thyroid cancer. Additional support for an early role for abnormalities of chromosomes 5 and 19 in thyroid tumorigenesis comes from the observation that these abnormalities are among the most common genetic abnormalities occurring in benign thyroid nodules, including nodular goiters and follicular adenomas. 32 Recently, Rippe and colleagues 33 have identified involvement of a novel KRAB zinc finger protein gene, designated ZNF331, at one of these sites (19q13.4) in follicular adenomas. This gene constitutes a potential candidate gene involved in thyroid tumorigenesis and merits further investigation. Additional abnormalities detected in our analysis that may represent early events in thyroid carcinogenesis include recurrent gains of 8q23 and losses of 22. Loss of 22 was common in our WDTC and PDTC. Deletions and translocations involving this chromosome have been commonly reported in goiters, follicular adenomas, and follicular carcinomas, 13,32 suggesting that this chromosome harbors a tumor suppressor gene that is inactivated early in thyroid tumorigenesis. Although chromosome 22 deletions were not detected in our ATC, Kitamura and colleagues 34 have shown that a significant number of ATCs exhibit small deletions on chromosome 22 that may be below the detection level of CGH, suggesting that this abnormality is relevant throughout the progression. Gain of 8q23 was very common in ATC but also recurrently detected in WDTC, suggesting that this abnormality may be an early event in thyroid carcinogenesis. However, work by Wilkens and colleagues 15 suggests that gains of 8q are involved in anaplastic transformation. The apparent paradoxical presence of this abnormality in two of our WDTC may be explained by the concept proposed by Vogelstein and colleagues 35 that it is not necessarily the order but rather the accumulation of particular events that drives cancer development and progression.
Several chromosomal abnormalities were exclusively detected in PDTC and ATC suggesting that they are transition events in the progression of WDTC to PDTC. Most commonly these included gains of 1p34–36, 17q25, and 20q and deletions of 1p11–31, 6q21, and 13q21–31. Several recent genetic studies confirm that these abnormalities are associated with aggressive thyroid cancers. 15,34,36,37 In addition, it is of particular interest that losses of 1p11–31, 6q, and 13q have been commonly described in development and progression of a broad range of endocrine tumors including familial and sporadic pheochromocytomas, pituitary adenomas, parathyroid tumors, and endocrine pancreatic tumors. 38-43 This suggests that common tumor suppressor genes are involved in the development and progression of various types of endocrine neoplasia.
Chromosomal abnormalities of 3p11–14 and 11q13 and deletion of 5q11–31 were exclusively associated with ATC. This suggests that these regions harbor genes that may drive the transformation from PDTC to ATC.
Several known genes may drive the selection for the aforementioned chromosomal abnormalities. Chromosomal regions 17q and 20q harbor multiple known genes that are associated with invasion and metastasis, including the matrix metalloproteinase genes MMP-9 and MMP-24 (20q), LAMBA5 (20q), Syndecan-4 (20q), and LGALS3BP (17q25). In addition, Fusco and colleagues 44 recently demonstrated that the SIR-T8 gene is located within 17q25 and highly overexpressed in thyroid cancer. Since aggressive thyroid cancer is commonly characterized by a loss of expression of thyroid-specific genes, 45 the chromosomal deletions at 1p, 6q, and 13q may target the thyroid-specific genes located in these regions including DIO1 (deiodinase, iodothyronine, Type 1), TSHα and TSHβ (thyroid stimulating hormone α and β subunits). This would help explain the phenotypic changes that are observed in the progression of WDTC. Another possible candidate gene located at 1p13 constitutes HTIF1, a gene that was recently identified as a novel translocation partner of RET (RET/PTC7) in radiation-induced thyroid cancers. 46 In addition to the abnormalities that were detected in PDTC and ATC, the abnormalities exclusively detectable in ATC harbor various interesting candidate genes. For example, intermediates of the Wnt signaling pathway (Wnt 11 at 11q13, Wnt 5A at 3p11–14, and APC at 5q), which is involved in aggressive thyroid cancer, 27 are located within these regions. Moreover, targets of 11q13 amplification may include cyclin D1 and FRA-1 since over expression of both is common in aggressive thyroid cancer. 47,48
Based on the frequency distribution of chromosomal abnormalities among WDTC, PDTC, and ATC, we identified abnormalities potentially involved in the progression of thyroid cancer. Although the majority of the most common abnormalities increase in frequency from WDTC to ATC, several abnormalities do not follow the classical models of genetic progression. 49 For example, gain of 1q was a common abnormality that was uniquely detected in PDTC. Some of these abnormalities may represent “noise” due to generalized chromosomal instability or may be representative of unique properties of the individual cases. Alternatively, the pattern of detection of these abnormalities may be influenced by the boundaries of the CGH resolution.
In conclusion, we report genetic differences among well-differentiated, poorly differentiated, and anaplastic carcinomas that may play a role in aggressive tumor behavior and potential tumor progression. The present report constitutes a starting point toward the identification of genes involved in this process.
Acknowledgments
We thank the Dutch Cancer Society and the foundations Dr. Hendrik Muller’s Vaderlandsch Fonds, Bekker La Bastide Fonds, Fundatie van de Vrijvrouwe van Renswoude, Schuurman Schimmel-van Outeren, Gerrit-Jan Mulder, and AA van Beek Fonds for their generous support of Volkert B. Wreesmann.
Footnotes
Address reprint requests to Bhuvanesh Singh, M.D., Laboratory of Epithelial Cancer Biology, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021. E-mail: singhb@mskcc.org.
V.B.W. and R.A.G. contributed equally to this work.
References
- 1.Venkatesh YS, Ordonez NG, Schultz PN, Hickey RC, Goepfert H, Samaan NA: Anaplastic carcinoma of the thyroid: a clinicopathologic study of 121 cases. Cancer 1990, 66:321-330 [DOI] [PubMed] [Google Scholar]
- 2.Carcangiu ML, Steeper T, Zampi G, Rosai J: Anaplastic thyroid carcinoma: a study of 70 cases. Am J Clin Pathol 1985, 83:135-158 [DOI] [PubMed] [Google Scholar]
- 3.Carcangiu ML, Zampi G, Rosai J: Poorly differentiated (“insular”) thyroid carcinoma: a reinterpretation of Langhans’ “wuchernde Struma”. Am J Surg Pathol 1984, 8:655-668 [DOI] [PubMed] [Google Scholar]
- 4.Sakamoto A, Kasai N, Sugano H: Poorly differentiated carcinoma of the thyroid: a clinicopathologic entity for a high-risk group of papillary and follicular carcinomas. Cancer 1983, 52:1849-1855 [DOI] [PubMed] [Google Scholar]
- 5.Rosai J, Saxen EA, Woolner L: Undifferentiated and poorly differentiated carcinoma. Semin Diagn Pathol 1985, 2:123-136 [PubMed] [Google Scholar]
- 6.Rosai J, Carcangiu ML, DeLellis RA: Tumors of the Thyroid Gland. 1992. Armed Forces Institute of Pathology Washington, DC
- 7.Ozaki O, Ito K, Mimura T, Sugino K: Anaplastic transformation of papillary thyroid carcinoma in recurrent disease in regional lymph nodes: a histologic and immunohistochemical study. J Surg Oncol 1999, 70:45-48 [DOI] [PubMed] [Google Scholar]
- 8.Spires JR, Schwartz MR, Miller RH: Anaplastic thyroid carcinoma: association with differentiated thyroid cancer Arch. Otolaryngol Head Neck Surg 1988, 114:40-44 [DOI] [PubMed] [Google Scholar]
- 9.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 10.Cahill DP, Kinzler KW, Vogelstein B, Lengauer C: Genetic instability and darwinian selection in tumors. Trends Cell Biol 1999, 9:57-60 [PubMed] [Google Scholar]
- 11.Antonini P, Venuat AM, Caillou B, Berger R, Schlumberger M, Bernheim A, Parmentier C: Cytogenetic studies on 19 papillary thyroid carcinomas. Genes Chromosomes Cancer 1992, 5:206-211 [DOI] [PubMed] [Google Scholar]
- 12.Pierotti MA, Bongarzone I, Borello MG, Greco A, Pilotti S, Sozzi G: Cytogenetics and molecular genetics of carcinomas arising from thyroid epithelial follicular cells. Genes Chromosomes Cancer 1996, 16:1-14 [DOI] [PubMed] [Google Scholar]
- 13.Hemmer S, Wasenius VM, Knuutila S, Franssila K, Joensuu H: DNA copy number changes in thyroid carcinoma. Am J Pathol 1999, 154:1539-1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kjellman P, Lagercrantz S, Hoog A, Wallin G, Larsson C, Zedenius J: Gain of 1q and loss of 9q21.3-q32 are associated with a less favorable prognosis in papillary thyroid carcinoma. Genes Chromosomes Cancer 2001, 32:43-49 [DOI] [PubMed] [Google Scholar]
- 15.Wilkens L, Benten D, Tchinda J, Brabant G, Potter E, Dralle H, von Wasielewski R: Aberrations of chromosomes 5 and 8 as recurrent cytogenetic events in anaplastic carcinoma of the thyroid as detected by fluorescence in situ hybridisation and comparative genomic hybridisation. Virchows Arch 2000, 436:312-318 [DOI] [PubMed] [Google Scholar]
- 16.Zhou XP, Hoang JM, Li YJ, Seruca R, Carneiro F, Sobrinho-Simoes M, Lothe RA, Gleeson CM, Russell SE, Muzeau F, Flejou JF, Hoang-Xuan K, Lidereau R, Thomas G, Hamelin R: Determination of the replication error phenotype in human tumors without the requirement for matching normal DNA by analysis of mononucleotide repeat microsatellites. Genes Chromosomes Cancer 1998, 21:101-107 [DOI] [PubMed] [Google Scholar]
- 17.Stoler DL, Datta RV, Charles MA, Block AW, Brenner BM, Sieczka EM, Hicks WL, Jr, Loree TR, Anderson GR: Genomic instability measurement in the diagnosis of thyroid neoplasms. Head Neck 2002, 24:290-295 [DOI] [PubMed] [Google Scholar]
- 18.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258:818-821 [DOI] [PubMed] [Google Scholar]
- 19.Singh B, Gogineni SK, Sacks PG, Shaha AR, Shah JP, Stoffel A, Rao PH: Molecular cytogenetic characterization of head and neck squamous cell carcinoma and refinement of 3q amplification. Cancer Res 2001, 61:4506-4513 [PubMed] [Google Scholar]
- 20.Kytola S, Rummukainen J, Nordgren A, Karhu R, Farnebo F, Isola J, Larsson C: Chromosomal alterations in 15 breast cancer cell lines by comparative genomic hybridization and spectral karyotyping. Genes Chromosomes Cancer 2000, 28:308-317 [DOI] [PubMed] [Google Scholar]
- 21.Ghadimi BM, Schrock E, Walker RL, Wangsa D, Jauho A, Meltzer PS, Ried T: Specific chromosomal aberrations and amplification of the AIB1 nuclear receptor coactivator gene in pancreatic carcinomas. Am J Pathol 1999, 154:525-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isola J, DeVries S, Chu L, Ghazvini S, Waldman F: Analysis of changes in DNA sequence copy number by comparative genomic hybridization in archival paraffin-embedded tumor samples. Am J Pathol 1994, 145:1301-1308 [PMC free article] [PubMed] [Google Scholar]
- 23.Zilmer M, Harris CP, Steiner DS, Meisner LF: Use of nonbreakpoint DNA probes to detect the t(X;18) in interphase cells from synovial sarcoma: implications for detection of diagnostic tumor translocations. Am J Pathol 1998, 152:1171-1177 [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Q, Schantz SP, Rao PH, Mo J, McCormick SA, Chaganti RS: Improving degenerate oligonucleotide primed PCR-comparative genomic hybridization for analysis of DNA copy number changes in tumors. Genes Chromosomes Cancer 2000, 28:395-403 [PubMed] [Google Scholar]
- 25.Hundahl SA, Fleming ID, Fremgen AM, Menck HR: A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U. S., 1985–1995. Cancer 1998, 83:2638-2648 [DOI] [PubMed] [Google Scholar]
- 26.Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA: Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J Clin Invest 1993, 91:1753-1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G: β-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and ctnnb1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol 2001, 158:987-996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H: Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol 2001, 3:433-438 [DOI] [PubMed] [Google Scholar]
- 29.Donehower LA, Godley LA, Aldaz CM, Pyle R, Shi YP, Pinkel D, Gray J, Bradley A, Medina D, Varmus HE: Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev 1995, 9:882-895 [DOI] [PubMed] [Google Scholar]
- 30.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL: Genetic alterations during colorectal-tumor development. N Engl J Med 1988, 319:525-532 [DOI] [PubMed] [Google Scholar]
- 31.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D: Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res 1996, 56:2488-2492 [PubMed] [Google Scholar]
- 32.Belge G, Roque L, Soares J, Bruckmann S, Thode B, Fonseca E, Clode A, Bartnitzke S, Castedo S, Bullerdiek J: Cytogenetic investigations of 340 thyroid hyperplasias and adenomas revealing correlations between cytogenetic findings and histology. Cancer Genet Cytogenet 1998, 101:42-48 [DOI] [PubMed] [Google Scholar]
- 33.Rippe V, Belge G, Meiboom M, Kazmierczak B, Fusco A, Bullerdiek J: A KRAB zinc finger protein gene is the potential target of 19q13 translocation in benign thyroid tumors. Genes Chromosomes Cancer 1999, 26:229-236 [PubMed] [Google Scholar]
- 34.Kitamura Y, Shimizu K, Tanaka S, Ito K, Emi M: Allelotyping of anaplastic thyroid carcinoma: frequent allelic losses on 1q, 9p, 11, 17, 19p, and 22q. Genes Chromosomes Cancer 2000, 27:244-251 [PubMed] [Google Scholar]
- 35.Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 36.Tung WS, Shevlin DW, Kaleem Z, Tribune DJ, Wells SA, Goodfellow PJ: Allelotype of follicular thyroid carcinomas reveals genetic instability consistent with frequent nondisjunctional chromosomal loss. Genes Chromosomes Cancer 1997, 19:43-51 [PubMed] [Google Scholar]
- 37.Segev DL, Saji M, Phillips GS, Westra WH, Takiyama Y, Piantadosi S, Smallridge RC, Nishiyama RH, Udelsman R, Zeiger MA: Polymerase chain reaction-based microsatellite polymorphism analysis of follicular and Hurthle cell neoplasms of the thyroid. J Clin Endocrinol Metab 1998, 83:2036-2042 [DOI] [PubMed] [Google Scholar]
- 38.Benn DE, Dwight T, Richardson AL, Delbridge L, Bambach CP, Stowasser M, Gordon RD, Marsh DJ, Robinson BG: Sporadic and familial pheochromocytomas are associated with loss of at least two discrete intervals on chromosome 1p. Cancer Res 2000, 60:7048-7051 [PubMed] [Google Scholar]
- 39.Dannenberg H, Speel EJ, Zhao J, Saremaslani P, van Der Harst E, Roth J, Heitz PU, Bonjer HJ, Dinjens WN, Mooi WJ, Komminoth P, de Krijger RR: Losses of chromosomes 1p and 3q are early genetic events in the development of sporadic pheochromocytomas. Am J Pathol 2000, 157:353-359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pei L, Melmed S, Scheithauer B, Kovacs K, Benedict WF, Prager D: Frequent loss of heterozygosity at the retinoblastoma susceptibility gene (RB) locus in aggressive pituitary tumors: evidence for a chromosome 13 tumor suppressor gene other than RB. Cancer Res 1995, 55:1613-1616 [PubMed] [Google Scholar]
- 41.Kytola S, Farnebo F, Obara T, Isola J, Grimelius L, Farnebo LO, Sandelin K, Larsson C: Patterns of chromosomal imbalances in parathyroid carcinomas. Am J Pathol 2000, 157:579-586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barghorn A, Speel EJ, Farspour B, Saremaslani P, Schmid S, Perren A, Roth J, Heitz PU, Komminoth P: Putative tumor suppressor loci at 6q22 and 6q23–q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. Am J Pathol 2001, 158:1903-1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Speel EJ, Scheidweiler AF, Zhao J, Matter C, Saremaslani P, Roth J, Heitz PU, Komminoth P: Genetic evidence for early divergence of small functioning and nonfunctioning endocrine pancreatic tumors: gain of 9q34 is an early event in insulinomas. Cancer Res 2001, 61:5186-5192 [PubMed] [Google Scholar]
- 44.de Nigris F, Cerutti J, Morelli C, Califano D, Chiariotti L, Viglietto G, Santelli G, Fusco A: Isolation of a SIR-like gene, SIR-T8, that is overexpressed in thyroid carcinoma cell lines and tissues. Br J Cancer 2002, 86:917-923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoang-Vu C, Dralle H, Scheumann G, Maenhaut C, Horn R, von Zur Muhlen A, Brabant G: Gene expression of differentiation and dedifferentiation markers in normal and malignant human thyroid tissues. Exp Clin Endocrinol 1992, 100:51-56 [DOI] [PubMed] [Google Scholar]
- 46.Klugbauer S, Rabes HM: The transcription coactivator HTIF1 and a related protein are fused to the RET receptor tyrosine kinase in childhood papillary thyroid carcinomas. Oncogene 1999, 18:4388-4393 [DOI] [PubMed] [Google Scholar]
- 47.Wang S, Lloyd RV, Hutzler MJ, Safran MS, Patwardhan NA, Khan A: The role of cell cycle regulatory protein, cyclin D1, in the progression of thyroid cancer. Mod Pathol 2000, 13:882-887 [DOI] [PubMed] [Google Scholar]
- 48.Chiappetta G, Tallini G, De Biasio MC, Pentimalli F, de Nigris F, Losito S, Fedele M, Battista S, Verde P, Santoro M, Fusco A: FRA-1 expression in hyperplastic and neoplastic thyroid diseases. Clin Cancer Res 2000, 6:4300-4306 [PubMed] [Google Scholar]
- 49.Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B: Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 1992, 355:846-847 [DOI] [PubMed] [Google Scholar]