

The pathogenesis of melanoma is beginning to be better understood due to a broad effort in defining clinically and pathohistologically the different stages of the disease. Cell culture and animal models have helped us to understand the dynamics of tumor progression. In recent years the field has begun systematic approaches to dissect the biological basis of melanoma. The molecular pathways and biological processes that are currently under investigation are expected to yield new markers for diagnosis and prognosis of melanoma, which currently solely rely on clinical and histopathological information. We also predict that the biological investigations will yield new target molecules for rational therapy. This review summarizes highlights of some of the most significant findings in recent years in melanoma biology (for additional reviews, see references 1-7 ).
Molecular Determinants in Melanoma Development and Progression
Mouse models of melanoma are beginning to help us understand the genetic and molecular events in melanoma. Lynda Chin summarized the work on the molecular pathways that are critical for melanoma and that have been dissected with mouse genetic models. The INK4a/ARF locus at the 9p21 chromosomal hotspot has a highly unusual genomic organization. It contains two upstream exons (1a and 1b) driven by separate promoters, splicing onto a common acceptor site of common downstream exons 2 and 3. Since the open reading frames used are different in the shared exon 2, two distinct protein products are encoded by this locus. One transcript is p16INK4a, a cyclin-dependent kinase inhibitor (INK4) that binds to cyclin-dependent kinases 4 and 6 (CDK4/6) and inhibits CDK4/6 phosphorylation of RB. The second transcript is p14ARF (or p19ARF in mouse), a negative regulator of cell growth that inhibits MDM2-mediated degradation of p53. Hence, the INK4a/ARF locus harbors two genes that impinge on the two major tumor suppression pathways, RB and p53. 7-9
A mouse model of cutaneous melanoma has been generated previously through the combined effects of Ink4a/Arf deficiency (null for p16INK4a and p19ARF) and melanocyte-specific expression of activated RAS (tyrosinase-driven H-RASV12G, Tyr-RAS). 10 In this model, it was demonstrated that loss of Ink4a/Arf function significantly reduces latency and increases incidence of melanoma development. Moreover, in the Ink4a/Arf heterozygous background, 100% of the melanomas that emerged sustained loss of heterozygosity (LOH) of the remaining wild-type allele, providing further genetic evidence for the requirement of Ink4a/Arf inactivation on the level of tumorigenesis. However, since the Ink4a/Arf locus encodes two distinct proteins, p16INK4a and p19ARF, both of which has been shown to demonstrate tumor suppressor activity in genetically distinct tumor suppressor pathways (ie, the “Rb pathway” for p16INK4a and the “p53 pathway” for p14ARF), the frequent loss of both products of the INK4a/ARF locus in melanoma raises the question as to which INK4a/ARF gene product functions to suppress melanomagenesis in vivo. The role of p16INK4a as a melanoma suppressor in humans is unequivocal. Most compelling is the presence of germline mutations that compromise p16INK4a, but preserve p14ARF function and confer a hereditary susceptibility to melanoma and pancreatic adenocarcinoma. 8 Until recently, germline mutations of exon 1b (targeting p14ARF only) have not been identified in melanoma-prone kindreds, 11,12 thus, raising a question regarding the importance of this axis of the p53 pathway in melanoma biology.
To address the relevance of the p19ARF-p53 axis in melanomagenesis, Chin and co-workers made use of the Tyr-RAS melanoma-prone transgenic allele to determine whether activated RAS could cooperate with p53 loss in melanomagenesis, whether such melanomas were biologically comparable to those arising in Ink4a/Arf−/− mice, and whether tumor-associated mutations emerged in the p16INK4a-RB pathway in such melanomas. It was found that p53 inactivation can cooperate with activated RAS to promote the development of cutaneous melanomas that are clinically indistinguishable from those of Ink4a/ARF−/− derived tumors. 13 As anticipated, p19ARF function is intact in these p53 mutant melanomas. Surprisingly, p16Ink4a was also found to be functionally intact. To uncover other potential RB-pathway lesions, the group used genome-wide screen with comparative genomic hybridization (CGH). The analyses revealed frequent gain of gene copy numbers on chromosome 15 in a region containing many interesting candidates, including Wnt1, Myc, PDGFβ, integrin α5, and integrin β7. Given the relative low resolution of conventional CGH, array-based CGH (aCGH) experiments were performed on these tumors using a genome-wide BAC arrays with 3 MB density coverage of the mouse genome (Spectral Genomics, Inc., Houston, TX). The aCGH profile of a RAS-induced p53 mutant melanoma identified a single BAC amplification. This BAC happened to contain the genomic sequence of a single gene, c-Myc, which was overexpressed in all p53 mutant melanomas from the model, 13 including those without c-Myc gene amplification. Given the known role of Myc in RB-regulated G1/S transition (see Ref. 13 and references therein). The observation suggested that Myc overexpression may serve as the RB-pathway lesion. Taken together, these data support the notion that dual inactivation of the RB and p53 pathways is important for melanomagenesis in the mouse.
To more specifically address the relative contribution of p16Ink4a and p19ARF in melanoma, the group next crossed the melanoma-prone Tyr-RAS transgenic mice onto p16INK4a and p19ARF specific KO. It was found that Tyr-RAS transgenic mice specifically deficient for either p16INK4a or p19ARF are prone to melanoma development with significantly shortened tumor latency compared to wild-type littermate controls (Figure 1) ▶ , demonstrating that loss of either p16INK4a or p19ARF in mice plays a causal role in development of melanoma. Preliminary molecular characterization of the RB and p53 pathways in p19ARF−/− or p16Ink4a−/− melanomas revealed evidence for their respective inactivation, further supporting the hypothesis that both RB and p53 pathways are important for melanoma suppression in vivo. Ongoing efforts in the laboratory focus on detailed molecular characterization of these melanomas, by genome-wide array-CGH approach as well as candidate gene survey.
Figure 1.

Relative importance of p16INK4a in melanoma. Transgenic mice for ras under the tyrosinase promoter were crossed with animals deficient for either p16INK4a or p19ARF and melanoma development was recorded over time.
Tumor Cell Plasticity Allows for Vasculogenic Mimicry by Aggressive Melanoma Tumor Cells
The concept of vasculogenic mimicry, presented by Mary Hendrix, describes the unique ability of aggressive melanoma cells to express vascular-associated genes and form tubular structures and patterned networks in three-dimensional culture. 14 This process, which mimics the pattern of embryonic vasculogenic networks, recapitulates the patterned networks seen in patients’ aggressive tumors. The functional significance of vasculogenic mimicry is unclear, but it may facilitate tumor perfusion and dissemination. A microarray analysis comparing aggressive and non-aggressive melanoma cells has yielded new markers associated with multiple phenotypes including the ability of aggressive melanoma cells to engage in vasculogenic mimicry and neovascularization. 15 One of the endothelial-specific genes expressed by aggressive melanoma cells with highest fidelity is vascular endothelial (VE)-cadherin (CD144 or cadherin 5). VE-cadherin is an adhesive protein, known to be expressed exclusively by endothelial cells, and belongs to the cadherin family of transmembrane proteins promoting homotypic cell-to-cell interaction. In a recent study, VE-cadherin was shown to be exclusively expressed by highly aggressive melanoma cells and was undetectable in the poorly aggressive cells, suggesting the possibility of a vasculogenic switch. 16 Based on the ability of aggressive melanoma cells to participate in vasculogenic mimicry, particularly their expression of endothelial-associated genes, the Hendrix group examined the plasticity of human metastatic cutaneous melanoma cells with respect to vascular function. Fluorescently labeled metastatic melanoma cells were challenged to an ischemic microenvironment surgically induced in the hindlimbs of nude mice. The data reveal the capability of these melanoma cells to express cell-fate determination molecules (belonging to the Notch family), normally expressed during embryonic vasculogenesis, and participate in the neovascularization of circulation-deficient muscle. 17 These results demonstrate the powerful influence of the microenvironment on the transendothelial differentiation of aggressive melanoma cells, and may provide new perspectives on tumor cell plasticity that could be exploited for novel therapeutic strategies.
Protein tyrosine kinases (PTKs) have been shown to play important and diverse roles in regulating cell adhesion, migration, and invasion. PTKs can be either transmembrane receptors for various growth factors or cytoplasmic kinases involved in relaying signals from the cell surface to its nucleus. Microarray analysis of aggressive melanoma cells compared with poorly aggressive melanoma cells revealed the differential expression of various protein tyrosine kinases. 15 One of the receptor PTKs that was up-regulated in the aggressive melanoma cells was EphA2 (Eck, epithelial cell kinase). Eph (ephrin-receptor) is comprised of a large family of 14 members. 18 Binding of ephrin-A1 (the EphA2 ligand) causes EphA2 to become phosphorylated; however, it has been found that EphA1 can also be constitutively phosphorylated in unstimulated cells. 19 EphA2 is not expressed in normal melanocytes but is often up-regulated in primary and metastatic melanomas. 20 However, the precise role of EphA2 in tumor progression remains enigmatic. Recent findings have shown the importance of EphA2 in melanoma cell vasculogenic mimicry. 21 In this study, EphA2 was expressed and phosphorylated exclusively in the highly aggressive melanoma cells, compared with no expression in the poorly aggressive cells. This expression profile is similar to the one observed for VE-cadherin. The localization of EphA2 in aggressive human melanoma tissues coincides with cell-cell contacts and is associated with VE-cadherin localization in patterned, vasculogenic-like networks. Transient knockout of EphA2 in vitro abrogated the ability of the highly aggressive melanoma cells to form vasculogenic-like networks. 21 Additional data suggest a possible interaction between EphA2 and VE-cadherin, assessed in immunoprecipitation experiments with cell lysates of highly aggressive melanoma cells. Based on the previous findings of VE-cadherin and EphA2 expression profiles in highly aggressive together with the immunoprecipitation data, the Hendrix group has proposed a hypothetical model for signaling during vasculogenic mimicry (Figure 2) ▶ . This model highlights possible cooperative interactions of VE-cadherin and EphA2, and suggests downstream signaling events involving PI3K and FAK, initiated by ephrin-A1 ligand binding. Collectively, these results suggest that VE-cadherin and EphA2 act together as a key regulatory element in the process of vasculogenic mimicry by aggressive melanoma tumor cells and illuminate a novel signaling pathway that could be potentially exploited for therapeutic intervention.
Figure 2.

Model for cooperation of VE-cadherin and EphA2 during vascular mimicry of melanoma cells. Hypothetical model for the regulation of EphA2 by VE-cadherin in aggressive melanoma cells. In this model, VE-cadherin association with other VE-cadherin molecules on adjacent cells facilitates the organization of EphA2, either by interacting directly or indirectly with EphA2, on the cell membrane. Once organized on the cell membrane, EphA2 is able to bind to its ligand, ephrin-A1, resulting in the phosphorylation of the receptor. Phosphorylated EphA2 can then bind to PI 3-kinase and lead to its activation. Furthermore, phosphorylated EphA2 can bind to phosphorylated FAK. The ability of EphA2 to interact with both FAK and PI 3-kinase may play an important role in the signaling pathways underlying melanoma cell vasculogenic mimicry. (Developed by A. Hess)
Optical Imaging for Visualization of the Dynamics of Pathological Changes in Melanoma
Recent advances in optical imaging modalities such as confocal and multiphoton scanning fluorescence microscopy, bioluminescence, optical coherence tomography, and spectral imaging have opened new avenues for visualizing and recording over time dynamic changes in genetic, developmental, and disease mechanisms that cannot be captured by conventional light microscopy. Using spectral imaging, Dorothea Becker reported on studies to demonstrate the feasibility of the technique to: 1) aid in the detection of melanoma evolving in atypical nevi, 2) capture gene expression profiles in melanocytic tissue specimens, and 3) visualize gene functions in melanoma xenografts in vivo.
Spectral imaging, defined as the application of spatially resolved spectroscopic analyses to macroscopic and microscopic samples, 22-24 allows a high-resolution spectrum, intensity as a function of wavelength, to be acquired at each pixel in an image. The result is an image cube that contains spectral as well as spatial information. The spectral information can be used to aid objective segmentation by classifying each pixel in an image according to its spectral signature. Instead of classifying each pixel absolutely, intensities can be modeled as a linear combination of spectra. Given a library of basic spectra, known to correspond to sample components, the percentage of each basic spectrum contributing to a pixel can be determined. Using these percentages and the total intensity of a pixel, the amount of each material from the spectral library in a specific pixel can be quantitatively measured and spatially resolved.
To determine whether macroscopic spectral imaging can detect in vivo melanoma at first presentation, the Becker group undertook a study in which 40 atypical nevi in patients with a clinical history of melanoma were subjected, noninvasively, to spectral imaging. Three of the 40 atypical nevi revealed spectrally segregated regions that were clearly distinct from the surrounding tissue. On surgical resection of the lesions, histological examination revealed that in two of them, the spectrally segregated areas corresponded to melanoma in situ, 25 and in one of them, to radial growth phase melanoma. 26
In contrast to primary vertical growth phase and metastatic melanomas, cells derived from atypical nevi, melanoma in situ, and radial growth phase melanoma are difficult to propagate in vitro. Thus, the only way to explore gene expression profiles in the early stages of melanoma development is by analysis of specimens. Regarding immunohistochemical and in situ hybridization analyses of tissue sections prepared from pigmented lesions, it is often difficult to detect a hybridization signal in cells producing melanin. Furthermore, the small size of nevocytic lesions and early-stage melanomas limits the number of sections that can be derived from these specimens and thus, the number of parameters that can be assessed. Microscopic spectral imaging can overcome these difficulties because it has the capacity to segment single cells, spectrally and spatio-temporally, localize and co-localize proteins and cellular features, count discrete objects on a cell-by-cell basis, and correlate expression of individual and multiple markers with tissue microarchitecture. Furthermore, microscopic Spectral imaging has the ability to determine, on a cell-by-cell basis, whether a protein resides in the cytoplasm or nucleus. 27 Using microscopic spectral imaging, it is currently possible to simultaneously visualize up to six different parameters in a 5-μm tissue section.
Gaining insights into the molecular and cellular mechanisms governing tumor angiogenesis has become one of the central themes in cancer biology, and the characterization of pro- and antiangiogenic molecules has led to a series of clinical trials to assess whether antiangiogenic treatment can be an effective strategy to restrict tumor growth. Given the recent advances in optical imaging combined with the application of cyanine-based fluorochromes that fluoresce in the near-infrared, 28,29 it has become possible to visualize noninvasively and in real time, dynamic changes in living biological systems that cannot be captured by conventional light microscopy. 30 Conducting noninvasive, dynamic fluorescence imaging of human melanoma xenografts, injected with human tyrosinase promoter-driven bFGF or FGFR-1 antisense vector constructs and fluorochrome-conjugated antibodies to a human melanoma and mouse blood vessel marker, data were presented that antisense targeting of bFGF and, likewise, FGFR-1 in only the melanoma cells is as effective in inhibiting tumor growth as blocking expression of bFGF and FGFR-1 simultaneously in the melanoma cells and the melanoma cell-interspersing blood vessels. In addition, the results of this macroscopic and microscopic cyanine fluorochrome-based optical imaging study provided first-time evidence that targeting bFGF and likewise, FGFR-1, in the melanoma cells induces extensive melanoma cell-specific apoptosis, a process the melanoma cells are unable to circumvent by activating or increasing expression of another growth factor/receptor.
Role of the Extracellular Matrix in Melanoma Proliferation
The switch from a low risk radial to high risk vertical growth phase primary melanoma is characterized by significant changes in the expression of molecules involved in cell-cell and cell-extracellular matrix (ECM) contact and in proteases. These changes typically involve a decrease in E-cadherin and increase in N-cadherin expression, 1 an increase in the expression of the integrins αvβ3 and α4β1 and the adhesion molecule MUC18/MCAM and an increase in the expression of several collagen degrading proteases, including matrix metalloproteinases (MMP). MMP consists of a large family of neutral endopeptidases with proteolytic activity for many proteins of the ECM but also for non-ECM proteins. 31,32 For a long time, the degradation of ECM proteins such as collagen by melanoma cells has been considered as a necessary step to remove a physical barrier to local invasion and metastasis. Yves DeClerk reported that proteolytic modification and remodeling of the ECM has three important consequences for tumor cell proliferation. First, because the ECM is a reservoir of many growth factors, its degradation has a positive effect on their bioavailability. 32,33 Secondly, the ECM is also a reservoir of biologically active peptides generated on proteolytic degradation of larger precursor molecules. For example, proteolytic processing of type XVIII collagen by MMP-12 generates endostatin, a 20-kd peptide inhibitor of angiogenesis. 34,35 Third, degradation of the ECM exposes cryptic binding domains in ECM proteins, which on contact with cell surface proteins affect cell growth and survival. For example, proteolytic degradation of collagen by melanoma cells generates new cryptic epitopes which on contact with the integrin αvβ3, protect cells from going into apoptosis. 26 The DeClerk laboratory has been interested in examining the molecular mechanisms by which ECM proteins can affect the proliferation of melanoma cells. Melanoma cells in which the collagenolytic activity has been suppressed by forced overexpression of a natural inhibitor of MMPs, TIMP-2, grew at a much slower rate than parental cells when plated in vitro in the presence of polymerized fibrillar collagen. In contrast, in the presence of denatured non-fibrillar collagen (gelatin), similar growth rates were observed between parent cells and TIMP-2 overexpressing cells, 37 suggesting that TIMP-2 inhibited tumor growth by its ability to prevent the degradation of fibrillar collagen in the tumor environment. On contact with of fibrillar collagen, melanoma cells are growth arrested with 90% of the cells being in G0/G1 phase in comparison to 51% when cells were in contact with gelatin. The inhibition of cell cycle entry on contact with fibrillar collagen was associated with an increase in p27KIP1 expression in the absence of changes in the expression of cyclin D1, cyclin E, Cdk2, p21WAF1/CIP1, and p57KIP2. 38 The increase in p27KIP1 resulted in an increased association with cyclin E and in a corresponding inhibition of cyclin E-associated kinase activity. This inhibitory effect was specific to type I collagen and was not observed in the presence of other ECM proteins such as laminin, fibronectin or vitronectin. 38 Treatment of melanoma cells plated in the presence of fibrillar collagen with bisindolylmaleimide I, an inhibitor of protein kinase C or Rottlerin, an inhibitor of protein kinase C δ, resulted in up-regulation of Skp2, with a corresponding down-regulation of p27KIP1 and inhibited entry into the cell cycle, without enhancing cell spreading. These data demonstrate that type I collagen has a profound effect on the expression of proteins that control cell cycle progression, Skp2 and p27KIP1 and that this effect depends on the physical structure of the protein (fibrillar versus non-fibrillar). When present in an intact fibrillar form, type I collagen exerts a negative effect on cell proliferation, whereas when denatured or proteolyzed, it has a positive effect. Degradation of fibrillar collagen by MMPs may therefore remove an important barrier not only against invasion but also proliferation (Figure 3) ▶ . The data also demonstrate that anchorage independent malignant cells remain responsive to growth regulatory signals that originate from their contact with the ECM.
Figure 3.

Collagen degradation and cell cycle progression. On contact with fibrillar collagen melanoma cells are growth inhibited, whereas they are stimulated on non-fibrillar collagen (gelatin). Activation of PKCδ by attachment to fibrillar collagen results in down-regulation of Skp2 and up-regulation of p27KIP1, whereas the effect is reversed when cells are cultured on denatured collagen (gelatin).
Proteoglycan/MMP Interactions in Melanoma Invasion
Membrane bound MMP (MT-MMPs) on the cell surface have a basic amino acid motif at the end of the propeptide that can be recognized and processed by furin or related proteinases to facilitate activation and cell surface expression. Among MT-MMPs, MT1-MMP is frequently expressed in invasive cancer cells. MT1-MMP directly degrades various ECM proteins such as collagens and fibronectin. It also activates other soluble MMPs such as MMP-2 and MMP-13. Thus, MT1-MMP is well placed to control pericellular proteolysis associated with cell growth and invasion by triggering the activation of multiple downstream proteases. MT1-MMP and MMP-2 are both up-regulated in human melanoma tissue samples, 39 and MT3-MMP and MT2-MMP have also been detected, 40 indicating that melanoma cells can overexpress multiple MT-MMPs for invasion. However, the potential importance of MT3-MMP in melanoma invasion and progression has not been previously documented or characterized.
James McCarthy and co-workers had previously shown that melanoma-associated chondroitin sulfate proteoglycan (MCSP), also termed high-molecular weight melanoma-associated antigen (MMW-MAA), is important for invasion through type I collagen and that it has gelatinolytic activity against denatured collagen. 41 MCSP activity has now been linked to MT3-MMP. 42 A construct encoding MT3-MMP in the antisense orientation could inhibit the invasion it stably transduced into cells. Inhibition of surface MT3-MMP expression led to correspondingly less gelatinolytic activity of melanoma cells, demonstrating that MT3-MMP could initiate gelatinolytic activity by melanoma cells. MT3-MMP and MCSP co-precipitated from detergent extracts of these cells. The association of these two molecules is chondroitin sulfate-dependent, since it was not observed in cells that had been treated to remove cell surface chondroitin sulfate, and recombinant MT3-MMP was shown to bind chondroitin sulfate affinity columns. The results suggest a model in which MCSP binds MT3-MMP on the cell surface, which leads to increased proteolysis of the ECM and tumor invasion (Figure 4) ▶ . It is based on a model for TIMP-2 mediated activation of proMMP-2 by MT1-MMP. 43 Since MMP-2 also binds chondroitin sulfate (J. McCarthy, unpublished observations), these data suggest that MCSP might act as a cell surface nucleation site for MT3-MMP and MMP-2, facilitating the activation of proMMP-2 by bringing it into proximity with MT3-MMP (Figure 4) ▶ . Whether or not TIMP-2 is important for this process remains an open question; however, the cells used in this study do express endogenous TIMP-2, suggesting that it may be involved in this mechanism. Understanding the mechanisms by which proteoglycans facilitate proteolysis will help to develop novel approaches for inhibiting MMP activation on melanoma cells, thereby limiting invasion and growth of the tumor.
Figure 4.

Model for MCSP mediated activation of MMPs on melanoma cells. Shown is a working model for how MCSP might act to stimulate melanoma invasion mediated by MT3-MMP. We have determined that MT3-MMP binds to MCSP via a CS-dependent mechanism. Based on other studies examining MT1-MMP mediated activation of pro-MMP-2, we propose that MCSP may act to assemble activation complexes of MT3-MMP, pro-MMP2 and possibly TIMP-2 on the cell surface, leading to activation of proMMP-2 and enhanced tumor invasion.
Apoptotic Signaling in Melanoma Cells
Failure of cells to undergo apoptotic cell death contributes to the pathogenesis of several cancers including melanoma. Work from Mark Nelson and co-workers demonstrate that deletion of chromosome band region 1p36 is frequent in malignant melanoma 44,45 and the Cdc2L locus encoding the PITSLRE protein kinases maps to 1p36.3. The PITSLRE protein kinases are part of the p34cdc2 family of cyclin dependent kinases and its cyclin partner appears to be Ania 6 (cyclin 1). 46 The Nelson group has shown that one allele of the Cdc2L locus on 1p36 is either deleted or translocated in melanoma cell lines 47 and decreased expression of the PITSLRE proteins has also been seen in melanoma cell lines and surgical specimens. Mutational analysis of the Cdc2L1 gene in germ line DNA samples from 1p36 linked melanoma kindreds and sporadic melanoma cell lines revealed that few genetic alterations occur within the coding region of the gene. However, polymorphisms in the putative promoter do occur. These data suggests that haploinsufficiency of Cdc2L locus may account for the decreased PITSLRE protein levels. 48
Diminution of PITSLRE kinases may also lead to deregulation of apoptosis in melanoma cells. The melanoma cell lines A375 (Cdc2L wild-type allele) and UACC 1227 (Cdc2L altered alleles) have a differential sensitivity to agonistic anti-Fas monoclonal antibodies. In A375 cells, cell death was evident as early as 24-hours post-treatment and maximal by 72 hours. On the other hand, UACC 1227 cells were resistant to Fas-mediated apoptosis. Induction of PITSLRE kinase activity was observed in A375 but not UACC cells. Also, PITSLRE protein kinase activity occurred before maximal morphological evidences of apoptosis. It has been reported that PITSLRE kinases are specifically cleaved in response to TNF by caspase 1 and 3 resulting in the activation of the PITSLRE kinase, both in vitro and in vivo. 49 The proteases responsible for processing and activation of PITSLRE kinase during apoptosis are the caspases 3 and 8. Caspase 3 and 8 inhibitors significantly reduced stimulation of PITSLRE kinase activity during Fas or staurosporine-mediated cell death and caspase inhibitors can block cleavage of PITSLRE. These results suggest that the PITSLRE protein kinases may be involved in apoptotic signaling in melanoma cells and that reduced levels of PITSLRE protein levels may result in resistance to apoptotic stimuli. 50 A key question still remains as to how exactly the PITSLRE protein kinases contribute to apoptosis. Current studies by the Nelson group suggests that caspase 3 cleavage of p110 PITSLRE results in generation of p60 and p46 PITSLRE fragments. The p46, which contains the kinase domain, appears to phosphorylate and regulate unknown downstream substrates, whereas the p60 PITSLRE fragment compromises other cellular functions. Thus, the PITSLRE protein kinases may be effectors in Fas- and staurosporine-induced apoptosis, acting at two distinct levels within a cell.
Conclusion
Progress in melanoma biology has been made in several areas that span from the role of oncogenes and tumor suppressor genes to the extracellular matrix. Although we still do not have unequivocal understanding of the genetic abnormalities that are the underlying cause of sporadic and familial melanoma, we now better understand molecular pathways related to cell growth. The p16INK4a gene may not structurally be altered in a high percentage of sporadic melanomas, but it is functionally not active. The p53 tumor suppressor gene is, similar to p16INK4a, structurally normal in most melanomas but not functionally active. 51 Apoptosis signaling can also be dysfunctional in melanoma, as studies on Apaf-1 52 have demonstrated. On the other hand, protein tyrosine kinase pathways that are activated in normal cells through growth factors are constitutively activated in metastatic cells through either autocrine or paracrine production of growth factors, 53 or through activating mutations in the B-Raf gene. 54 Thus, activation of critical pathways for melanoma cell survival, growth and invasion can occur through distinct interconnected mechanisms (Figure 5) ▶ . The PI3 and MAP kinase pathways appear critical for melanoma cells. The convergence of signaling from CAM molecules, integrins, and receptor tyrosine kinases expressed abundantly by melanoma cells through these pathways provide powerful stimulation for the malignant cells, particularly if B-Raf mutations in 70% of all melanomas are present.
Figure 5.
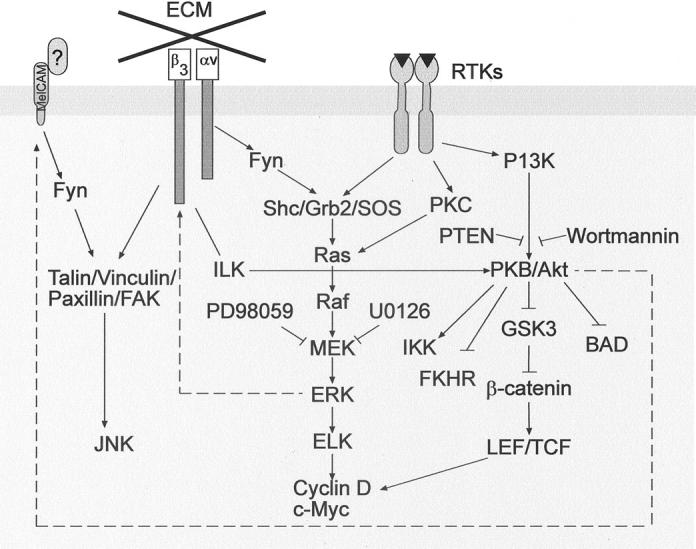
Convergence of signaling pathways. CAM, integrin, and receptor tyrosine kinase signaling can converge in melanoma cells. Mutations in B-Raf can lead to constitutive activation of the ERK/MAP kinase pathway. AKT activation may occur through PTKs or integrins, and there appears to be a signaling feedback to CAM. Broken lines indicate that intermediates are not yet known.
The plasticity of melanoma cells appears unique among all human tumors. The malignant cells share not only many markers with endothelial cells, fibroblasts and monocytes, they can also take over some of the differentiated functions of stromal cells. The remarkable ability of melanoma cells to invade distant organs and proliferate in a difficult environment suggests that the malignant cells can effectively interact with any scaffolding structures to which they are exposed. They not only attach to matrix, digest it, and then migrate over it, they create their own microenvironment in which matrix-degrading enzymes, adhesion receptors, and growth factor receptors combine to functional units with strong biological activities.
What are the “tender spots” of melanoma cells that provide vulnerability to antagonists? We can only speculate at this time and have to target each pathway individually to get a better understanding. We know already that keratinocytes can control melanoma cells when proper E-cadherin-mediated attachment is enforced. 55 Keratinocytes achieve their dominance by decreasing the expression of invasion-related molecules such as MUC18/Mel-CAM and αvβ3 vitronectin receptor. Such experiments suggest that a better understanding of the mechanisms of the normal homeostatic balance will help us to develop new strategies for therapy of metastatic disease.
Footnotes
Address reprint requests to Dr. Meenhard Herlyn, The Wistar Institute, 3601 Spruce Street, Philadelphia, PA 19104. E-mail: herlynm@wistar.upenn.edu.
The Workshop was held January 28, 2002 in Keystone, CO.
References
- 1.Herlyn M, Berking C, Li G, Satyamoorthy K: Lessons from melanocyte development for naevus and melanoma formation. Melanoma Res 2000, 10:303-312 [DOI] [PubMed] [Google Scholar]
- 2.Herlyn M, Satyamoorthy K: Molecular biology of cutaneous melanoma. DeVita VT, Jr Hellma S Rosenberg SA eds. Cancer: Principles and Practice of Oncology, ed 6 2000:pp 2003-2012 Lippincott Williams & Wilkins, Philadelphia
- 3.Li G, Satyamoorthy K, Herlyn M: Dynamics of cell interactions and communications during melanoma development. Crit Rev Oral Biol Med 2002, 13:62-70 [DOI] [PubMed] [Google Scholar]
- 4.Gruss C, Herlyn M: Role of cadherins and matrixins in melanoma. Curr Opin Oncol 2001, 13:117-123 [DOI] [PubMed] [Google Scholar]
- 5.Ruiter D, Bogenrieder T, Elder DE, Herlyn M: Melanoma-stroma interactions: structural and functional aspects. Lancet Oncol 2002, 3:35-43 [DOI] [PubMed] [Google Scholar]
- 6.Herlyn M: Emerging concepts and technologies in melanoma research. Melanoma Res 2002, 12:3-8 [DOI] [PubMed] [Google Scholar]
- 7.Satyamoorthy K, Herlyn M: Cellular and molecular biology of human melanoma. Cancer Biol Ther 2002, 1:14-17 [DOI] [PubMed] [Google Scholar]
- 8.Ruas M, Peters G: The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta 1998, 1378:F115-F177 [DOI] [PubMed] [Google Scholar]
- 9.Sharpless NE, DePinho RA: The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev 1999, 9:22-30 [DOI] [PubMed] [Google Scholar]
- 10.Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, Horner JW, DePinho RA: Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev 1997, 11:2822-2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Randerson-Moor JA, Harland M, Williams S, Cuthbert-Heavens D, Sheridan E, Aveyard J, Sibley K, Whitaker L, Knowles M, Bishop JN, Bishop DT: A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet 2001, 10:55-62 [DOI] [PubMed] [Google Scholar]
- 12.Rizos H, Puig S, Badenas C, Malvehy J, Darmanian AP, Jimenez L, Mila M, Kefford RF: A melanoma-associated germline mutation in exon 1beta inactivates p14ARF. Oncogene 2001, 20:5543-5547 [DOI] [PubMed] [Google Scholar]
- 13.Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, Chin L: Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol 2001, 21:2144-2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, Trent JM, Meltzer PS, Hendrix MJC: Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol 1999, 155:739-752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V, Hayward N, Trent J: Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406:536-540 [DOI] [PubMed] [Google Scholar]
- 16.Hendrix MJC, Seftor EA, Meltzer PS, Gardner LMG, Hess AR, Kirschmann DA, Schatteman GC, Seftor REB: Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci USA 2001, 98:8018-8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrix MJC, Seftor REB, Seftor EA, Gruman LM, Lee LML, Nickoloff BJ, Miele L, Sheriff DD, Schatteman GC: Transendothelial function of human metastatic melanoma cells: role of the microenvironment in cell-fate determination. Cancer Res 2002, 62:665-668 [PubMed] [Google Scholar]
- 18.Lindberg RA, Hunter T: cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol Cell Biol 1990, 10:6316-6324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao H, Burnett E, Kinch M, Simon E, Wang B: Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nature Cell Biol 2000, 2:62-69 [DOI] [PubMed] [Google Scholar]
- 20.Easty DJ, Ganz SE, Farr CJ, Lai C, Herlyn M, Bennett DC: Novel and known protein tyrosine kinases and their abnormal expression in human melanoma. J Invest Dermatol 1993, 101:579-584 [DOI] [PubMed] [Google Scholar]
- 21.Hess AR, Seftor EA, Gardner LMG, Carles-Kinch K, Schneider GB, Seftor REB, Kinch MS, Hendrix MJC: Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer Res 2001, 61:3250-3255 [PubMed] [Google Scholar]
- 22.Farkas DL, Baxter G, DeBasio RL, Gough A, Nederlof MA, Pane D, Pane J, Patek DR, Ryan KW, Taylor DL: Multimode light microscopy and the dynamics of molecules. Annu Rev Physiol 1993, 55:785-817 [DOI] [PubMed] [Google Scholar]
- 23.Farkas DL, Ballou BT, Fisher GW, Taylor DL: From in vitro to in vivo by dynamic multi-wavelength imaging. Proc SPIE 1995, 2386:138-149 [Google Scholar]
- 24.Farkas DL, Ballou BT, Fisher GW, Fishman D: Microscopic and mesoscopic spectral bioimaging. Proc SPIE 1996, 2678:200-209 [Google Scholar]
- 25.Yang P, Farkas DL, Kirkwood JM, Abernethy JL, Edington HD, Becker D: Macroscopic spectral imaging and gene expression analysis of the early stages of melanoma. Mol Med 1999, 5:785-794 [PMC free article] [PubMed] [Google Scholar]
- 26.Farkas DL, Becker D: Applications of spectral imaging for melanoma and its precursors. Hearing V eds. Pigment Cell Research. Review: Innovative Technology. 2001:pp 2-8 Munksgaard Publishers [DOI] [PubMed]
- 27.Kirkwood JM, Farkas DL, Chakraborty A, Dyer KF, Tweardy DJ, Abernethy JL, Edington HD, Donnelly SS, Becker D: Systemic interferon-α (IFN-α) treatment leads to Stat3 inactivation in melanoma precursor lesions. Mol Med 1999, 5:11-20 [PMC free article] [PubMed] [Google Scholar]
- 28.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS: Cyanine labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjugate Chem 1993, 4:105-108 [DOI] [PubMed] [Google Scholar]
- 29.Ballou B, Fisher GW, Farkas DL, Hakala TR: Tumor detection and visualization using cyanine fluorochrome-labeled antibodies. Biotechnol Prog 1997, 13:649-658 [DOI] [PubMed] [Google Scholar]
- 30.Farkas DL, Du C, Fisher GW, Lau C, Niu WN, Wachman ES, Levenson RM: Non-invasive image acquisition and advanced processing in optical bioimaging. Comput Med Imag Graphics 1998, 22:89-102 [DOI] [PubMed] [Google Scholar]
- 31.McCawley LJ, Matrisian LM: Matrix metalloproteinases: they’re not just for matrix anymore. Curr Opin Cell Biol 2001, 13:534-540 [DOI] [PubMed] [Google Scholar]
- 32.Chang C, Werb Z: The many faces of metalloproteases: cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol 2001, 11:S37-S43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin DC, Fowlkes JL, Babic B, Khokha R: Insulin-like growth factor II signaling in neoplastic proliferation is blocked by transgenic expression of the metalloproteinase inhibitor TIMP-1. J Cell Biol 1999, 146:881-892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Reilly MS, Wiederschain D, Stetler-Stevenson WG, Folkman J, Moses MA: Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem 1999, 274:29568-29571 [DOI] [PubMed] [Google Scholar]
- 35.Wen W, Moses MA, Wiederschain D, Arbiser JL, Folkman J: The generation of endostatin is mediated by elastase. Cancer Res 1999, 59:6052-6056 [PubMed] [Google Scholar]
- 36.Petitclerc E, Boutaud A, Prestayko A, Xu J, Sado Y, Ninomiya Y, Sarras MP, Hudson BG, Brooks PC: New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo J Biol Chem 2000, 275:8051-8061 [DOI] [PubMed] [Google Scholar]
- 37.Montgomery AM, Mueller BM, Reisfeld RA, Taylor SM, DeClerck YA: Effect of tissue inhibitor of the matrix metalloproteinases-2 expression on the growth and spontaneous metastasis of a human melanoma cell line. Cancer Res 1994, 54:5467-5473 [PubMed] [Google Scholar]
- 38.Henriet P, Zhong ZD, Brooks PC, Weinberg KI, DeClerck YA: Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27KIP1. Proc Natl Acad Sci USA 2000, 97:10026-10031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hofmann UB, Westphal JR, Zendman AJ, Becker JC, Ruiter DJ, van Muijen GN: Expression and activation of matrix metalloproteinase-2 (MMP-2) and its co-localization with membrane-type I matrix metalloproteinase (MT1-MMP) correlate with melanoma progression. J Pathol 2000, 191:245-256 [DOI] [PubMed] [Google Scholar]
- 40.Ohhashi Y, Tajima S, Ishibashi A: Coordinate expression of membrane type-matrix metalloriteinase-2 and -3 (MT2-MMP and MT3-MMP and matrix metalloproteinase-2 (MMP-2) in primary and metastatic melanoma cells. Eur J Dermatol 2001, 11:420-423 [PubMed] [Google Scholar]
- 41.Iida J, Meijne AM, Spiro RC, Roos E, Furcht LT, McCarthy JB: Melanoma chondroitin sulfate proteoglycan regulates matrix metalloproteinase-dependent human melanoma invasion into type I collagen. J Biol Chem 2001, 276:18786-18794 [DOI] [PubMed] [Google Scholar]
- 42.Iida J, Pei D, Kang T, Simpson MA, Herlyn M, Furcht LT, McCarthy JB: Melanoma chondroitin sulfate proteoglycan regulates matrix metalloproteinase-dependent human melanoma invasion into type I collagen. J Biol Chem 2001, 276:18786-18794 [DOI] [PubMed] [Google Scholar]
- 43.Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI: Mechanism of cell surface activation of 72 kDa type IV collagenase. J Biol Chem 1995, 270:5331-5338 [DOI] [PubMed] [Google Scholar]
- 44.Thompson FH, Emerson J, Olson S, Weinstein R, Leavitt SA, Leong SP, Emerson S, Trent JM, Nelson MA, Salmon SE: Cytogenetics of 158 patients with regional or disseminated melanoma. Subset analysis of near-diploid and simple karyotypes Cancer Genet Cytogenet 1995, 83:93-104 [DOI] [PubMed] [Google Scholar]
- 45.Nelson MA, Radmacher MD, Simon R, Aickin M, Yang J, Panda L, Emerson J, Roe D, Adair L, Thompson F, Bangert J, Leong SP, Taetle R, Salmon S, Trent JM: Chromosome abnormalities in malignant melanoma: clinical significance of nonrandom chromosome abnormalities in 206 cases. Cancer Genet Cytogenet 2000, 122:101-109 [DOI] [PubMed] [Google Scholar]
- 46.Berke JD, Sgambato V, Zhu PP, Lavoir B, Vincent M, Krause M, Hymen SE: Dopamine and glutamate induce distinct striatal splice forms of Ania-6, an RNA polymerase II-associated cyclin. Neuron 2000, 32:277-287 [DOI] [PubMed] [Google Scholar]
- 47.Nelson MA, Ariza ME, Yang J-M, Thompson FH, Taetle R, Trent JM, Wymer J, Massey-Brown K, Broome-Powell M, Easton J, Lahti JM, Kidd VJ: Abnormalities in the p34cdc2-related PITSLRE protein kinase gene complex (CDC2L) on chromosome band 1p36 in melanoma. Cancer Genet Cytogenet 1999, 108:91-99 [DOI] [PubMed] [Google Scholar]
- 48.Feng Y, Shi J, Goldstein AM, Tucker MA, Nelson MA: Analysis of mutations and identification of several polymorphisms in the putative promoter region of the p34cdc2-related Cdc2l1 gene located at 1p36 in melanoma cell lines and melanoma families. Int J Cancer 2002, 99:834-838 [DOI] [PubMed] [Google Scholar]
- 49.Bayaert R, Kidd VJ, Cornelis S, Van de Craen M, Denecker G, Lahti JM, Gururajan R, Vandenabeele P, Fiers W: Cleavage of PITSLRE kinases by ICE/CASP-1 and CPP32/CASP-3 during apoptosis induced by tumor necrosis factor. J Biol Chem 1997, 272:11694-11697 [DOI] [PubMed] [Google Scholar]
- 50.Ariza ME, Broome-Powell M, Lahti JM, Kidd VJ, Nelson MA: Fas-induced apoptosis in human malignant melanoma cell lines is associated with the activation of the p34(cdc2)-related PITSLRE protein kinases. J Biol Chem 1999, 274:28505-28513 [DOI] [PubMed] [Google Scholar]
- 51.Satyamoorthy K, Cheha NH, Waterman MJF, Lien MC, El-Deiry W, Herlyn M, Halazonetis TD: Aberrant regulation and function of wild-type p53 in radioresistant melanoma cells. Cell Growth Differ 2000, 11:467-474 [PubMed] [Google Scholar]
- 52.Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, Mccombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW: Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001, 409:207-211 [DOI] [PubMed] [Google Scholar]
- 53.Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M: IGF-1 induces survival and growth in biologically early melanoma cells through both the MAP kinase and β-catenin pathways. Cancer Res 2001, 61:7318-7324 [PubMed] [Google Scholar]
- 54.Davies H, Bignell GR, Co C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnet MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JWC, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow L, Paterson H, Marals R, Marshall CJ, Wooster R, Stratton MR, Futreal A: Mutations of the BRAF gene in human cancer. Nature 2002, 417:949-954 [DOI] [PubMed] [Google Scholar]
- 55.Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE, Herlyn M: E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol 2000, 156:1515-1525 [DOI] [PMC free article] [PubMed] [Google Scholar]