

Abstract
We describe the clinical, genetic, biochemical, and molecular characterization of a mouse that arose in the first generation (G1) of a random mutagenesis screen with the chemical mutagen ethyl-nitrosourea. The mouse was observed to have skeletal abnormalities inherited with an X-linked dominant pattern of inheritance. The causative mutation, named Skeletal abnormality 1 (Ska1), was shown to be a single base pair mutation in a splice donor site immediately following exon 8 of the Phex (phosphate-regulating gene with homologies to endopeptidases located on the X-chromosome) gene. This point mutation caused skipping of exon 8 from Phex mRNA, hypophosphatemia, and features of rickets. This experimentally induced phenotype mirrors the human condition X-linked hypophosphatemia; directly confirms the role of Phex in phosphate homeostasis, normal skeletal development, and rickets; and illustrates the power of mutagenesis in exploring animal models of human disease.
Rickets is a disorder of the developing skeleton, with major effects on the growth of endochondral-derived long bones. Osteomalacia occurs in bones that have undergone epiphyseal closure. Rickets and osteomalacia occur secondary to low concentrations of calcium or phosphate in the extracellular fluid, associated with an increase in the unmineralized extracellular matrix of bone. Vitamin D has a major role in calcium homeostasis and vitamin D deficiency remains a common cause of rickets. During skeletal growth, vitamin D deficiency leads to expansion and disordered arrangement of chondrocytes at the growth plate, because of failure of hypertrophic cartilage extracellular matrix to induce angiogenesis and calcification. Phosphate deficiency occurs less commonly, but produces a similar phenotype. 1
X-linked hypophosphatemia (XLH) is a dominantly inherited disease characterized by rickets and osteomalacia, dwarfism, hypophosphatemia, and elevated serum alkaline phosphatase. 2 The Phex (phosphate-regulating gene with homologies to endopeptidases on the X-chromosome) gene, which encodes a secreted endopeptidase, is mutated in XLH 3 and a number of different mutations have been found in different kindreds with XLH. 3-8 Defective Phex function causes a decrease in extracellular phosphorus and calcium levels, leading to reduced bone mineralization. Phex protein levels are highest in growing bone 9,10 and Phex is thought to act on a circulating substrate that represses phosphate resorption by the renal sodium phosphate transporter in the proximal convoluted tubule of the kidney. 11 Recent evidence suggests that a key substrate may be FGF-23. 12
A detailed understanding of complex biological processes such as bone development requires knowledge about the roles of individual genes. The advantage of using random mutagenesis and classical genetics to identify relevant genes involved is that no previous knowledge concerning gene function is required, or assumed. 13 As the genetically malleable organisms Drosophila melanogaster and Caenorrhabditis elegans do not undergo bone formation, genetic analysis of this process must be undertaken using fish or mice. Mice are attractive because much is already known about murine development, growth, and physiology. Many of the genes in the mouse are conserved in sequence, function, and relative location in humans. Thus, understanding murine gene function through analysis of mutations may allow inferences to be made about corresponding human genes and diseases. 13 Two strains of mutant mice harboring deletions that encompass part of the Phex gene, Hyp and Gy mice, have been described. We describe the phenotype of a third mouse harboring a mutation, termed Ska1, which arose during the course of mutagenesis experiments using ethyl-nitrosourea (ENU). Rather than harboring a large genetic deletion, as is the case in Gy and Hyp mice, Ska1 is a point mutation that results in aberrant splicing of the Phex gene, hypophosphatemia, and skeletal abnormalities consistent with rickets.
Materials and Methods
Mutagenesis
A single 250-mg/kg dose of ENU (Sigma Aldrich, St. Louis, MO, USA) was injected intravenously into 8-week-old male C57BL/6 mice as described previously. 14 Mice were left for 4 weeks to recover fertility and then crossed to untreated C57BL/6 females. G1 mice were screened for obvious external abnormalities. Animals with a phenotype of interest were mated to wild-type C57BL/6 mice to generate progeny, which were examined to ascertain whether the phenotype was heritable. Mutations of interest were maintained on an inbred C57BL/6 background.
Radiology
Mice were X-rayed using a Senographic DMR Specialized Mammography Unit (General Electrics, Fairfield, CT, USA) and Kodak MR2000–1 film (Eastman-Kodak, Rochester, NY) at a magnification factor of 1.8.
Skeletal Preparations and Histology
Mice were sacrificed and soft tissue was removed. The remaining tissue was dissolved in 1% KOH and 20% glycerol for 30 days. The skeleton was stained with alizarin red and stored in glycerol. To measure bones, skeletons were scanned using an Agfa 1236 scanner. The unmagnified image sizes were quantitated using Adobe Photoshop 5.5. The resulting data were analyzed using the statistical package “R” (http://www.r-project.org/).
For light microscopy, tissues from 8-week-old mice were fixed in 3.7% formalin/phosphate-buffered saline (PBS) (pH 7.4), and stored in fixative at 4°C. In some cases tissues were decalcified in 10% formalin/PBS (pH 7.4) containing 20% ethylenediaminetetraacetic acid. Paraffin blocks were prepared by standard histological procedures. Sections were cut and stained with Safranin O and fast green. For undecalcified sections, tibiae were collected at 8 weeks of age, fixed in 3.7% formaldehyde in PBS, and embedded in methylmethacrylate. Sections were then cut and stained by a modified von Kossa technique as described previously. 15
Serum Biochemistry
Blood was obtained from mice by axillary bleeding and allowed to clot for 1 to 2 hours. Serum was isolated by centrifugation at 340 × g for 5 minutes and stored at 4°C. Biochemical tests were performed by IDEXX Central Veterinary Diagnostic Laboratories, Mount Waverley, Victoria, Australia.
Mapping
A male C57BL/6 mouse carrying the Ska1 mutation was crossed to a wild-type BALB/C female. The resultant F1 females were backcrossed to BALB/C to produce 164 N2 generation mice. N2 mice were classed as affected or unaffected by their radiological phenotype. Mice were genotyped using polymorphic microsatellite markers located across the X-chromosome.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Poly(A+) RNA was isolated from mouse calvaria as described 16 and reverse-transcribed using an oligo d(T) primer (Promega Corporation, Madison, WI, USA) and avian myeloblastosis virus reverse transcriptase (Roche Molecular Biochemicals, Mannheim, Germany) under standard conditions. 17 A fragment encompassing Phex exons 1 to 10 was amplified from the cDNA using primers 5′-GAGACCAGCCACCAAACCACGAAA-3′ and 5′-GA-ATAAACCATTCTCCACACACTAAA-3′ and sequenced with the same primers using a BigDye Terminator kit (Applied Biosystems, Foster City, CA, USA). PCR was initiated by incubation of the reaction at 96°C for 2 minutes, followed by 30 cycles of incubation at 96°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute and was completed by incubation of the reaction at 72°C for 10 minutes.
Long-Range PCR
The Expand Long Template PCR System (Roche) was used according to the manufacturer’s instructions with primers 5′-GGTGGACACTGCTGTGCT-TTTAGGAGC-3′ and 5′-CCACATTCTCCGAGGGACCA-ATGTCTT-3′, complementary to Phex exons 7 and 9, respectively. PCR products were sequenced with primer 5′-TATGGAAATCCTATTGCACACACA-3′.
Southern Blotting
Primers 5′-CATGGTAGAGGAAAAGGGCACTCT-3′ and 5′-TATGGAAATCCTATTGCACACACA-3′, complementary to Phex intron 7 and intron 8, respectively, were used to amplify a 527-bp fragment of Phex from genomic DNA. This was subcloned and used as a radiolabeled probe on Southern blots of genomic DNA digested with Scr FI as described previously. 16
Results
An ENU-Induced, X-Linked Dominant Mutation Affecting the Skeleton in Ska1 Mice
In the course of conducting a random ENU mutagenesis screen, a female first-generation (G1) mouse was observed to have an abnormal gait, with bowing of the hind limbs (Figure 1) ▶ . The affected female mouse was mated to a wild-type C57BL/6 male and was observed to give rise to affected male and female progeny. The causative mutation in this pedigree was termed Skeletal abnormality 1 (Ska1). To establish the colony we mated affected females with wild-type males and reciprocally, affected males with wild-type females. During this process it became apparent that although affected females gave rise to approximately equal numbers of affected and unaffected male and female progeny, affected males sired only unaffected male and affected female progeny, suggesting that the skeletal abnormality was inherited in an X-linked dominant manner (Table 1) ▶ . Moreover, because affected males sired no unaffected females, we can conclude that the mutation is fully penetrant.
Figure 1.

Affected male mice (Ska1/Y) are distinguished from wild-type animals (+/Y) by the abnormal angulation of the hips and knees.
Table 1.
Phenotypes of Progeny Derived from Mating Affected and Unaffected Mice
| Parents | Progeny | ||||
|---|---|---|---|---|---|
| Unaffected | Affected | ||||
| Mother | Father | Female | Male | Female | Male |
| Affected | Unaffected | 34 | 28 | 41 | 27 |
| Unaffected | Affected | 0 | 28 | 38 | 0 |
To determine whether homozygous affected female (Ska1/Ska1) mice could be generated, obligate heterozygous affected female (Ska1/+) mice were mated with affected hemizygous (Ska1/Y) males. As expected, all female progeny and approximately half the male progeny of this cross were affected (data not shown). Of the female offspring, we predicted half to be homozygous for the Ska1 mutation. When crossed to unaffected males, 2 of these females generated more than 10 progeny, all of which were affected. The Ska1 mutation has since been maintained by mating homozygous females with hemizygous males and as expected all progeny are affected.
Ska1 Mutant Mice Are Hypophosphatemic and Display Skeletal Features of Rickets
To determine the basis for the abnormality in gait, male mice hemizygous for the Ska1 mutation (Ska1/Y), female mice heterozygous and homozygous for the Ska1 mutation and wild-type male and female mice were X-rayed. X-ray analyses of 8-week-old mice revealed that not only was the pelvis and acetabular angle of affected animals abnormal and likely to contribute to the gait of the mice, but other bones were also affected (Figure 2 ▶ ; Table 2 ▶ ). Notably X-rays showed that in all affected mice many bones appeared shorter than in wild-type mice and the vertebrae of affected mice were elliptical. In 30 to 50% of affected hemizygous male mice, heterozygous female mice and homozygous female mice, exostoses on the ribs were also observed.
Figure 2.

In comparison to wild-type females (left, +/+), X-rays reveal multiple skeletal abnormalities in the Ska1 heterozygous (middle, Ska1/+) and homozygous (right, Ska1/Ska1) female mice. These defects include shortened and distorted long bones in the hind limbs, rib exostoses at the costochondral junctions, a smaller pelvis with medial displacement of the acetabular cavity, cortical thinning, and reduced bone density in the pelvic bones and shaft of the proximal femur and elliptical tail vertebrae.
Table 2.
Prevalence of Radiological Phenotypes in Ska1 Mice
| Parameter | X-ray phenotype, fraction affected | ||||
|---|---|---|---|---|---|
| Males | Females | ||||
| +/Y | Ska1/Y | +/+ | Ska1/+ | Ska1/Ska1 | |
| Pelvis | 0/56 | 31/31 | 0/31 | 71/71 | 6/6 |
| Femoral shortening | 0/56 | 31/31 | 0/31 | 71/71 | 6/6 |
| Elliptical vertebrae | 0/56 | 31/31 | 0/31 | 71/71 | 6/6 |
| Rib exostoses | 0/56 | 16/31 | 0/31 | 22/71 | 2/6 |
To examine further the bone defects, cleared skeletons were prepared from affected and unaffected male and female mice and the length and/or width of nine bones were measured. One- and two-way linear models with the presence or absence of a mutant allele and sex as explanatory variables were fitted with analysis of variance. This was possible because of the normality of the data. Because of nonorthogonality of the experimental design the fitting order of the factors produced different fits. We chose the most parsimonious model in all cases. Ten of the 11 bone measurements were significantly shorter in mutant mice and appeared independent of sex. Only the mandible length was not significantly shorter. Scatterplots for the 11 variables show the tight correlations in most cases and also distinct clustering according to presence or absence of the mutant allele (Figure 3) ▶ . The tight correlations between most variables indicate that these bones are equally affected by the mutation. The obvious exception to this is the mandible that appears to be less significantly affected by the Ska1 mutation than other bones. These results are summarized in Table 3 ▶ .
Figure 3.
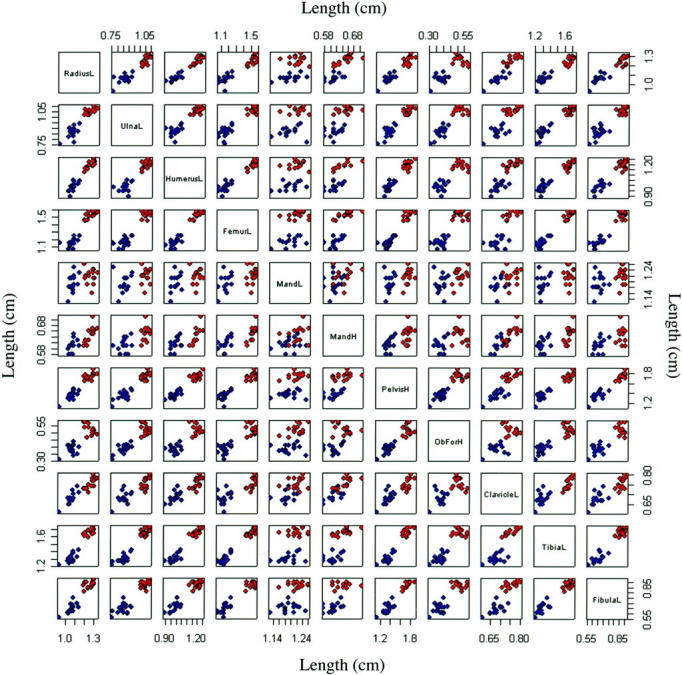
Comparison of the bone lengths from heterozygous females (Ska1/+) and hemizygous males (Ska1/Y) with their wild-type counterparts (+/+ and +/Y). Bone measurements were compared pairwise to determine whether they were correlated with each other depending on genotype (blue, affected Ska1/Y and Ska1/+ mice; red, unaffected +/Y and +/+ mice). The identity of the measurements is shown in the boxes on the diagonal (L, length; H, height; Mand, mandible; ObFor, obturator foramen.
Table 3.
Comparison of Bone Dimensions in Ska1 and Wild-Type Male and Female Mice
| Variable | Model | F test statistic | P value |
|---|---|---|---|
| Radius length | RadiusL = μ + Mutation + ε | 195.89 | 1.09e-14 |
| Ulna length | UlnaL = μ + Mutation + ε | 209.47 | 4.55e-15 |
| Tibia length | TibiaL = μ + Mutation + ε | 451.93 | <2.2e-16 |
| Fibula length | FibulaL = μ + Mutation + ε | 296.97 | <2.2e-16 |
| Femur length | FemurL = μ + Mutation + ε | 412.08 | <2.2e-16 |
| Clavicle length | ClavicleL = μ + Mutation + ε | 52.77 | 5.33e-08 |
| Mandible height | MandH = μ + Mutation + ε | 18.06 | 2.14e-04 |
| Mandible length | MandL = μ + ε | 6.98 | 0.01 |
| Obturator foramen height | ObForH = μ + Mutation + Sex + ε | 372.87 1 | <2.2e-16 |
| 54.25 2 | 5.08e-08 | ||
| Humerus length | HumerusL = μ + Mutation + ε | 212.57 | 1.32e-14 |
| Pelvis height | PelvisH = μ + Mutation + ε | 196.23 | 1.93e-14 |
Bones of affected mice (13 Ska1/Y males and 14 Ska1/ + females) and unaffected mice (5 +/Y males and 7 +/ + females) were measured. The data was analyzed using the statistical package R. All 11 variables were tested for normality and found to be adequately modeled with normal distributions (Figure 3) ▶ . One- and two-way linear models with the presence or absence of a mutant allele and sex as explanatory variables were fitted with analysis of variance, where μ represents the overall mean, and ε the error in the model. This was possible because of the normality of the data. Because of nonorthogonality of the experimental design the fitting order of the factors produced different fits. We chose the most parsimonious model in all cases. All testing was adjusted using a Bonferroni correction which is the most stringent adjustment, by assuming independence of tests, for 11 tests. This reduces the single test significance level from 0.05 to 0.0046. Ten of the 11 response variables had significant F tests after fitting the presence or absence of one parameter. The height of the obturator foramen also showed a significant sex effect and in this case the F statistic for the fitting of the mutation term 1 is shown above the F statistic measuring the significance of fitting the sex term. 2
A histological examination of decalcified and undecalcified bone sections was performed to examine the defects in Ska1 mice in greater detail. Examination of hematoxylin and eosin-stained sections of ribs from wild-type and affected Ska1 mice confirmed the presence of exostoses, which were initially observed radiologically (Figure 4) ▶ .
Figure 4.

Safranin O and Fast Green sections of the ribs of female homozygous (Ska1/Ska1) and wild-type mice (+/+) showing exostoses.
Undecalcified Von Kossa-stained sections from wild-type and affected mice (Figure 5, a and b) ▶ demonstrated characteristic features of hypophosphatemic rickets including increased width of unmineralized epiphyseal growth plate, reduced bone width in the central metaphysis, and increased trabecular bone volume in the proximal metaphysis. High-power images at the growth plate reveal delayed cartilage calcification in the hypertrophic zone and epiphyseal end of the growth plate in affected Ska1 mice compared to wild-type (Figure 5, c and d) ▶ , and a substantial increase in hypertrophic zone width in affected mice. This increase in hypertrophic zone width is consistent with a reduction in bone resorption, 18 also suggested by increased cartilage remnants within the secondary spongiosa in Ska1 mice (data not shown).
Figure 5.
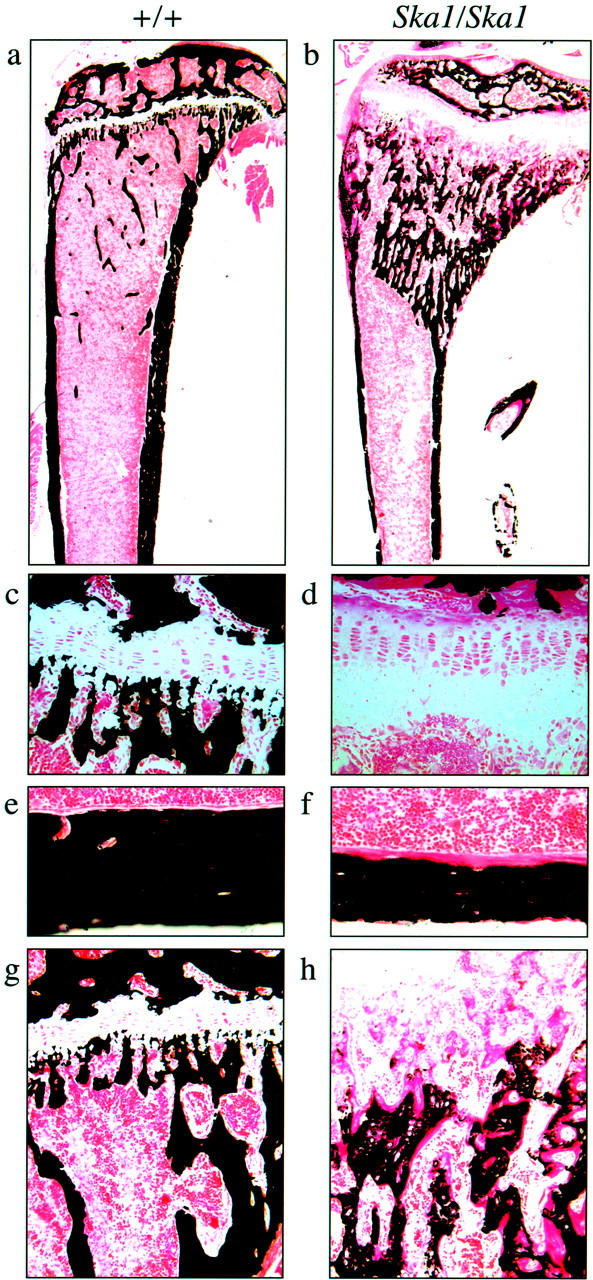
Von Kossa staining of undemineralized sections of the tibiae of homozygous (Ska1/Ska1) females (b, d, f, and h) in comparison to wild-type (+/+) females (a, c, e, and g), demonstrates an increase in the width of the hypertrophic zone (c, d), decreased thickness of the cortical bone (e, f), and delayed mineralization at the growth plate (g, h).
Cortical bone thickness was reduced in affected Ska1 mice, compared to wild-type mice, and osteoid width was dramatically increased (Figure 5, e and f) ▶ . The slow rate of osteoid calcification was also indicated by the irregular and low density of calcification at the mineralization front in affected Ska1 mice, compared with the clearly delineated mineralization front in wild-type mice. Although in wild-type mice cortical osteoid was covered with osteoblasts, cortical osteoid in Ska1 mice lacked coverage by osteoblasts, as previously observed in human biopsies. 19 Primary and secondary spongiosa in wild-type mice were densely mineralized and displayed a low percentage of osteoid surface with narrow osteoid seams. In contrast, mineralization was delayed in Ska1 mutant mice (Figure 5, g and h) ▶ . In Ska1 mice mineralization did not begin until after cartilage resorption, vascularization, and generation of marrow space had commenced at the growth plate. Mineralization progressed gradually, with only sparse calcification in the most proximal regions. Throughout this region, very wide osteoid seams were observed, indicating prolonged mineralization lag time. Chondrocytes and remnants of the growth plate were present in the mineralized trabeculae, indicating impaired osteoclastic bone resorption.
Serum phosphate, calcium, and alkaline phosphatase levels were compared in cohorts of wild-type mice and hemizygous, heterozygous, and homozygous affected Ska1 mice. All three genotypes of Ska1 mice had markedly reduced serum phosphate levels and markedly elevated serum alkaline-phosphatase levels (Table 4) ▶ . In addition, modest reductions in serum calcium were also observed. Comparison of these parameters between heterozygous and homozygous affected females revealed that although there was no significant difference in either serum calcium or phosphorous levels, the serum alkaline-phosphatase levels observed in homozygous females were significantly higher than those of heterozygous females.
Table 4.
Comparison of Serum Phosphate, Calcium, and Alkaline Phosphatase in Wild-Type (+/ + and +/Y), Hemizygous (Ska1/Y), Heterozygous (Ska1/ +), and Homozygous (Ska1/Ska1) Mice
| Sex | Genotype | Number | Calcium (mmol/L) mean ± SD | Phosphorous (mmol/L) mean ± sd | Alkaline phosphatase (IU/l) mean ± SD |
|---|---|---|---|---|---|
| Male | +/Y | n = 19 | 2.47 ± 0.09 | 2.33 ± 0.21 | 156 ± 23 |
| Male | Ska1/Y | n = 14 | 2.41 ± 0.11 | 1.41 ± 0.18* | 494 ± 221* |
| Female | +/ + | n = 18 | 2.47 ± 0.05 | 2.47 ± 0.05 | 192 ± 15 |
| Female | Ska1/ + | n = 7 | 2.34 ± 0.09 | 1.15 ± 0.22* | 338 ± 61* |
| Female | Ska1/Ska1 | n = 15 | 2.27 ± 016* | 1.21 ± 0.17* | 497 ± 121*† |
P values were calculated using the Student’s two-tailed t-test with unequal variance. Comparisons of serum calcium, phosphorous, and alkaline phosphatase levels were made between affected (Ska1/Y) and unaffected (+/Y) males, between affected heterozygous (Ska1/+) and unaffected (+/+) females and between homozygous (Ska1/Ska1) and heterozygous (Ska1/+) females. All testing was adjusted using a Bonferroni correction, which is the most stringent adjustment, by assuming independence of tests, for 12 tests. This reduces the single test significance level from 0.05 to 0.0041. For comparisons to unaffected mice * denotes P < 0.0041 and for comparisons between homozygous and heterozygous female mice † indicates P < 0.0041.
Ska1 Is an Allele of Phex that Affects Gene Splicing
The location of the causative mutation on the X-chromosome was mapped using a standard backcross. Mice (164 N2 generation) were genotyped for 19 microsatellite markers located across the X-chromosome. Informative recombinations narrowed the candidate interval containing the mutation to between DXMit98 (65.2 cM) and DXMit179 (65.5 cM) (Figure 6) ▶ . The Phex gene lies in this region of the genome and has been associated with a similar skeletal phenotype in Hyp and Gy mice. 20,21
Figure 6.

Maternal X-chromosome haplotypes of the four most informative affected male (A) and unaffected male (U) backcross (N2 generation) mice. ▪, C57BL/6 allele; □, Balb/C allele.
The Phex coding region was amplified by RT-PCR from mRNA isolated from the bone of Ska1 and wild-type mice and sequenced. This showed that 84 nucleotides, homologous to exon 8 in human Phex (GenBank accession number Y10196), were absent from the Phex mRNA in Ska1 mice (data not shown). A region extending across intron 8 of Phex was amplified from genomic DNA prepared from wild-type and Ska1 male mice. Sequencing of this product revealed the presence of a CG to TA point mutation at the first nucleotide pair of intron 8 (Figure 7) ▶ . This mutation destroys the splice donor sequence and likely causes skipping of the previous exon during pre-RNA splicing (Figure 7) ▶ . 22 This mutation also destroys an Scr FI site, allowing homozygous and heterozygous Ska1 mice to be distinguished from wild-type mice by Southern blotting (data not shown).
Figure 7.

The coding strand sequence around the Phex intron 8 splice donor site in (top left) wild-type and (bottom left) Ska1 mice. The CG→TA change destroys the canonical splice donor sequence leading to splicing out of exon 8 (compare top and bottom right panels).
Discussion
In this study we describe a novel, mutagen-induced allele of the Phex gene, termed Ska1. The PhexSka1 allele contains a point mutation in the splice donor site of intron 8 leading to an in-frame deletion of exon 8 from Phex mRNA. The deleted region is in the predicted extracellular domain of the Phex protein and encompasses a potential N-linked glycosylation site. 23 The loss of exon 8 from the human Phex gene, resulting in deletion of 28 identical amino acids, has been observed in an XLH patient [PHEX database (PHEXdb), PHEXdb Web Site, Montreal Children’s Hospital, Quebec, Canada (URL: http://www.data.mch. mcgill. ca/phexdb)], suggesting that this region is normally required for Phex function in both humans and mice.
In addition to a number of documented mutations in the human Phex gene, two other mouse mutants, Gy (Gyro) and Hyp (Hypophosphatemic), harbor mutations in Phex. Unlike the allele described in this study, the Gy and Hyp mutations are large deletions, which in Gy mice also extend to neighboring gene(s). In contrast to Ska1 mice, Gy mice exhibit behavioral abnormalities and male Gy mice are sterile and have reduced viability. 24 These nonskeletal effects suggest that deletion of gene(s) near Phex impacts on the phenotype of Gy mice. The skeletal abnormalities of all three strains appear to be similar and are presumably because of loss of function of the Phex gene. Like the hemizygous male mice, female mice homozygous for the PhexSka1 allele are viable and fertile, allowing a comparison with heterozygous females. All homozygous and heterozygous females examined had radiological evidence of disease and a similar proportion showed evidence of exostoses on the ribs. Serum biochemical analyses revealed that although phosphorous and calcium levels were comparable, alkaline phosphatase levels were significantly elevated in homozygous females compared with their heterozygous counterparts. This suggests that for some aspects of the phenotype, the PhexSka1 allele exerts a dominant effect, whereas for other aspects it exerts a semidominant effect. The relationship between phenotypic severity of disease and gene dosage may be explored further using PhexSka1 mice and will be aided by the generation of other Phex alleles by gene targeting and through random mutagenesis.
Given that Phex is a putative transmembrane protease expressed on osteoblasts and not directly involved in renal phosphate transport, the systemic effects of altered Phex function may result from failure to process a circulating substrate. A candidate substrate for Phex has recently been identified as FGF-23. 13 Mutations in FGF-23 prevent proteolytic cleavage by Phex and cause autosomal dominant hypophosphatemia. 13,25 Furthermore, oncogenic hypophosphatemic osteomalacia is very similar to XLH and appears to be caused by tumors that secrete FGF-23. 26 These observations suggest that increased net biological activity of FGF-23 and consequent renal phosphate wasting is the common link between XLH, oncogenic hypophosphatemic osteomalacia, and autosomal dominant hypophosphatemia. We have characterized an experimentally induced XLH phenotype and using a combination of classical genetics and gene mapping, identified a point mutation that disrupts Phex function by deleting exon 8. These findings directly confirm the role of Phex in phosphate homeostasis and rickets and will provide further insights into the molecular interactions between Phex, FGF-23, and phosphate regulation in skeletal development and bone disease.
Acknowledgments
We thank Phillip Wood of the Royal Melbourne Hospital for assistance with X-rays.
Footnotes
Address reprint requests to Dr. Douglas James Hilton, The Walter and Eliza Hall Institute of Medical Research, PO Royal Melbourne Hospital, Victoria 3050 Australia. E-mail: hilton@wehi.edu.au.
Supported by the Cooperative Research Centre for Cellular Growth Factors; the Cooperative Research Centre for Discovery of Genes for Common Human Diseases; the National Health and Medical Research Council of Australia; the Reid Family Trusts; the Anti-Cancer Council of Victoria, Melbourne, Australia; AMRAD Corporation Pty. Ltd., Melbourne, Australia; the J. D. and L. Harris Trust; and by an Undergraduate Research Opportunities Program Scholarship from the Cooperative Research Centre for Cellular Growth Factors and The Cooperative Research Centre for Discovery of Genes for Common Human Diseases (to M. R. C.).
References
- 1.Di Meglio LA, Econs MJ: Hypophosphatemic rickets. Rev Endocr Metab Disorders 2001, 2:165-173 [DOI] [PubMed] [Google Scholar]
- 2.Rasmussen H, Tenenhouse HS: Hypophosphatemias. Scriver C Beaudet A Sly W Valle D eds. The Metabolic Basis of Inherited Diseases. 1989:pp 2581-2604 McGraw-Hill, Baltimore
- 3.Francis F, Hennig S, Korn B, Reinhardt R, de Jong P, Poustka A, Lehrach H, Rowe PNS, Goulding JN, Summerfield T, Mountford R, et al: A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 1995, 11:130-136 [DOI] [PubMed] [Google Scholar]
- 4.Dixon P, Christie P, Wooding C, Trump D, Grieff M, Holm I, Gertner JM, Schmidtke J, Shah B, Shaw N, Smith C, Tau C, Schlessinger D, Whyte MP, Thakker RV: Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab 1998, 83:3615-3623 [DOI] [PubMed] [Google Scholar]
- 5.Filisetti D, Ostermann G, von Bredow M, Strom T, Filler G, Ehrich J, Pannetier S, Garnier JM, Rowe P, Francis F, Julienne A, Hanauer A, Econs MJ, Oudet C: Non-random distribution of mutations in the PHEX gene, and under-detected missense mutations at non-conserved residues. Eur J Human Genet 1999, 7:615-619 [DOI] [PubMed] [Google Scholar]
- 6.Holm IA, Huang X, Kunkel LM: Mutational analysis of the PEX gene in patients with X-linked hypophosphatemic rickets. Am J Hum Genet 1997, 60:790-797 [PMC free article] [PubMed] [Google Scholar]
- 7.Francis F, Strom TM, Hennig S, Boddrich A, Lorenz B, Brandau O, Mohnike KL, Cagnoli M, Steffens C, Klahes S, Borzym K, Pohl T, Oudet C, Econs MJ, Rowe PS, Reinhardt R, Meitinger T, Lehrach H: Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res 1997, 7:573-585 [DOI] [PubMed] [Google Scholar]
- 8.Rowe PS, Oudet CL, Francis F, Sinding C, Pannetier S, Econs MJ, Strom TM, Meitinger T, Garabedian M, David A, Macher MA, Questiaux E, Popowska E, Pronicka E, Read AP, Mokrzycki A, Glorieux FH, Drezner MK, Hanauer A, Lehrach H, Goulding JN, O’Riordan JL: Distribution of mutations in the PEX gene in families with X-linked hypophosphataemic rickets (HYP). Hum Mol Genet 1997, 6:539-549 [DOI] [PubMed] [Google Scholar]
- 9.Frota Ruchon A, Tenenhouse HS, Marcinkiewicz M, Siegfried G, Aubin JE, DesGroseillers L, Crine P, Boileau G: Developmental expression and tissue distribution of Phex protein: effect of the Hyp mutation and relationship to bone markers. J Bone Miner Res 2000, 15:1440-1450 [DOI] [PubMed] [Google Scholar]
- 10.Frota Ruchon AF, Marcinkiewicz M, Siegfried G, Tenenhouse HS, Des Groseillers L, Crine P, Boileau G: Pex mRNA is localized in developing mouse osteoblasts and odontoblasts. J Histochem Cytochem 1998, 46:459-468 [DOI] [PubMed] [Google Scholar]
- 11.Tenenhouse H, Werner A, Biber J, Ma S, Martel J, Roy S, Murer H: Renal Na(+)-phosphate cotransport in murine X-linked hypophosphatemic rickets. J Clin Invest 1994, 93:671-676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowe AE, Finnegan R, Jan de Beur SM, Cho J, Levine MA, Kumar R, Schiavi SC: FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Comm 2001, 284:977-981 [DOI] [PubMed] [Google Scholar]
- 13.Justice MJ, Zheng B, Woychik RP, Bradley A: Using targeted large deletions and high-efficiency N-ethyl-N-nitrosourea mutagenesis for functional analyses of the mammalian genome. Methods: a Companion to Methods Enzymol 1997, 13:423-436 [DOI] [PubMed] [Google Scholar]
- 14.Bode VC: Ethylnitrosourea mutagenesis and the isolation of mutant alleles for specific genes located in the T region of mouse chromosome 17. Genetics 1984, 108:457-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sims NA, Clement-Lacroix P, Da Ponte F, Bouali Y, Binart N, Moriggl R, Goffin V, Coschigano K, Gaillard-Kelly M, Kopchick J, Baron R, Kelly PA: Bone homeostasis in growth hormone receptor null mice is restored by IGF-1 treatment but independent of STAT5. J Clin Invest 2000, 106:1095-1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander WS, Metcalf D, Dunn AR: Point mutations within the dimer interface homology domain of c-Mpl induce constitutive receptor activity and tumorigenicity. EMBO J 1995, 14:5569-5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metcalf D, Willson TA, Hilton DJ, Di Rago L, Mifsud S: Production of hematopoietic regulatory factors in cultures of adult and fetal mouse organs: measurement by specific bioassays. Leukemia 1995, 9:1556-1564 [PubMed] [Google Scholar]
- 18.Gazit D, Tieder M, Liberman UA, Passi-Even L, Bab IA: Osteomalacia in hereditary hypophosphatemic rickets with hypercalciuria: a correlative clinical-histomorphometric study. J Clin Endocrinol Metab 1991, 72:229-235 [DOI] [PubMed] [Google Scholar]
- 19.Marie PJ, Glorieux FH: Stimulation of cortical bone mineralization and remodeling by phosphate and 1,25-dihydroxyvitamin D in vitamin D-resistant rickets. Metab Bone Dis Relat Res 1981, 3:159-164 [DOI] [PubMed] [Google Scholar]
- 20.Du L, Desbarats M, Cornibert S, Malo D, Ecarot B: Fine genetic mapping of the Hyp mutation on mouse chromosome X. Genomics 1996, 32:177-183 [DOI] [PubMed] [Google Scholar]
- 21.Meyer RA, Jr, Henley CM, Meyer MH, Morgan PL, McDonald AG, Mills C, Price DK: Partial deletion of both the spermine synthase gene and the Pex gene in the X-linked hypophosphatemic, gyro (Gy) mouse. Genomics 1998, 48:289-295 [DOI] [PubMed] [Google Scholar]
- 22.Berget SM: Exon recognition in vertebrate splicing. J Biol Chem 1995, 270:2411-2414 [DOI] [PubMed] [Google Scholar]
- 23.Turner AT, Tanzawa K: Mammalian membrane metallopeptidases: nEP, ECE, KELL, and PEX. EMBO J 1997, 11:355-364 [DOI] [PubMed] [Google Scholar]
- 24.Lyon MF, Scriver CR, Baker LR, Tenenhouse HS, Kronick J, Mandla S: The Gy mutation: another cause of X-linked hypophosphatemia in mouse. Proc Natl Acad Sci USA 1986, 83:4899-4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White KE, Evans WE, O’Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM: Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF-23. Nat Genet 2000, 26:345-34811062477 [Google Scholar]
- 26.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T: Cloning and characterization of FGF-23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 2001, 98:6500-6505 [DOI] [PMC free article] [PubMed] [Google Scholar]