

Abstract
Axonal loss, already present in the acute and first relapse phases of experimental allergic encephalomyelitis (EAE) in the ABH mouse, only becomes apparent in the third relapse in the interleukin-12 model of relapsing EAE in the Lewis rat. Caspase-1 immunostaining in the spinal cord of Lewis rats was mainly localized to inflammatory cuffs with the greatest proportion of active caspase-1-positive cells detected during the first and second relapses, correlating with enzyme activity and protein on Western blots. However, in the spinal cord of ABH mice during acute EAE, caspase-1 immunostaining was localized both on inflammatory and neuronal cells, again correlating with enzyme activity and protein production. In contrast, caspase-3 expression in the spinal cord of Lewis rats did not increase significantly until the third relapse when inflammatory and neuronal cells and axons became positive in line with a significant increase in caspase activity. In ABH mice active caspase-3 was already immunolocalized on axons and apoptotic neurons in the spinal cord during the acute stage of EAE. Because caspase-3 is a downstream cell death signal it may be possible to reduce apoptosis by selectively blocking caspase-3 and therefore provide a therapeutic target for EAE and potentially, multiple sclerosis.
We have previously shown that interleukin (IL)-12, a proinflammatory cytokine, can induce serial paralytic relapses of experimental allergic encephalomyelitis (EAE) in the Lewis rat 1 with the appearance of myelin basic protein-laden macrophages, indicating myelin processing. 1,2 Signs of limited axonal death together with localization of tissue plasminogen activator (tPA) were observed on axons by the third relapse. 2 In ABH mice, however, chronic relapsing EAE (CREAE) can be induced by a single sensitization with spinal cord homogenate and animals undergo relapsing-remitting disease. This is characterized by progressive inflammatory demyelination but also by axonal loss even during the acute stage. 3 Interestingly, Lewis rats showed no clinical signs of disease between the relapses whereas ABH mice showed cumulative deficits after each bout of disease.
Caspases, a family of cysteine proteases mainly involved in the apoptotic pathway 4 are essential for the control of cell populations not only during development but also in pathology. 5,6 Apoptosis is recognized by distinct morphological changes, including cell shrinkage, nuclear condensation, and fragmentation. 7 Caspases are synthesized as inactive proenzymes releasing active subunits when they are catalytically activated. 8-12 The active caspases hydrolyze a number of key structural and housekeeping proteins in the progression of apoptosis. 8,13,14 Among these, caspase-1, also known as IL-1β-converting enzyme, is less involved in the apoptotic cascade but prominent in inflammation in which it plays a key role in regulating cellular export of proinflammatory cytokines such as IL-1β. 15 However, overexpression of caspase-1 can lead to apoptotic cell death and may mediate microglial apoptosis after inflammatory microglial activation in vitro. 16-18 Caspase-3 or CPP32, another member of the caspase family, appears to be the main effector caspase involved in neuronal apoptosis. 19,20 Activation of caspase-3 has been shown to be a critical event in neuronal apoptosis in the brain during development and after acute injury and after exposure to the cytokines produced from activated microglia. 17,19,20
Because there is limited axonal death in Lewis rats after three relapses of EAE and ABH mice undergo axonal damage and demyelination, particularly after the first relapse, the aim of the study was to test the hypothesis that the axonal death observed might be mediated by caspases, caspase-1 and in particular caspase-3.
Materials and Methods
Induction of Active EAE
Female Lewis rats (180 to 200 g) (Charles River, Kent, UK) were immunized in each hind foot with a mixture of purified guinea pig myelin basic protein (final concentration, 1 mg/ml), emulsified in Freund’s complete adjuvant (myelin basic protein-complete Freund’s adjuvant) containing Mycobacterium tuberculosis H37Ra (final concentration, 5 mg/ml; Difco Laboratories, Detroit, MI) in a final volume of 50 μl. Animals were weighed and monitored daily for clinical signs of EAE using the earlier published criteria. 2
CREAE was induced in 6- to 8-week-old Biozzi ABH mice (bred at the Institute of Neurology) by injection with mouse spinal cord homogenate emulsified in complete Freund’s adjuvant on days 0 and 7. 3 Animals with complete hindlimb paralysis were examined during the initial acute paralytic attack on day 18 after immunization and the first clinical relapse on days 37 to 42 after immunization Animals were also examined during the first clinical remission on days 27 to 28 after immunization in which animals exhibited only minimal tail paresis and chronically affected remission animals after three relapses on days 80 to 100, in which animals had residual hindlimb paresis. 21,22
IL-12 Administration to Lewis Rats
Recombinant murine IL-12 (Genetics Institute, Cambridge, MA) was administered intraperitoneally as described before. 2
Immunocytochemistry
Rats and mice were culled at different stages after immunization and the brain and spinal cord were removed and rapidly frozen on solid CO2. Longitudinal frozen sections were cut, adhered to Vectabond-coated slides (Vector Laboratories, Peterborough, UK) and processed for immunohistochemistry using the following antibodies to caspases: rabbit anti-caspase-1 (p10 subunit, diluted 1:200) and goat anti-caspase-3 (1:200) (both from Santa Cruz Biotechnology Inc., Santz Cruz, CA). Tissues were fixed in ethanol for 1 minute at room temperature and washed twice in phosphate-buffered saline (PBS) followed by incubation with the appropriate primary antibody diluted in PBS, pH 7.4. For caspase-3 immunostaining an additional step was included in which tissues were permeabilized in 0.1% Triton X-100 for 5 minutes after fixation. After incubation with the primary antibody sections were washed in PBS before incubation with either biotinylated anti-goat or anti-rabbit IgG (Vector Laboratories) diluted 1:200 for 30 minutes at room temperature. Peroxidase-labeled avidin-biotin complex solution was added to the sections (Vector Laboratories) for 45 minutes, washed, and peroxidase activity detected by placing the slides in a solution of 0.5 mg/ml of 3,3′-diaminobenzidine (Sigma, Poole, UK) in PBS containing 0.01% hydrogen peroxide for 5 minutes. For caspase-3 immunostaining, nickel chloride was added to the 3,3′-diaminobenzidine and hydrogen peroxide solution and sections were not counterstained. Other sections were rinsed and counterstained in Mayer’s hematoxylin (30 seconds), dehydrated through a graded series of alcohols, and mounted in DPX (BDH, Poole, UK). Appropriate antibody control sections were included with each run, including preabsorption of caspase-1 and -3 with the appropriate blocking peptide (Santa Cruz Biotechnology) for 2 hours at room temperature before incubating with sections.
Identification of the Cell Types Expressing Cleaved Caspase-3 by Double-Immunofluorescent Staining
Fixed sections were permeabilized with 1% Triton X-100 for 10 minutes followed by three washes on PBS for 5 minutes each before incubating with polyclonal cleaved caspase-3 antibody (1:100 dilution in PBS containing 1% bovine serum albumin; Cell Signaling Technology, Beverly, MA). Sections were then washed in PBS and incubated with fluorescein isothiocyanate-conjugated anti-rabbit IgG (Sigma). Sections were allowed to react with one of the following monoclonal antibodies: ED-1 (macrophage), OX-42 (macrophages/microglia), glial fibrillary acidic protein (astrocytes), SMI-32 (1:1000, neurons/axons), OX-34 (1:500, T cells); or the following polyclonal antibodies: CNPase (1:1000, gift from Dr. T. J. Sprinkle, Veterans Affairs Medical Centre, Augusta, USA) and neurofilament 200 (1:500, neurons/axons, Sigma) or rat anti-mouse CD4 (1:50, CD4 T cells), F4/80 (1:50, macrophages) (Serotec). Appropriate secondary antibodies (diluted 1:50 in PBS) used for the above markers were: tetramethyl-rhodamine isothiocyanate-conjugated anti-mouse, anti-rat, anti-rabbit, and anti-goat IgG (all from Sigma). Finally sections were mounted using Citifluor (Citifluor Ltd., London, UK) and viewed under the Radiance 2100 confocal laser microscope using Lasersharp 2000 software system (Bio-Rad, Hertfordshire, UK). Captured images were converted to TIFF files using Confocal Assistant (Version 4.02, Todd Clark Brelje) and assembled using Adobe Photoshop (Adobe Systems Inc., San Jose, CA). Appropriate control sections were included in each run.
Western Blotting
Chopped spinal cord tissue was homogenized for 25 seconds in cold lysis buffer consisting of: 20 mmol/L Tris/acetate (pH 7), 1 mmol/L EGTA, 10 mmol/L sodium β-glycerophosphate (pH 7.4), 1 mmol/L sodium orthovanadate, 5% glycerol, 1% Triton X-100, and 0.27 mol/L sucrose with 1 μmol/L microcystin LR, 1 mmol/L benzamidine, 4 μg/ml leupeptin, and 0.1% β-mercaptoethanol (Bio-Rad, UK). The tissue suspension was centrifuged at 1500 rpm at 4°C and the supernatant and pellet separated. Supernatant protein was assayed using the Lowry method with bovine serum albumin as a standard.
Fifty μg of the supernatant protein including sodium dodecyl sulfate containing sample buffer was run on either 7.5% (Bio-Rad), 12%, or 16% (both from Autogen Bioclear, Wiltshire, UK) Tris-glycine precast gels and then transferred onto Immobilon P polyvinylidene difluoride membrane (Millipore, Bedford, MA). The membrane was blocked in 2% bovine serum albumin in Tween-Tris-buffered saline (T-TBS) and incubated with secondary antibody for 2 hours diluted 1:2000 in 2% bovine serum albumin T-TBS. Membranes were washed in T-TBS before incubating with horseradish peroxidase-labeled anti-mouse or rabbit IgG. After further washes in T-TBS, membranes were developed using an enhanced chemiluminescent kit (Amersham, Buckingham, UK). For control experiments, caspase-1 or -3 antibody was preblocked as described above before applying onto Western blots.
Detection of Active Caspases by CaspaTag
CaspaTag detection kits (Intergen Company, Oxford, UK) were used to detect activated caspase-1 and -3 in sections from experimental animals 23 according to the manufacturer’s instructions. Sections were incubated with the appropriate detection kit in a humidified chamber at 37°C for 1 hour in the dark. Sections were washed three times in PBS followed by fixation for 10 minutes and two further washes in PBS. Sections were then counterstained with Hoechst 33342 to indicate apoptotic cells and viewed under a fluorescent microscope at 490 excitation to observe green fluorescence of caspase-positive cells and a UV filter to view Hoechst staining.
The number of CaspaTag-positive cells in spinal cord sections from Lewis rats were counted using a 100-μm2 graticule in five different random fields for each section. A total of two sections from three animals in each group were counted and the results expressed as the mean ± SD. The results were analyzed statistically using GraphPad Prism software (GraphPad Prism, San Diego, CA). Sample means were analyzed using a one-way analysis of variance with Bonferroni’s post hoc testing.
Detection of Fragmented DNA in Cells Indicative of Apoptosis
The Fluorescein-FragEL (QIA-39; Oncogene Research Products, San Diego, CA) system was used to evaluate DNA fragmentation in apoptotic cells according to the manufacturer’s instructions. Briefly, sections were fixed in 4% formaldehyde, permeabilized with proteinase K, and incubated with a fluorescent-labeled terminal deoxynucleotidyl transferase. Finally, sections were washed and mounted using the media provided and viewed under a confocal laser-scanning microscope (Bio-Rad).
Results
Disease Course and Pathology
The samples for this study came from the same spinal cord tissue used in the previous report of IL-12-induced relapses in the Lewis rat. 2 Tissues from the representative acute and the three subsequent relapses of EAE were used in this study. The clinical course and pathology of CREAE in the ABH mouse has been described. 3 Here we selected animals during the peak of acute EAE, after remission from acute attack, first relapse, and atypical chronic stages.
Caspase-1 Immunostaining
Caspase-1 immunostaining in Lewis rats was localized to a few cells in the spinal cord of control rats (not shown), none of which were positive by the CaspaTag-1 for active caspase-1. In acute EAE, caspase-1-positive cells were restricted to inflammatory cuffs (Figure 1a) ▶ with some also staining with CaspaTag-1 (Figure 1b) ▶ . During IL-12-induced relapses, numbers of both immunopositive and CaspaTag-1-positive cells were localized in inflammatory cuffs as well as individual cells in the parenchyma, peaking during the second relapse (Figure 1, c and d ▶ , respectively, and Figure 2 ▶ ). During the second and third relapse, immunopositive astrocytes were localized (Figure 1e) ▶ in close proximity to the inflammatory cuffs (Figure 1f) ▶ and also further into the parenchyma, whereas caspase-1-positive neurons were observed in third relapse animals (Figure 1g) ▶ .
Figure 1.
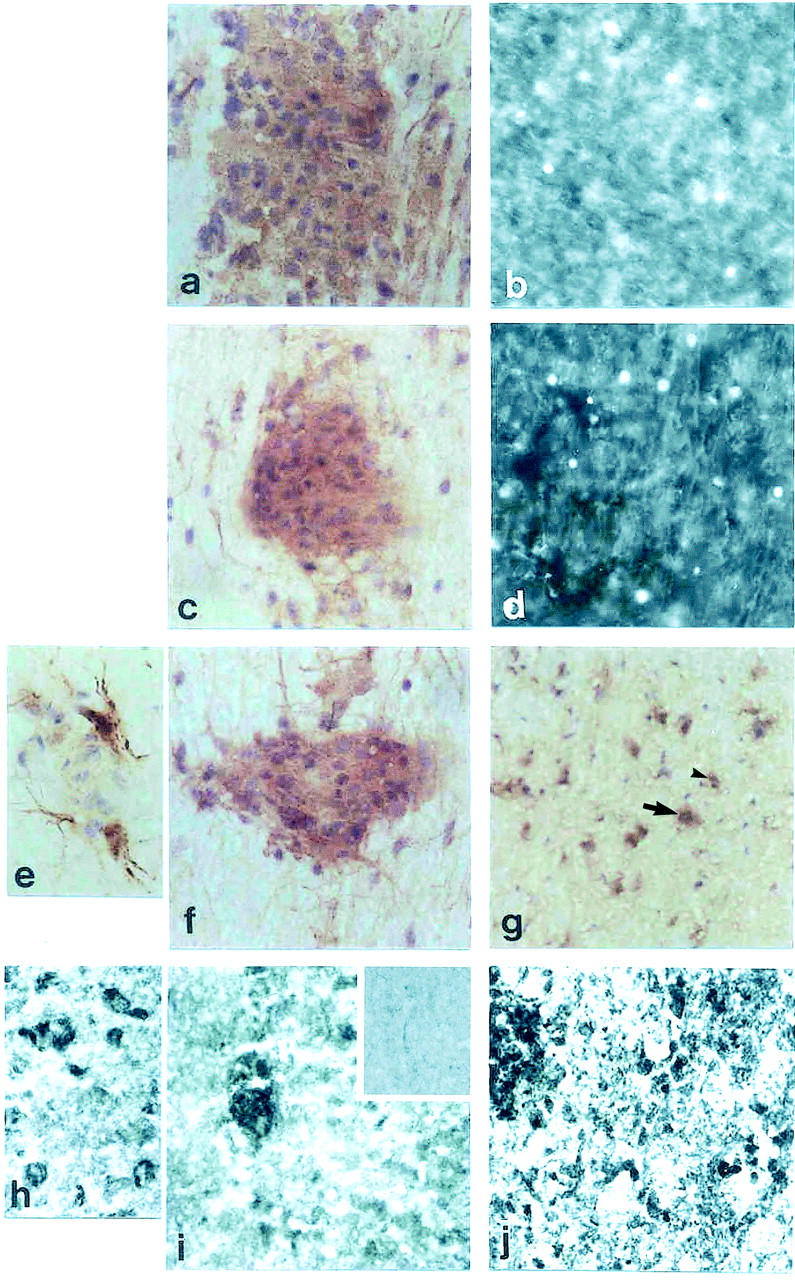
Caspase and CaspaTag-1 immunostaining. a: Spinal cord section from Lewis rat acute EAE immunostained with caspase-1. b: CaspaTag-1 immunostaining in a serial section. c: First relapse. d: Caspatag-1 in a serial section. e: Caspase-1 immunostaining in parenchymal astrocytes. f: White matter inflammatory cuff. g: Gray matter neurons of third relapse animals. h: Caspase-1-positive neurons in gray matter of ABH mice. i: Inflammatory cuff in acute EAE. j: Immunostaining in first relapse animals. Original magnifications: ×500 (a–d, f, g, i, and j); ×1000 (e and h). Inset to i shows caspase-1-negative staining in control mice.
Figure 2.

Quantitation of the number of Caspatag-1 and -3+ cells in the spinal cord of Lewis rats during the relapses. ***, P < 0.0001 versus acute EAE.
In contrast, Caspase-1 immunostaining in CREAE mice was abundant even during the acute phase within inflammatory cells (Figure 1i) ▶ and neurons (Figure 1h) ▶ . Caspase-1-positive immunostaining was also observed on axonal surfaces predominantly during the relapse phase as well as neurons (arrow) and inflammatory cells (Figure 1j ▶ , arrowhead). CaspaTag-1 immunostaining for active caspase-1 activity was also strongly localized on axons and cells in all of the different phases studied, however it was difficult to distinguish between cellular and axonal staining (not shown). Weak caspase-1 and CaspaTag-1 immunostaining was also present in control ABH mice (not shown). Control sections were negative.
Caspase-3 Immunostaining
During acute EAE there were immunopositive cells in inflammatory cuffs (Figure 3a) ▶ with only a few of these cells positive for active caspase-3 in a serial section (Figure 3b) ▶ whereas very few cells were positive during the first and second relapse with caspase-3 and its active component (not shown). However, during the third relapse numerous microglial cells in white matter (Figure 3c) ▶ and neuronal cells in gray matter (Figure 3d) ▶ were stained positive with caspase-3 and CaspaTag-3, together with small sections of axons in white matter and gray matter (Figure 3, c and d ▶ , arrowheads). Control animals did not show any immunostaining for caspase or CaspaTag-3.
Figure 3.
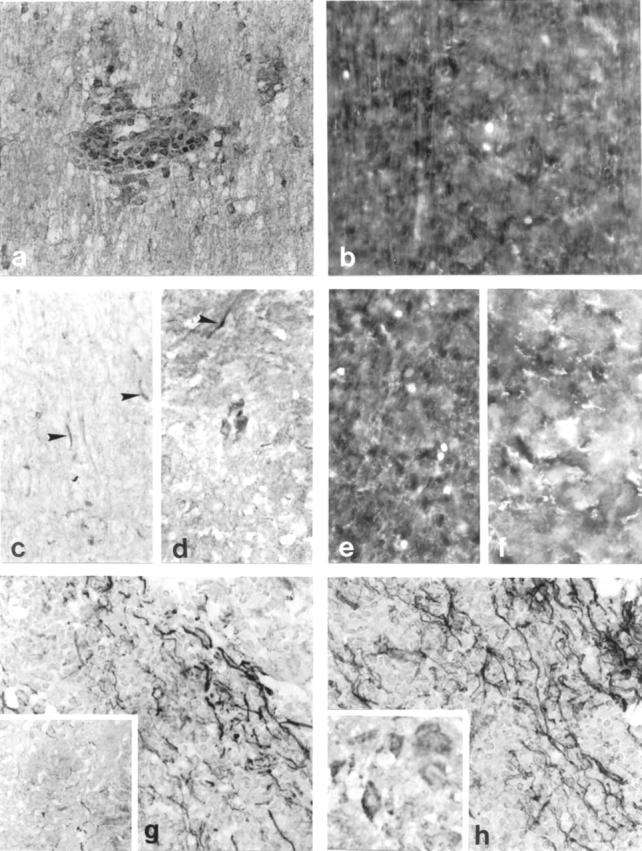
Caspase-3 immunostaining in Lewis rat spinal cords from EAE (a), third relapse white matter (c), and third relapse (d) gray matter neuronal cells and axons (arrowhead). The corresponding CaspaTag-3 immunostaining is shown in b, e, and f, respectively. g and h: Caspase-3-immunostained axons in ABH mice acute EAE and first relapse animals, respectively. Inset to h shows caspase-3-positive immunostaining in neurons in gray matter of ABH mice. Original magnifications: ×500 (a–h); ×1000 (inset). Inset to g shows caspase-3-positive immunostaining in control mice.
Caspase-3 immunostaining in ABH mice showed that small sections of axons were immunopositive even in control animals (Figure 3g ▶ , inset). Once disease was induced, many white matter axons were immunopositive for caspase-3 (Figure 3g) ▶ and CaspaTag-3 activity was also localized to these axons (not shown). After remission from acute EAE small sections of axons remained positive but by the first relapse phase, there was extensive detection of axonal and neuronal caspase-3 (Figure 3h ▶ and inset). Active caspase-1- and -3-positive cells could not be counted in ABH mice because there was such overwhelming positive immunostaining on axons such that it was difficult to decipher individual cells. In all CaspaTag-1- and -3-positive cells, bright pyknotic nuclei, indicative of apoptosis, were observed when sections were counterstained with Hoechst 33342 (not shown).
Double-immunofluorescent staining for activated caspase-3 and a range of cell markers showed similar cell-type localization from the spinal cord of Lewis rats in the third relapse and in ABH mouse EAE. In the Lewis rat, inflammatory cells including macrophages (Figure 4; a, b, and c) ▶ and neuronal cells (Figure 4, d and f) ▶ in the gray matter of the spinal cord were immunopositive for activated caspase-3. Similarly, inflammatory cells including CD4+ T cells (Figure 4, g and h) ▶ were immunopositive for activated caspase-3, localized in both in the parenchyma and in inflammatory cuffs in the spinal cord of the mouse, with the greatest amount of positive staining during the relapse stage of mouse EAE. Additionally there was prominent activated caspase-3 staining in axons (Figure 4; j, k, and l) ▶ and oligodendrocytes (Figure 4; m, n, and o) ▶ , particularly during the relapse stage in the mouse. Control spinal cord sections from rat and mouse were negative (see supplemental figures).
Figure 4.
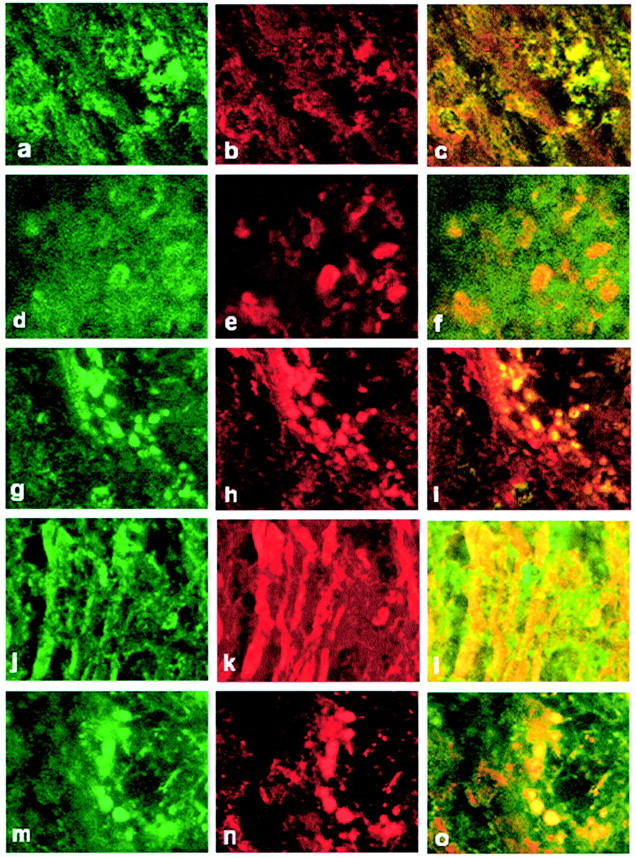
Phenotypic characterization of active caspase-3+ cells. a and d show double staining for active caspase-3 in ED-1+ macrophages (b) and NF+ neuronal cells (e) and c and f are merged images of a and b and d and e, respectively, in the spinal cord of Lewis rats from the third relapse. g, j, and m show activated caspase-3 in CD4+ T cells (h), NF+ axons (k), and CNPase+ oligodendrocytes (n). i, l, and o are merged images of g and h, j and k, and m and n, respectively. Original magnifications, ×1500. White matter in all panels except d–f.
Presence of Fragmented DNA within Cells Indicating Apoptosis
Fragmented DNA was present only during the third relapse in Lewis rat spinal cords localized within inflammatory (Figure 5a) ▶ and neuronal (Figure 5b) ▶ cells. However, many more inflammatory (Figure 5, c and e) ▶ and neuronal (Figure 5, d and e) ▶ cells with fragmented DNA were localized in the mouse spinal cord with the highest number present during the relapse stage of mouse EAE. Sections from the spinal cord of control mice (Figure 5g) ▶ showed rare weakly positive cells whereas control rat samples were totally negative (Figure 5h) ▶ .
Figure 5.
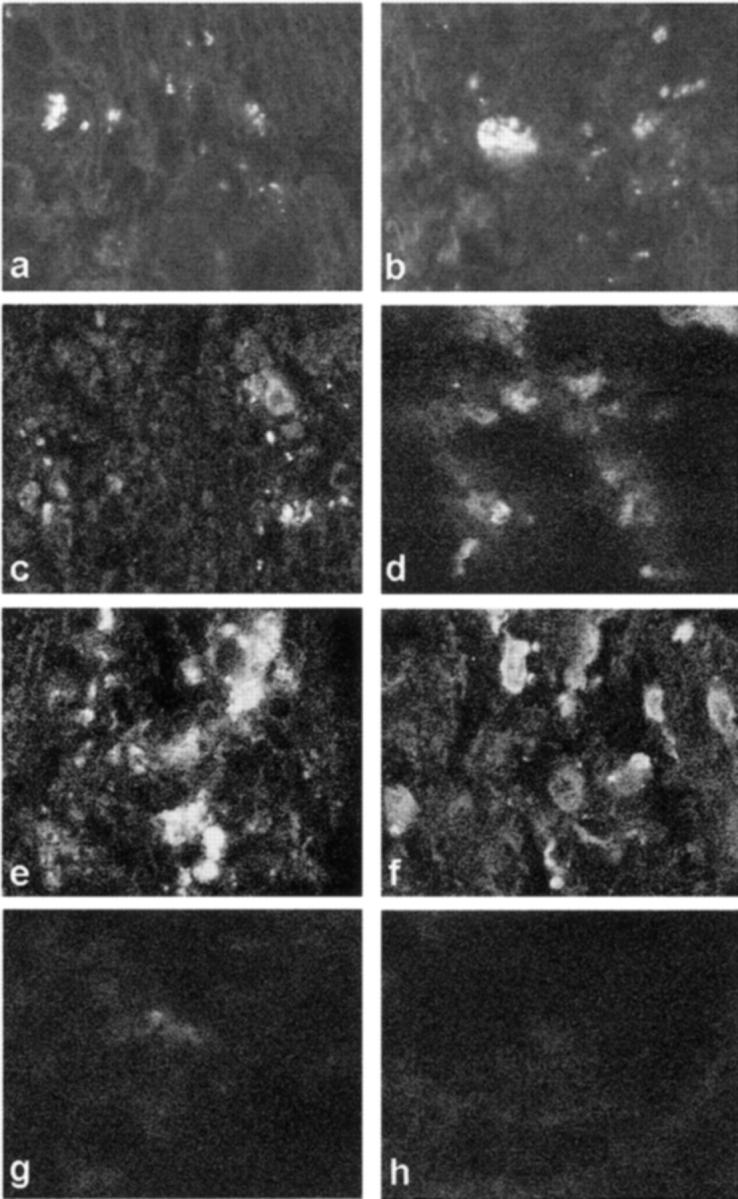
a, c, e, f, and h: White matter sections. b, d, and f: Gray matter section. a, b, and h: Lewis rat spinal cord. c–g: ABH mice. a, c, and e show fragmented DNA in inflammatory cells in white matter and b, d, and f show neuronal cells in gray matter from the spinal cords of third relapse Lewis rat and acute and relapse ABH mice, respectively. g and h: Negative control for DNA fragmentation in the spinal cord of normal animals. Original magnifications, ×1500.
Changes in Caspase-1 and -3 Protein in Lewis Rat and ABH Mouse
Immunohistochemical changes were reflected in Western blots of spinal cord samples during the different stages of EAE in the two models. Caspase-1 protein was induced during acute EAE in Lewis rats (Figure 6a) ▶ and increased further after the relapses peaking during the second relapse and finally decreasing during the third relapse. However, the greatest amount of caspase-3 together with 12-kd cleaved active caspase-3 protein was found during the third relapse in Lewis rats compared with any other stage of disease (Figure 6a) ▶ . Caspase-1 and caspase-3 proteins were undetectable in control Lewis rat spinal cords and peptide blocking eliminated staining in the test samples.
Figure 6.

a: Western Blot of caspase-1 and -3 protein expression during the relapses in Lewis rat EAE and a corresponding graph to show the relative amounts of protein in each band determined by gel analysis using ScionImage 4.0.2 (Scion Corporation, MD). b: Western blot in ABH mice and a graph to show the relative amounts of protein in each band. Actin blots of the same gels are shown for control of protein loading.
In contrast to the Lewis rat model, some caspase-1 and -3 protein was detected even in normal control ABH mouse spinal cord (Figure 6b) ▶ . However, the greatest amount of caspase-1 protein was found during acute EAE whereas that of caspase-3 protein and the 20- and 12-kd active caspase fragments was found during the first relapse (Figure 6b) ▶ .
Discussion
In this study, we have shown that caspase-1 protein and activity were induced in inflammatory cells during the initial acute episodes of EAE in both the Lewis rat (mainly inflammatory) and ABH mouse (mainly degenerative) models. By the third relapse in the Lewis rat, neuronal cells in the spinal cord were positive for caspase-1 whereas even during the acute phase in the ABH mouse, immunostained neurons were detectable that increased during relapses. In agreement with this the greatest amount of caspase activity was observed during the inflammatory stages of the disease (first and second relapse in the Lewis rat and the acute EAE stage in the ABH mouse). In contrast caspase-3 protein and activity did not increase significantly until the third relapse in the Lewis rat with immunostaining initially restricted to macrophages and T cells in inflammatory cuffs and extending to neuronal cells at later stages. However, in the ABH mouse, caspase-3 expression and activity was already high in acute EAE and during the first relapse, predominantly localized to axons and neuronal cells but also in oligodendrocytes.
These results are in conformity with the idea that caspase-1 has dual biological roles in that it can participate in the cellular induction of apoptosis as well as the processing of pro-IL-1β to the active form. 17,24 In our experiments, it is suggested that in the inflammatory model, caspase-1 may primarily function as a processor of pro-IL-1β because there was no evidence from our earlier study using electron microscopic criteria 2 of cellular or axonal death during first and second relapse. It has been reported that during maximum EAE severity in the mouse, expression of caspase-1 mRNA mirrors that of proinflammatory cytokines such as IL-1β, IL-6, tumor necrosis factor-α, and interferon-γ. 15 In the same study, a reduction of the incidence and severity of EAE was observed both in caspase-1-deficient mice (−/−) and with pharmacological blockers, suggesting a key involvement of the enzyme in the development of EAE. 15 However, the observation of caspase-1-positive astrocytes in second and third relapse of Lewis rats suggests that these cells undergo apoptosis during this stage of the disease. Furthermore, caspase-1-positive neurons in the spinal cord of third relapse animals in the Lewis rat and immediately after disease induction in the ABH mouse suggests that caspase-1 can contribute to apoptotic cell death by blocking DNA repair mechanisms. 16
The localization of caspase-3 in axons and neurons with increased enzyme activity in the third IL-12-induced relapse of EAE in the Lewis rat correlates with the limited axonal loss observed in our earlier study, 2 implicating a caspase-3-mediated mechanism. This is strengthened further by observations in the ABH mouse in which the highest amount of active caspase-3 was found in the spinal cord of relapse animals with immunostaining predominantly localized to axons and neurons but also in oligodendrocytes. This correlates well with a previous study in which axonal loss together with demyelination was found to be prominent during the first relapse compared to acute EAE. 3 Caspase-3 has been shown to be the major executioner caspase involved in neuronal apoptosis and in vitro when microglia are activated. 17,19,20 Procaspases are also found within axons and caspase-3 can be activated within the axon itself or transported from the neuronal cell body. 25,26 Activation of caspase-3 can occur by the release from mitochondria of cytochrome c causing hydrolysis of key structural and housekeeping genes which leads to DNA fragmentation and apoptosis. 8,13,14 The degenerative changes in axons through activation of caspase-3 can therefore be attributed to the cytochrome c release with consequent dysfunction of mitochondria that are distributed along axons as well as in the neuronal soma. 27,28 Although demyelination is predominantly executed by inflammatory macrophages, the detection of active caspase-3 oligodendrocytes in the ABH mouse suggests additional mechanisms of myelin breakdown.
The fundamental difference between the two models is that in EAE in the ABH mouse, extensive axonal pathology and demyelination are observed whereas myelin basic protein-induced EAE in Lewis rats is predominantly inflammatory. Even after three relapses of EAE induced by IL-12, only limited axonal death could be observed in the Lewis rat model. 2 ABH mice however, may be particularly susceptible to EAE because both caspase-1 and caspase-3 protein and immunostaining are already detected in control animals. This may represent a primed response system or enhanced gene transcription of these cell death proteins in the ABH mice such that in the early stages of EAE, abundant caspase-1 and -3 protein can be synthesized. Furthermore, the axonal localization of caspases in the ABH mouse during EAE reinforces the disease susceptibility in this strain, in which the production of caspases was many fold higher than in the Lewis rat.
In conclusion, the current study suggests that caspase-1 and caspase-3 can mediate axonal damage and cell death in EAE, especially in degenerative CREAE in the ABH mouse where axons are particularly susceptible. Caspase-3, being a downstream executioner of neuronal cell death could be an important target for specific caspase-3 inhibitors, reducing axonal damage and degeneration in multiple sclerosis and in EAE and is being investigated further.
Footnotes
Address reprint requests to Prof. L. Cuzner, Department of Neuroinflammation, Institute of Neurology, University College London, 1 Wakefield St., London, WC1N 1PJ, UK. E-mail: l.cuzner@ion.ucl.ac.uk.
Supported by the Brain Research Trust, UK.
References
- 1.Smith T, Hewson AK, Kingsley CI, Leonard JP, Cuzner ML: Interleukin-12 induces relapse in experimental allergic encephalomyelitis in the Lewis rat. Am J Pathol 1997, 150:1909-1917 [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed Z, Gveric D, Pryce G, Baker D, Leonard JP, Cuzner ML: Myelin/axonal pathology in interleukin-12 induced serial relapses of experimental allergic encephalomyelitis in the Lewis rat. Am J Pathol 2001, 158:2127-2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker D, O’Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL: Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J Neuroimmunol 1990, 28:261-270 [DOI] [PubMed] [Google Scholar]
- 4.Thornberry NA, Lazebnik Y: Caspases: enemies within. Science 1998, 281:1312-1316 [DOI] [PubMed] [Google Scholar]
- 5.Arends MJ, Wyllie AH: Apoptosis: mechanisms and roles in pathology. Int Rev Exp Pathol 1991, 32:223-254 [DOI] [PubMed] [Google Scholar]
- 6.Raff MC: Social controls on cell survival and cell death. Nature 1992, 356:397-400 [DOI] [PubMed] [Google Scholar]
- 7.MacFarlane M, Cain K, Sun XM, Alnemri ES, Cohen GM: Processing/activation of at least four interleukin-1beta converting enzyme-like proteases occurs during the execution phase of apoptosis in human monocytic tumor cells. J Cell Biol 1997, 137:469-479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen GM: Caspases: the executioners of apoptosis. Biochem J 1997, 326:1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ: Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol 1999, 144:281-292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu Y, Wu J, Faucheu C, Lalanne JL, Diu A, Livingston DJ, Su MS: Interleukin-1 beta converting enzyme requires oligomerization for activity of processed forms in vivo. EMBO J 1995, 14:1923-1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darmon AJ, Nicholson DW, Bleackley RC: Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 1995, 377:446-448 [DOI] [PubMed] [Google Scholar]
- 12.Walker NP, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR, Franklin S, Ghayur T, Hackett MC, Hammill LD: Crystal structure of the cysteine protease interleukin-1 beta-converting enzyme: a (p20/p10)2 homodimer. Cell 1994, 78:343-352 [DOI] [PubMed] [Google Scholar]
- 13.Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC: Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA 1996, 93:8395-8400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver VM, Carson CE, Walker PR, Chaly N, Lach B, Raymond Y, Brown DL, Sikorska M: Degradation of nuclear matrix and DNA cleavage in apoptotic thymocytes. J Cell Sci 1996, 109:45-56 [DOI] [PubMed] [Google Scholar]
- 15.Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, Desina G, Comi G, Flavell R, Su MS, Adorini L: Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol 1999, 163:2403-2409 [PubMed] [Google Scholar]
- 16.Kumar S: ICE-like proteases in apoptosis. Trends Biochem Sci 1995, 20:198-202 [DOI] [PubMed] [Google Scholar]
- 17.Kingham PJ, Cuzner ML, Pocock JM: Apoptotic pathways mobilized in microglia and neurones as a consequence of chromogranin A-induced microglial activation. J Neurochem 1999, 73:538-547 [DOI] [PubMed] [Google Scholar]
- 18.Kingham PJ, Pocock JM: Microglial apoptosis induced by chromogranin A is mediated by mitochondrial depolarization and the permeability transition but not by cytochrome c release. J Neurochem 2000, 74:1452-1462 [DOI] [PubMed] [Google Scholar]
- 19.Bredesen DE: Apoptosis: overview and signal transduction pathways. J Neurotrauma 2000, 17:801-810 [DOI] [PubMed] [Google Scholar]
- 20.Eldadah BA, Faden AI: Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma 2000, 17:811-829 [DOI] [PubMed] [Google Scholar]
- 21.Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, Layward L: Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature 2000, 404:84-87 [DOI] [PubMed] [Google Scholar]
- 22.Allen SJ, Baker D, O’Neill JK, Davison AN, Turk JL: Isolation and characterization of cells infiltrating the spinal cord during the course of chronic relapsing experimental allergic encephalomyelitis in the Biozzi AB/H mouse. Cell Immunol 1993, 146:335-350 [DOI] [PubMed] [Google Scholar]
- 23.Kingham PJ, Pocock JM: Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem 2001, 76:1475-1484 [DOI] [PubMed] [Google Scholar]
- 24.Schumann RR, Belka C, Reuter D, Lamping N, Kirschning CJ, Weber JR, Pfeil D: Lipopolysaccharide activates caspase-1 (interleukin-1-converting enzyme) in cultured monocytic and endothelial cells. Blood 1998, 91:577-584 [PubMed] [Google Scholar]
- 25.Krupinski J, Lopez E, Marti E, Ferrer I: Expression of caspases and their substrates in the rat model of focal cerebral ischemia. Neurobiol Dis 2000, 7:332-342 [DOI] [PubMed] [Google Scholar]
- 26.Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC: Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci 2000, 20:1333-1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Springer JE, Azbill RD, Knapp PE: Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med 1999, 5:943-946 [DOI] [PubMed] [Google Scholar]
- 28.Van Vliet BJ, Sebben M, Dumuis A, Gabrion J, Bockaert J, Pin JP: Endogenous amino acid release from cultured cerebellar neuronal cells: effect of tetanus toxin on glutamate release. J Neurochem 1989, 52:1229-1239 [DOI] [PubMed] [Google Scholar]