

Abstract
Angiotensin (Ang) II promotes renal infiltration by immunocompetent cells in double-transgenic rats (dTGRs) harboring both human renin and angiotensinogen genes. To elucidate disease mechanisms, we investigated whether or not dexamethasone (DEXA) immunosuppression ameliorates renal damage. Untreated dTGRs developed hypertension, renal damage, and 50% mortality at 7 weeks. DEXA reduced albuminuria, renal fibrosis, vascular reactive oxygen stress, and prevented mortality, independent of blood pressure. In dTGR kidneys, p22phox immunostaining co-localized with macrophages and partially with T cells. dTGR dendritic cells expressed major histocompatibility complex II and CD86, indicating maturation. DEXA suppressed major histocompatibility complex II+, CD86+, dendritic, and T-cell infiltration. In additional experiments, we treated dTGRs with mycophenolate mofetil to inhibit T- and B-cell proliferation. Reno-protective actions of mycophenolate mofetil and its effect on dendritic and T cells were similar to those obtained with DEXA. We next investigated whether or not Ang II directly promotes dendritic cell maturation in vitro. Ang II did not alter CD80, CD83, and MHC II expression, but increased CCR7 expression and cell migration. To explore the role of tumor necrosis factor (TNF)-α on dendritic cell maturation in vivo, we treated dTGRs with the soluble TNF-α receptor etanercept. This treatment had no effect on blood pressure, but decreased albuminuria, nuclear factor-κB activation, and infiltration of all immunocompetent cells. These data suggest that immunosuppression prevents dendritic cell maturation and T-cell infiltration in a nonimmune model of Ang II-induced renal damage. Ang II induces dendritic migration directly, whereas in vivo TNF-α is involved in dendritic cell infiltration and maturation. Thus, Ang II may initiate events leading to innate and acquired immune response.
Tubulointerstitial infiltration of macrophages and lymphocytes is a common feature in nonimmune renal diseases; however, the mechanisms involved are imperfectly defined. 1 An infiltration of macrophages and lymphocytes in the interstitium has been observed in angiotensin (Ang) II-induced hypertension and two-kidney one-clip hypertension that correlated approximately with the increase in blood pressure. 2,3 Possible mechanisms of how Ang II might promote the accumulation of immunocompetent cells include hemodynamic effects, stimulation of chemokines and adhesion molecules by direct effects on cells capable of releasing these mediators, stimulation of reactive oxygen species (ROS) production, or by immunological mechanisms that are as yet imperfectly defined. 1 Ang II induces the expression of adhesion molecules, activates the nuclear transcription factor kappa B (NF-κB), and stimulates the synthesis of monocyte chemoattractant protein 1 and osteopontin. 4-7 We have studied a primarily nonimmune double-transgenic rat model (dTGR) with increased circulating and local Ang II levels. The rats develop hypertension, severe inflammatory vascular damage, and die of renal and cardiac failure by age 7 weeks. We addressed the possibility of hemodynamic effects and found that blood-pressure lowering with hydralazine, hydrochlorothiazide, and reserpine delayed, but did not ameliorate, the disease. 8 In contrast, AT1 receptor blockade or inhibition of NF-κB markedly attenuated end-organ damage in this model. 9,10 To address the role of immune cells more directly, we have now used anti-immunocompetent cell strategies by treating the dTGRs with dexamethasone (DEXA), etanercept, or mycophenylate mofetil (MMF).
Materials and Methods
Experiments were conducted in 4-week-old male dTGRs and age-matched Sprague-Dawley (SD) rats after due approval. The DEXA dTGR group (n = 15), received DEXA for 3 weeks (0.5 mg/kg/day i.p.) whereas control dTGRs (n = 15) received vehicle. To investigate the role of tumor necrosis factor (TNF)-α on dendritic cell maturation and renal damage, we treated in an additional protocol dTGRs (n = 15) with the sTNF receptor, entanercept (1.25 mg/kg/day i.p.; kindly provided by Dr. K. Mohler, Immunex, Seattle, WA) for 20 days. Saline-treated dTGRs (n = 15) and nontransgenic SD rats (n = 10) were used as controls. To address the issue of T-cell and B-cell proliferation further, dTGRs (n = 9) were treated with MMF (40 mg/kg/day by gavage). Untreated dTGRs (n = 10) and SD rats (n = 10) received carboxymethylcellulose vehicle. Rats that died could not be used for histology because of autolysis and their data are not included in the analysis. Furthermore, blood and urine could not be obtained from these animals. Thus, the differences between the groups exhibiting mortality and those that did not are probably greater than we estimate. Urine was collected throughout a 24-hour period. Albumin was determined by enzyme-linked immunosorbent assay (CellTrend, Luckenwalde, Germany). Rats were killed at age 7 weeks. The kidneys were washed with ice cold saline, blotted dry, and weighed. For Western blot and NF-κB analysis, the tissues were snap-frozen in liquid nitrogen, for immunohistochemistry in isopentane (−35°C), and stored at −80°C.
Immunohistochemistry
Ice-cold acetone-fixed cryosections (6 μm) were stained by immunofluorescence or the alkaline phosphatase-anti-alkaline phosphatase technique as described earlier. 8-10 The sections were incubated with the following monoclonal antibodies: anti-CD4 (PharMingen, Germany), anti-ICAM-1, anti-ED-1, anti-ED-2, anti-CD8 (all Serotec, Germany), anti-CD86, anti-Ox62 and anti-Ox6 MHC II (all BD Pharmingen, Germany), anti-macrophage inhibitory factor (MIF) (R&D Systems, Germany) and polyclonal antibodies anti-fibronectin (Paesel, Germany), anti-collagen IV (Southern Biotechnology, Birmingham, AL), anti-interleukin (IL)-10 (R&D Systems, Germany), and anti-p22phox (generated by J Kreuzer). Preparations were analyzed under a Zeiss Axioplan-2 microscope (Carl Zeiss, Germany) and digitally photographed using the AxioVision 2 multi-channel image-processing system (Carl Zeiss, Germany). Semiquantitative scoring of infiltrated (ED-1+, CD4+, CD8+, Ox62+, Ox6+) cells in the kidney was performed using a computerized cell count program (KS 300 3.0, Zeiss, Germany). Fifteen different cortical areas of each kidney (n = 5 in all groups) were analyzed. For quantification of perivascular macrophage infiltration, all selected view fields included a small vessel in their analysis. Quantification of CD4+ T cells, and MHC II+ and CD86+ cells was performed periglomerularly, whereas CD8+ cells were quantified interstitially. The samples were examined without knowledge of the rat’s identity. Collagen IV and fibronectin expressions were assessed semiquantitatively by two independent observers, who were blind to the treatments. The data are expressed in arbitrary units (0 to 5) based on the staining intensity.
Western Blot Analysis
Western blot analysis was performed after standard procedure. Briefly, 30 μg of total kidney extract was incubated with the same primary antibodies used in immunohistochemical studies. Three different organs per group were analyzed in three independent blots and quantified. Human leukocyte extract was used as positive control for p22phox and rat pituitary gland extract for MIF.
Electrophoretic Mobility Shift Assay
Tissue preparation for electrophoretic mobility shift assay was performed as described earlier. 9 Nuclear extracts (5 μg) were incubated in binding reaction medium with 0.5 ng of 32P-dATP end-labeled oligonucleotide, containing the NF-κB binding site from the MHC-enhancer (H2K, 5′-gatcCAGGGCTGGGGATTCCCCATCTCCACA-GG). The DNA-protein complexes were analyzed on a 5% polyacrylamide gel and 0.5% Tris buffer, dried, and autoradiographed. Unlabeled H2K oligonucleotides (50 ng) were used in competition assays.
Oxidative Fluorescent Microtopography
The redox-sensitive fluorophore hydroethidine (Molecular Probes, Eugene, OR) was used to evaluate the production of O2.− in situ as described by Fleming and colleagues 11 Freshly cut cryosections (6 μm) were placed on glass slides and topically incubated with hydroethidine (5 μmol/L) for 30 minutes. After a 30-minute incubation period during which hydroethidine was oxidized to the fluorophore ethidium, images were obtained using a Zeiss Axioplan-2 microscope (Carl Zeiss) and digitally photographed using AxioVision 2 multi-channel image-processing system (Carl Zeiss).
Intracellular Redox State Assay
Intracellular ROS production was measured in living cells with flow cytometry. 12 Briefly, 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFH-DA) (Molecular Probes) is a nonpolar compound that is converted into a nonfluorescent polar derivative (H2DCFH) by cellular esterases after incorporation into cells. H2DCFH is rapidly oxidized to the highly fluorescent 2′,7′-dichlorofluorescein in the presence of intracellular hydrogen peroxide and peroxidases. U937 cells were grown in RPMI medium and incubated 30 minutes at 37°C with 5 μmol/L of H2DCF-DA in Hanks’ balanced salt solution-Hepes. Cells were stimulated and the fluorescence intensity was measured four times by flow cytometry (Cytofluorograph 50H; Ortho Diagnostics, USA). Cells (n = 10,000) were preincubated with the AT1 receptor blocker losartan (1 μmol/L), active and inactivated catalase (1000 U/ml), diphenylene iodonium (10 μmol/L), and DEXA (1 to 1000 nmol/L) where indicated and stimulated after 24 hours with Ang II (0.1 μmol/L). Fluorescent emissions at 510 to 550 nm were recorded.
In Vitro Generation of Dendritic Cells
Dendritic cells were obtained by in vitro differentiation from CD34+ stem cells of human cord blood. In brief, mononuclear cord blood cells were recovered by Ficoll-Hypaque gradient centrifugation (density, 1.077 g/ml; Eurobio, France) and subjected to immunomagnetic bead purification using the MACS system (Miltenyi Biotec, Germany). CD34+ cells were obtained by two cycles of selection and selected cells were analyzed by flow cytometry. Purity was 83 to 90%. CD34+ cells (0.3 to 0.5 × 106 cells/ml) were cultured in StemSpan serum-free medium (StemCell Technologies Inc., Canada) with 100 ng/ml of stem cell factor, 50 ng/ml of FLT3 ligand, 20 ng/ml of thrombopoietin, and 10 ng/ml of hyper-IL-6. 13 After 10 to 14 days culture, cells were induced to differentiate into dendritic cells in RPMI medium (0.5 × 106 cells/ml) supplemented with 10% fetal calf serum, 2 mmol/L l-glutamine, 0.1 mmol/L mercaptoethanol, 100 U/ml penicillin and streptomycin (GIBCO-BRL), 500 U/ml GM-CSF, and 500 U/ml IL-4 with or without Ang II (1 μmol/L, Sigma) for 6 days. TNF-α (10 ng/ml) was added for an additional 24 hours at day 6 to induce dendritic cell maturation. Alternatively, cells were simultaneously treated with 10 ng/ml of TNF-α and DEXA (1 μmol/L, Sigma) at day 6 for 24 hours. To monitor dendritic cell maturation, the expression of specific surface markers was analyzed by flow cytometry. 14 The following antibodies were used: CD1a (NA1/34), MHC class II HLA-DR (clone CR3/43, both from DAKO, Germany), CD34 (anti-HPCA-1, clone My10; Becton Dickinson, Germany), CD80 (B70/B7-2, clone MAB104), CD83 (HB15A, both from Immunotech, Germany), and CCR7 (kindly provided by Dr. M. Lipp and Dr. R. Förster, MDC, Germany). Data were processed and analyzed by CELLQuest software (Becton Dickinson, Germany).
Dendritic Cell Chemotaxis Assay
Chemotaxis assay was performed as described by Kellermann and colleagues 15 with minor modifications. Briefly, dendritic cells were incubated for 2 to 4 hours in serum-free RPMI medium with 500 U/ml GM-CSF and 500 U/ml IL-4. Before analysis, transwell inserts (5-μm pore size; Costar, UK) were preincubated in RPMI medium containing 1% bovine serum albumin, and 2 × 105 cells were seeded in the upper compartment in 100-μl RPMI medium and 1% bovine serum albumin. Ang II (10 μmol/L) or Epstein-Barr virus-induced molecule 1 ligand chemokine (100 ng/ml; Preprotech, London, UK) were added to the lower compartment to analyze migration toward gradient. Ninety minutes later 1 × 104 Dynabeads (15-μm diameter; Dynal Polymers AS, Lillestrom, Norway) were added to the lower compartment to normalize for variations in the experimental procedure. Cells were stained with fluorescein-di-acetate (5 μg/ml, Sigma). Cells and beads were then recovered and analyzed by flow cytometry. By gating on beads, the ratio beads:fluorescein-di-acetate-positive cells was determined and allowed a precise calculation of transmigrated cells.
Statistics
Data are presented as means ± SEM. Statistically significant differences in mean values were tested by analysis of variance, and blood pressure and albuminuria by repeated measures analysis of variance and the Scheffé test. Mortality was examined with a Kaplan-Meier analysis. A value of P < 0.05 was considered statistically significant. The data were analyzed using Statview statistical software.
Results
Dexamethasone Ameliorates Renal Damage Independent of Blood Pressure
In the vehicle dTGR group, 7 of 15 rats died before sacrifice, whereas in the DEXA group, the mortality was zero (P < 0.001). No SD rat died (Figure 1A) ▶ . This dramatic result was clearly independent of blood pressure, because DEXA did not lower blood pressure (n = 6 to 10; P = 0.95; Figure 1B ▶ ). dTGRs showed increased 24-hour albuminuria and plasma creatinine compared to SD rats, respectively. DEXA reduced albuminuria by 85% (n = 6 to 8; P < 0.01; Figure 1C ▶ ) and normalized creatinine (n = 6 to 8; P < 0.01; Figure 1D ▶ ). DEXA had dramatic effects on matrix protein deposition. Collagen IV (n = 5, Figure 2A ▶ ) and fibronectin (n = 5; data not shown) staining in DEXA-treated dTGRs were reduced almost to the levels observed in SD rats. Vehicle-treated dTGRs, on the other hand, showed prominent staining for these matrix proteins (grade 1+ for DEXA-treated dTGRs versus 5+ for vehicle-treated dTGRs versus 1+ for SD rats, respectively).
Figure 1.

DEXA reduced mortality and ameliorated end-organ damage independent of blood pressure in dTGRs. A: Vehicle-treated dTGRs showed >50% mortality at week 7. In contrast, no DEXA-treated dTGRs and nontransgenic SD rat died before sacrifice. B: Initially after 1 week of DEXA treatment, blood pressure increased significantly compared to vehicle-treated dTGRs. Thereafter, DEXA-treated and vehicle-treated dTGRs had similar blood pressure levels that were increased compared to SD rats. C and D: DTGR showed increased 24-hour albuminuria and plasma creatinine concentrations compared to SD rats, respectively. DEXA reduced albuminuria by 85% and normalized creatinine. Results are mean ± SEM (*, P < 0.05, dTGRs versus DEXA; #, P < 0.05, SD rats versus dTGRs and dTGRs + DEXA).
Figure 2.
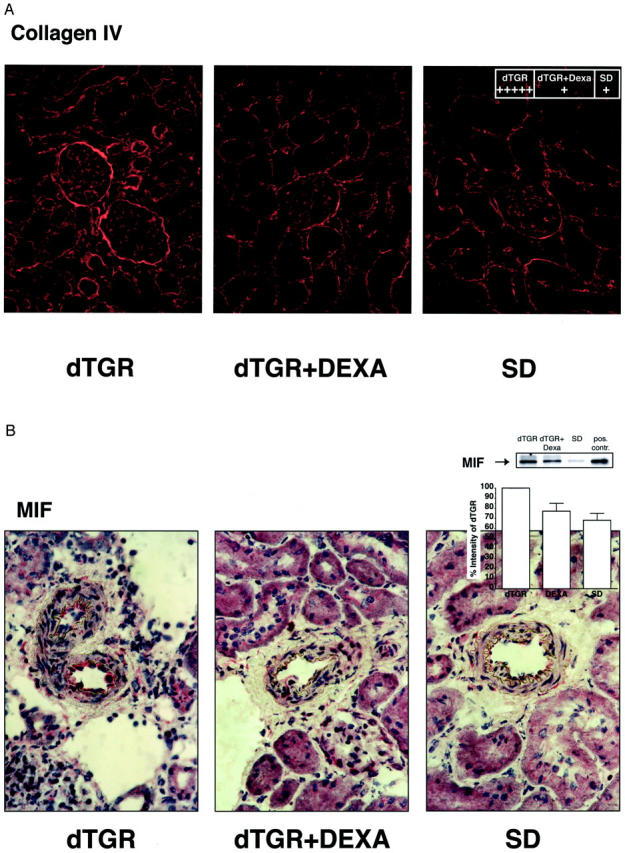
DEXA reduced renal fibrosis and MIF expression. A: Collagen IV in the glomerulus and glomerular basal membrane as well as peritubular area was markedly increased in dTGRs. B: MIF was markedly increased in the endothelium of damaged renal vessels as well as in infiltrated leukocytes. DEXA treatment reduced collagen IV, and MIF to control levels. Western blot of MIF from total kidney extracts confirmed the results. Rat pituitary gland extracts were used as a positive control (pos. contr.).
Dexamethasone Reduces MIF
Vehicle-treated dTGRs showed increased MIF immunostaining in the endothelium as well as in infiltrated cells. DEXA reduced endothelial MIF (n = 5, Figure 2B ▶ ) and MIF expression of perivascular-infiltrated cells. Low-dose DEXA is known to induce MIF in cell culture. Therefore, we also investigated the effect of Ang II and DEXA in U937 macrophages. However, we found no significant up-regulation of MIF mRNA after Ang II stimulation, whereas DEXA treatment slightly increased MIF mRNA levels (data not shown). TNF-α stimulation also slightly stimulated MIF (data not shown). Thus, we believe that reduced MIF immunostaining by DEXA treatment is most likely an indirect effect because of its potency in reducing cell infiltration.
Dexamethasone Reduces Renal NF-κB Activity
DEXA-treated dTGRs showed markedly decreased NF-κB activation, compared to vehicle-treated dTGRs (n = 3, Figure 3A ▶ ). Supershifts for p65 and p50 NF-κB subunits demonstrated the specificity. In vitro, NF-κB mediates the maturation of dendritic cells. Therefore, we performed co-localization studies for dendritic cells and p65. Renal sections show Ox62+ dendritic cells (circumferential green staining indicated by white arrows, Figure 3B ▶ ). A higher magnification (Figure 3C) ▶ demonstrated that dendritic cells (green) co-localized with nuclear p65 staining (red) marked by white arrows. Semiquantification showed that more than 80% of dendritic cells infiltrated in vehicle-treated dTGR kidneys expressed p65 (n = 4). In contrast, the very few infiltrated Ox62+ cells in DEXA-treated and nontransgenic rats did not express the NF-κB subunit. Nuclear p65 expression was also detected in tubules (T) and glomeruli (G) of vehicle-treated dTGRs (gray arrowheads, Figure 3C ▶ ). Nuclear p65 expression was markedly reduced by DEXA toward SD rat levels (data not shown).
Figure 3.
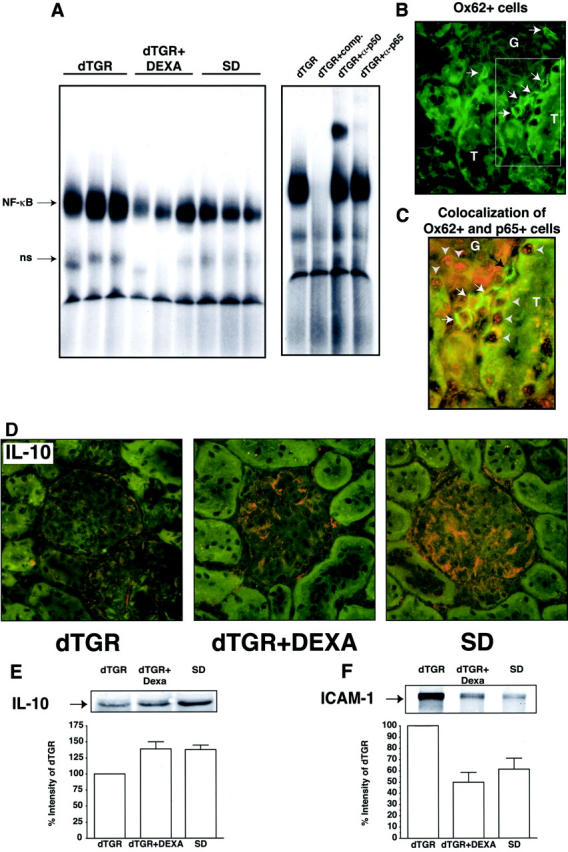
Renal NF-κB activity and co-localization of dendritic cells and p65. A: DNA-binding activity for NF-κB was markedly induced in dTGRs. Nontransgenic SD rats showed no NF-κB activity. DEXA reduced NF-κB activity toward SD rats. Competition and supershift experiments with antibodies against p50 and p65 showed specificity of the NF-κB activity. B: Renal Ox62+ dendritic cell infiltration (green staining indicated by white arrows). C: A higher magnification demonstrated that dendritic cells (green) co-localized with nuclear p65 staining (red) marked by white arrows. Tubules (T) and glomeruli (G) of vehicle-treated dTGRs showed also nuclear p65 staining (gray arrowheads). D: IL-10 was expressed in glomeruli of DEXA-treated and nontransgenic rats. E and F: Representative Western blot analysis of total kidney extracts confirmed the IL-10 and ICAM-1 immunohistochemical results, respectively.
We next studied renal IL-10 and ICAM-1 expression. IL-10 was expressed in glomeruli (n = 5, Figure 3D ▶ ) and smooth muscles cells (data not shown) of DEXA-treated and nontransgenic rats. In contrast to the endogenous NF-κB inhibitor IL-10, ICAM-1 was markedly increased in the endothelium and glomeruli of vehicle-treated dTGRs (data not show). Western blot analysis of total kidney extracts confirmed the IL-10 and ICAM-1 immunohistochemical results (Figure 3, E and F ▶ , respectively).
Dexamethasone Reduces p22phox-Positive Cells and ROS in the Kidney
P22phox expression (n = 5, Figure 4A ▶ ) was co-localized with all infiltrated macrophages (ED-2+ cells). ED-1+ cells were partially positive for p22phox indicating that only differentiated macrophages expressed p22phox. P22phox was also co-localized in CD4+ and CD8+ cells. In contrast to macrophages, the CD4+ and CD8+ cells were only partially positive for p22phox. Granulocytes, the majority of infiltrated cells in the glomerulus, also expressed p22phox (data not shown). P22phox immunohistochemical staining and Western blotting showed markedly reduced p22phox expression in DEXA-treated kidneys (Figure 4B) ▶ .
Figure 4.
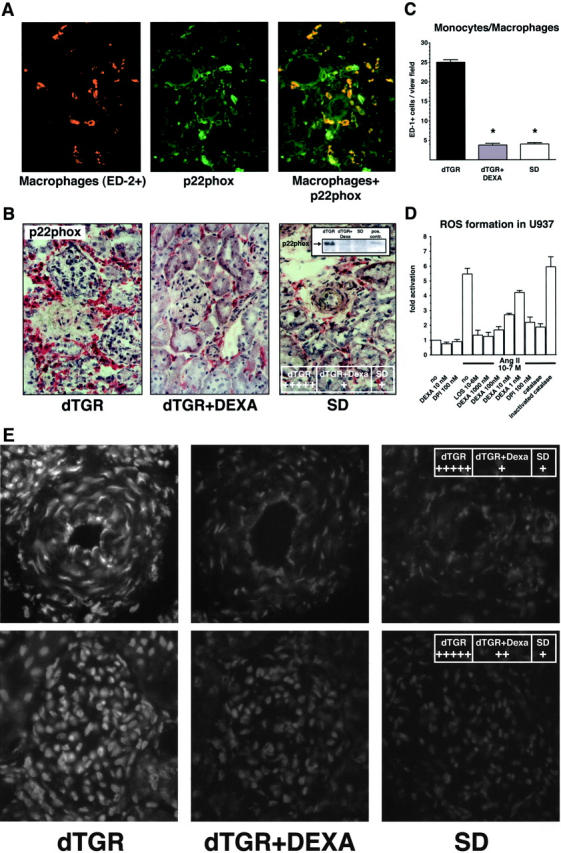
Macrophage infiltration, p22phox NAD(P)H subunit, and in situ ROS production in renal sections. A: Co-localization of macrophages (red) and p22phox (green) and the merged picture (orange). Almost all perivascular-infiltrated macrophages express p22phox. B: Representative photomicrograph of p22phox showed increased expression in dTGRs because of infiltrated leukocytes. DEXA reduced expression levels to SD rat controls. Representative p22phox Western blot from total kidney extracts confirmed the results. TNF-α-treated leukocytes were used as positive control (pos. contr.). C: Semiquantification of renal sections revealed that DEXA reduced infiltrated monocytes/macrophages (ED-1+) to nontransgenic level. D: Fluorescence signal of U937 macrophages preincubated with DCFH-DA was directly induced by Ang II. Preincubation with catalase (1000 U/ml), diphenylene iodonium (10 μmol/L), and DEXA (1 to 1000 nmol/L) inhibited the Ang II-induced fluorescence intensity. None of the inhibitors changed the baseline of ROS formation. E: Vehicle-treated dTGRs show a fluorescent ethidium signal in the endothelium, smooth muscle cells, adventitia of the vessel wall, infiltrated cells, as well as in the glomerulus, which was markedly attenuated by DEXA.
Semiquantification of monocytes/macrophages (n = 5, Figure 4C ▶ ) and granulocytes (n = 5; data not shown) demonstrated a complete prevention of cell infiltration in DEXA-treated dTGRs. Ang II directly induced ROS generation in cultured U937 macrophages using flow cytometry with DCFH-DA (Figure 4D) ▶ . Quiescent U937 showed only a weak DCFH fluorescence intensity. Ang II dose-dependently induced an increase in intracellular fluorescence (data not shown). Addition of catalase reduced the Ang II-induced fluorescence intensity to basal levels. These findings indicate that H2O2 participates in the oxidation of DCFH. The addition of the AT1 receptor blocker losartan, DEXA, and flavin-containing enzyme inhibitor diphenylene iodinium inhibited the increase of oxidized intracellular state by Ang II. These results demonstrate that the NAD(P)H oxidase was involved in Ang II-induced ROS formation.
Renal sections were incubated with hydroethidine to investigate in situ ROS production. Vehicle-treated dTGRs show a fluorescent ethidium signal in the endothelium, smooth muscle cells, and adventitia of the vessel wall as well as in the glomeruli and infiltrated cells. Chronic DEXA treatment markedly attenuated the fluorescent signal (n = 5, Figure 4E ▶ ). These results strongly support the notion that DEXA reduced cell infiltration and therefore NAD(P)H oxidase activity and renal ROS formation in dTGRs.
Dexamethasone Suppresses T-Cell Infiltration and MHC II and CD86 Expression
Vehicle-treated dTGR kidneys showed a significantly increased infiltration of CD4+ and CD8+ T cells that was reduced by DEXA to SD rat levels (n = 5, Figure 5A ▶ ). We were surprised by the fact that infiltrated T cells were frequently adjacent to dendritic cells. Therefore, we performed serial kidney sections (Figure 5B) ▶ of vehicle-treated dTGRs demonstrating that dendritic cells (Figure 5B ▶ , top) express MHC II (Figure 5B ▶ , bottom) and are adjacent to CD4+ cells (Figure 5B ▶ , middle) indicated by the black arrows. Vehicle-treated dTGRs show a profound perivascular and periglomerular infiltration not only of MHC II+, but also CD86+ cells. DEXA suppressed MHC II and CD86 expression below nontransgenic level (n = 5, Figure 5, C and D ▶ ).
Figure 5.
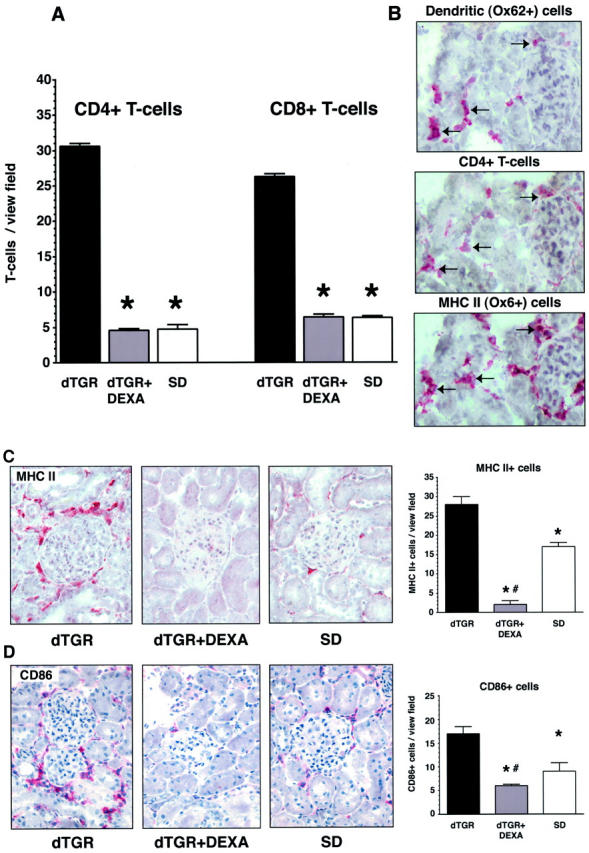
DEXA suppresses immune cells. A: Vehicle-treated dTGR kidneys showed a significantly increased infiltration of CD4+ and CD8+ T cells. DEXA-treated and SD rats showed a similar number of CD4+ and CD8+ T cells. B: Serial kidney sections (6 μm) of vehicle-treated dTGRs demonstrated that dendritic cells (top) express MHC II (bottom) and are adjacent to CD4+ cells (middle) indicated by the black arrows. C and D: Vehicle-treated dTGRs show perivascular and periglomerular infiltration of MHC II+ and CD86+ cells, respectively. DEXA reduced MHC II and CD86 expression below nontransgenic level.
Effect of Ang II on Dendritic Cells in Vivo and in Vitro
Semiquantification of dendritic cells in the kidney (n = 5, Figure 6A ▶ ) demonstrated a complete prevention of infiltration in DEXA-treated dTGRs. We next tested the hypothesis that Ang II might directly induce dendritic cell maturation. Human CD34-derived dendritic cells were differentiated in immature dendritic cells. The addition of Ang II for 6 days did not alter the expression of CD80, CD83, and MHC II. In contrast, TNF-α for 24 hours led to the induction of the surface molecules and could be blocked by the preincubation with DEXA (Figure 6B) ▶ . However, both TNF-α and Ang II induced CCR7 expression. Therefore, we investigated the ability of Ang II to induce dendritic cell migration (Figure 6C) ▶ . Both Ang II and Epstein-Barr virus-induced molecule 1 ligand chemokine, the ligand for CCR7, increased the number of transmigrated cells. The effect was more pronounced in mature compared to immature dendritic cells. These data suggest that in the damaged kidney dendritic cells exist in a mature state. Maturation of the dendritic cell is not induced directly by Ang II. Possibly, TNF-α secreted by macrophages promotes dendritic cell maturation. To test the hypothesis that TNF-α is involved in dendritic cell maturation and renal damage, we treated dTGRs chronically with the soluble TNF-α receptor etanercept.
Figure 6.

Effect of Ang II on dendritic cell in vivo and in vitro. A: Vehicle-treated dTGRs showed increased dendritic cell infiltration into the kidney that was reduced even below SD rat level by DEXA. B: FACS analysis of human dendritic cells showed that Ang II was unable to promote maturation. In contrast, TNF-α induced dendritic cell maturation. This effect was blocked by DEXA. In addition, Ang II and TNF-α both induced CCR7 expression. C: Chemotaxis assay of immature dendritic cells at day 7 of differentiation and mature dendritic cells. For maturation, cells were cultured at day 6 for 24 hours with TNF-α. Both, Ang II and Epstein-Barr virus-induced molecule 1 ligand chemokine, the ligand for CCR7 as positive control, increased cell migration. Cell migration was more pronounced in mature, compared to immature dendritic cells.
Etanercept Inhibits Dendritic Cells, Inflammation, and Renal Damage
Chronic sTNF receptor treatment reduced mortality from 40% in untreated dTGRs (6 of 15 dTGRs) to 8% (1 of 12 etanercept). Etanercept did not affect systolic blood pressure (n = 6 to 8; P = 0.98 etanercept versus dTGRs; Figure 7A ▶ ). In contrast, albuminuria (n = 6 to 8; P < 0.05 etanercept versus dTGRs; Figure 7B ▶ ) and renal fibronectin expression (n = 5; grade 3+ for etanercept-treated dTGRs versus 5+ for vehicle-treated dTGRs versus 1+ for SD rats, respectively; data not shown) were significantly reduced. Infiltration of dendritic, MHC II+, and CD86+cells (n = 5, Figure 7C ▶ ) in the kidney was reduced by sTNF receptor treatment toward SD rat levels (P < 0.001 etanercept versus dTGRs), whereas macrophage and T-cell infiltration (n = 5, Figure 7D ▶ ) was partially reduced (P < 0.05 etanercept versus dTGRs and SD rats). Electrophoretic mobility shift assay showed that renal NF-κB and AP-1 DNA-binding activity were also reduced by sTNF receptor treatment (n = 3; data not shown). Thus, these data suggest that TNF-α is involved in infiltration of inflammatory and immune cells as well as in the infiltration and maturation of dendritic cells in Ang II-induced renal damage.
Figure 7.

Chronic treatment with a soluble TNF-α receptor (sTNF receptor) reduced macrophage, dendritic, and T-cell infiltration; expression of MHC II and CD86; and ameliorated renal damage independent of blood pressure in dTGRs. A: Systolic blood pressure was not affected by sTNF receptor treatment. B: In contrast, sTNF receptor treatment reduced albuminuria. C: Infiltration of dendritic, MHC II+, and CD86+ cells in the kidney was reduced by sTNF receptor treatment toward SD rat level. D: In contrast, macrophage and T-cell infiltration was partially reduced. Results are mean ± SEM (*, P < 0.05, dTGR versus dTGR + sTNF receptor and SD; #, P < 0.05 SD versus dTGR + sTNF receptor).
MMF Ameliorates Ang II-Induced Renal Damage
To inhibit immunocompetent cells by a DEXA-independent mechanism, we treated dTGRs chronically with MMF. MMF reduced mortality to zero (0 of 9 MMF-treated dTGRs) versus 54% mortality in dTGRs (12 of 22, P < 0.001). With MMF treatment, systolic blood pressure decreased somewhat (n = 8 to 14; P < 0.001; Figure 8A ▶ ), whereas albuminuria was completely abolished (n = 7 to 11; P < 0.001; Figure 8B ▶ ). MMF also reduced fibronectin and collagen IV expression (n = 5; grade 2+ for MMF-treated dTGRs versus 5+ for vehicle-treated dTGRs versus 1+ for SD rats, respectively). Inflammatory response, as well as infiltration of immune cells in the kidney, was abolished by MMF. Semiquantification of dendritic cell, MHC II+ cell, and CD86+ cell infiltration in the kidney revealed that MMF reduced infiltration to nontransgenic levels (n = 5; P < 0.01; Figure 8C ▶ ). In addition, vehicle-treated dTGRs show significantly increased infiltration of macrophages, CD4+ and CD8+ T cells, which was normalized after MMF (n = 5; P < 0.001; Figure 8D ▶ ). Thus, inhibition of T-cell proliferation improved renal damage.
Figure 8.

Chronic MMF treatment reduced macrophage, dendritic, and T-cell infiltration; expression of MHC II and CD86; and ameliorated renal damage. A and B: MMF partially reduced systolic blood pressure, whereas albuminuria was normalized. C and D: Infiltration of dendritic, MHC II+, CD86+, and T cells, and macrophages in the kidney was reduced by MMF treatment toward SD rat level. Results are mean ± SEM (*, P < 0.05, dTGR versus dTGR + MMF and SD; *, P < 0.05 SD versus dTGR + MMF).
Discussion
Our study provides additional evidence on the role of immunocompetent cells in a nonimmune, Ang II-induced, renal disease. We found that ROS were generated by Ang II exposure in vivo and in vitro, that NF-κB was activated, and that inflammatory cells infiltrated the tissues. Furthermore, we were interested to observe T cell, macrophage, and dendritic cell infiltration in our model. MHC class II+ and CD86+ cells were present in abundance. A conventional anti-inflammatory and immunosuppressive approach with DEXA treatment ameliorated the disease independent of blood pressure reduction, confirming that Ang II does not act by means of hemodynamic mechanisms in this model. A role for TNF-α in the responses was documented by etanercept treatment, which was somewhat less effective compared to DEXA, but also functioned independent of blood pressure reduction. We were intrigued to observe that Ang II did not influence dendritic cell maturation, but did influence dendritic cell migration via CCR7. The maturation was instead directed by TNF-α. Finally, we sought additional support for our hypothesis that Ang II-induced vascular damage is mediated primarily via immunocompetent cells by treating dTGRs with MMF. The MMF results were similar in kind to those obtained with DEXA.
To separate actions of Ang II from hemodynamic effects is difficult. We admit that we have not succeeded completely in doing so. In an earlier study, we attempted to control for blood pressure by treating dTGRs with a triple-regimen, independent of the renin-angiotensin system. 8 We lowered blood pressure in that study reasonably effectively, but delayed end-organ damage by only 1 week. Unfortunately, normalizing blood pressure in this model without blocking the renin-angiotensin system is extremely difficult. We believe our effects are primarily independent of hemodynamics because DEXA markedly ameliorated end-organ damage without lowering blood pressure to any detectable degree. Conceivably, glomerular arteriolar constriction may have induced a postglomerular ischemic change that was responsible for inflammation. However, we believe that Ang II-induced postglomerular constriction, as an initiating factor for inflammation in our study is unlikely. In an earlier study, we measured glomerular filtration rate, renal blood flow, and pressure-natriuresis-diuresis curves in these rats before end-organ damage had occurred. 16 The experiments were performed with and without renal denervation, and with and without infusions of vasopressin, norepinephrine, 17-OH-corticosterone, and aldosterone to block other potential influences. Pressure-natriuresis-diuresis curves were shifted rightward in dTGRs compared to controls. Renal blood flow was indeed less; however, glomerular filtration rate was preserved. Had marked postglomerular arteriolar constriction been present, we would have expected the glomerular filtration rate to be increased in dTGRs compared to controls. Blockade of the renin-angiotensin system with either an angiotensin-converting enzyme inhibitor or an AT1 receptor blocker restored pressure-natriuresis-diuresis curves to normal in dTGRs. 17
To further explore the inflammatory nature of this model, we directed our attention at MIF. MIF is an immunoregulatory cytokine involved in T-cell activation and delayed-type hypersensitivity and is elaborated by immune cells; 18,19 however, MIF can also be produced by endothelial and vascular smooth muscle cells. 20 The cytokine seems to be required by macrophages to assure response to TNF-α. A deficiency is characterized by a down-regulated Toll-like receptor and decreased activation of NF-κB. 21 We reasoned that Ang II might influence MIF production directly in U937 cells, but that proved not to be the case. DEXA treatment down-regulated MIF, as did treatment with etanercept. These findings, as well as recent observations in plaques from patients with atherosclerosis, suggest that MIF plays a role in mediating inflammatory vascular disease. 22 Ang II and TNF-α are closely interrelated in our model. Ang II modulates TNF-α and IL-6 production in the kidney. 23 TNF-α can activate the angiotensinogen gene by an NF-κB-related mechanism. 24,25 Furthermore, many effects in renal disease are mediated by TNF-α after activation of NF-κB activity. 26
Our results confirm the important role of ROS generated by Ang II through activation of NAD(P)H oxidase and other oxidases. 27-30 ROS generation is presumably primarily responsible for NF-κB activation. 31 NF-κB in turn activates a host of genes important to innate immunity. 32 Furthermore, the transcription factors AP-1 and NF-AT are activated subsequent to AT1 receptor stimulation in this model and in models presented by other investigators. 33 The multiplicity of the signaling involved has enabled a series of interventions that have also been applied by others and us in Ang II-dependent models of end-organ damage. In addition to angiotensin-converting enzyme inhibition and AT1 receptor blockade, antioxidant treatments, mineralocorticoid receptor blockade, interference with endothelin signaling, I-κB kinase inhibition with aspirin, and NF-AT inhibition with cyclosporin have all been effective, independent of blood pressure reduction, whereas blood pressure reduction alone, on the other hand, was not effective. 34
We believe that our study underscores the role of immunocompetent cells and provides support for both the participation of innate and acquired immunity in Ang II-induced vascular injury. 1 Convincing proof would require identification of the molecules recognized by predetermined receptors for the former and the antigens actually presented by dendritic cells for the latter. This proof is not forthcoming from our studies. Nevertheless, others have also forwarded evidence supporting a role for adaptive immunity. Nataraj and colleagues 33 drew attention to this possibility when they showed that Ang II promotes proliferation of splenic lymphocytes via a calcineurin-dependent pathway. They further demonstrated that Ang II modulates immune responses in a mouse model of cardiac transplantation by activating the AT1 receptor. We were intrigued by these results in light of the success we had with ameliorating renal damage with cyclosporin in this model earlier. 35 Participation of T lymphocytes and macrophages was a prominent feature in that study.
DEXA is a highly complex and nonspecific form of immunosuppression that by no means allows us to conclude that lymphocytes alone are responsible for the vascular injury. DEXA treatment was highly effective even though TNF-α has recently been shown to modulate the expression of the glucocorticoid receptor isoforms α and β in such a manner that glucocorticoid resistance may occur. 36 DEXA activates the cytoplasmic glucocorticoid α-receptor that then binds to activated NF-κB and prevents the transcription factor from binding to κB sites on the genes important to inflammation. This interaction may occur in the cytoplasm and/or the nucleus. 37 Glucocorticoids also increase transcription of the gene for I-κB-α, thereby increasing formation of the protein that binds to activated NF-κB in the nucleus. The I-κB-α protein induces the dissociation of NF-κB from κB sites on target genes and causes NF-κB to move into the cytoplasm. 38 This mechanism has been observed in T cells and monocytes, but may not occur in all types of cells. 39 We cannot state for certain which cell types contributed most to the NF-κB activity in our model. We believe that the overall NF-κB activity stems from infiltrated cells including dendritic cells, as well as a contribution from other cells in the kidney indicated by nuclear p65 staining in glomerular and tubular cells.
In our dTGRs model, DEXA affected renal dendritic cell infiltration and maturation. DEXA significantly reduced MHC class II+ and CD86+ cells that co-localized with dendritic cells. DEXA treatment also partially restored IL-10 expression, whereas ICAM-1 expression was concomitantly decreased. IL-10 is a pleiotropic cytokine produced by both T cells and macrophages and possesses both anti-inflammatory and immunosuppressive properties. 40 IL-10 can influence dendritic cells to induce anergic CD4 and CD8 T cells. 41 On the other hand, CD40 ligand-activated dendritic cells can promote the generation of CD8 T regulatory cells that produce IL-10. 42 Vehicle-treated dTGRs showed nuclear staining of the NF-κB subunit p65, which co-localized with OX62+ dendritic cells in our model.
We investigated whether or not Ang II directly promotes dendritic cell maturation in vitro. Ang II did not alter CD80, CD83, and MHC II expression. In contrast, TNF-α induced dendritic cell maturation; the effect was inhibited by DEXA preincubation. On the other hand, Ang II directly increased CCR7 expression and in vitro cell migration. Vehicle-treated dTGRs show severe infiltration of proinflammatory cytokine-producing macrophages. This finding led us to the hypothesis that Ang II-induced effects on dendritic cell maturation in vivo are indirect and may involve TNF-α. We treated dTGRs with etanercept, a recombinant dimer of two p75-soluble TNF receptor molecules fused to the Fc fragment of human IgG. Blockade of TNF signaling reduced infiltration of all inflammatory cells, including dendritic, CD86+, and MHC II+ cells. These data suggest that Ang II induces dendritic cell infiltration directly, whereas the maturation is mediated by TNF-α. CCR7 is also responsible for B- and T-cell migration. Possible direct effects of Ang II on migration in these cells have not yet been tested.
We sought to suppress lymphocytes by a DEXA-independent mechanism to strengthen our results. We considered crossing our dTGRs into rat lines devoid of T cells. However, the genetic background of the rats would be altered and the features of the T-cell-depleted model might not be retained. Thus, we settled on MMF, admittedly also not a T-cell-specific treatment. MMF provides mycophenolic acid, which blocks the action of inosine monophosphate dehydrogenase, resulting in the inhibition of the novo purine synthesis. MMF has an anti-proliferative effect on T and B cells and also inhibits the glycosylation of cell surface adhesion proteins involved in cell-cell contact and in the recruitment of circulating leukocytes to sites of tissue damage and inflammation. Recently, Nieto and colleagues 43 used mercuric chloride to induce an autoimmune nephritic syndrome in rats. This model features the production of autoantibodies, as we had a host of responses similar to those reported here. MMF had a strong effect on innate and acquired immune responses in that model. Furthermore, the fact that mercuric chloride induced autoimmunity through cytotoxic actions suggests that chronic Ang II might act in an analogous manner. As a matter of fact, recent studies involving rats with 5/6 nephrectomy and rats given a chronic Ang II infusion support that view. 44,45
The future perspectives of our study extend beyond the pharmacological successes of DEXA, etanercept, and MMF treatment of Ang II-induced vascular injury. The signal transduction by which Ang II stimulated dendritic cell migration and CCR7 expression are unknown. Our data show that macrophages, antigen-presenting cells expressing MHC class II and CD86, and T cells are all involved in Ang II-induced renal damage. However, what antigen is being presented here is totally unclear. Whether or not the activation is confined to T lymphocytes is also unknown. Conceivably, Ang II could initiate antibody production by B cells. Activating antibodies to the AT1 receptor have been described in preeclampsia, a disease state that features increased sensitivity to Ang II. 46 We believe the notion that Ang II not only modulates innate immunity, but also initiates adaptive immunity. The elucidation of the processes involved will open a hitherto unappreciated mechanism of Ang II-induced end-organ damage.
Footnotes
Address reprint requests to Friedrich C. Luft, Franz Volhard Clinic, Wiltberg Strasse 50, 13125 Berlin, Germany. E-mail: luft@fvk-berlin.de.
References
- 1.Rodriguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ: Role of immunocompetent cells in nonimmune renal diseases. Kidney Int 2001, 59:1626-1640 [DOI] [PubMed] [Google Scholar]
- 2.Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ: Salt-sensitive hypertension develops after short-term exposure to Angiotensin II. Hypertension 1999, 33:1013-1019 [DOI] [PubMed] [Google Scholar]
- 3.Mai M, Geiger H, Hilgers KF, Veelken R, Mann JF, Dammrich J, Luft FC: Early interstitial changes in hypertension-induced renal injury. Hypertension 1993, 22:754-765 [DOI] [PubMed] [Google Scholar]
- 4.Grafe M, Auch-Schwelk W, Zakrzewicz A, Regitz-Zagrosek V, Bartsch P, Graf K, Loebe M, Gaehtgens P, Fleck E: Angiotensin II-induced leukocyte adhesion on human coronary endothelial cells is mediated by E-selectin. Circ Res 1997, 81:804-811 [DOI] [PubMed] [Google Scholar]
- 5.Krejcy K, Eichler HG, Jilma B, Kapiotis S, Wolzt M, Zanaschka G, Gasic S, Schutz W, Wagner O: Influence of angiotensin II on circulating adhesion molecules and blood leukocyte count in vivo. Can J Physiol Pharmacol 1996, 74:9-14 [PubMed] [Google Scholar]
- 6.Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Suzuki Y, Mezzano S, Plaza JJ, Egido J: Role of the renin-angiotensin system in vascular diseases: expanding the field. Hypertension 2001, 38:1382-1387 [DOI] [PubMed] [Google Scholar]
- 7.Luft FC, Mervaala E, Muller DN, Gross V, Schmidt F, Park JK, Schmitz C, Lippoldt A, Breu V, Dechend R, Dragun D, Schneider W, Ganten D, Haller H: Hypertension-induced end-organ damage: a new transgenic approach to an old problem. Hypertension 1999, 33:212-218 [DOI] [PubMed] [Google Scholar]
- 8.Mervaala E, Muller DN, Schmidt F, Park JK, Gross V, Bader M, Breu V, Ganten D, Haller H, Luft FC: Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension 2000, 35:587-594 [DOI] [PubMed] [Google Scholar]
- 9.Muller DN, Mervaala EM, Dechend R, Fiebeler A, Park JK, Schmidt F, Theuer J, Breu V, Mackman N, Luther T, Schneider W, Gulba D, Ganten D, Haller H, Luft FC: Angiotensin II (AT(1)) receptor blockade reduces vascular tissue factor in angiotensin II-induced cardiac vasculopathy. Am J Pathol 2000, 157:111-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC: NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 2000, 35:193-201 [DOI] [PubMed] [Google Scholar]
- 11.Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R: Endothelium-derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res 2001, 88:44-51 [DOI] [PubMed] [Google Scholar]
- 12.Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M: Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol 1983, 130:1910-1917 [PubMed] [Google Scholar]
- 13.Ju X-S, Zenke M: Differentiation of human antigen presenting dendritic cells from CD34+ hematopoietic stem cells in vitro. Körholz D eds. Methods in Molecular Medicine. Cytokines and Colony Assays. 2001. Humana Press, Totowa [DOI] [PubMed]
- 14.Diebold SS, Lehrmann H, Kursa M, Wagner E, Cotten M, Zenke M: Efficient gene delivery into human dendritic cells by adenovirus polyethylenimine and mannose polyethylenimine transfection. Hum Gene Ther 1999, 10:775-786 [DOI] [PubMed] [Google Scholar]
- 15.Kellermann SA, Hudak S, Oldham ER, Liu YJ, McEvoy LM: The CC chemokine receptor-7 ligands 6Ckine and macrophage inflammatory protein-3 beta are potent chemoattractants for in vitro- and in vivo-derived dendritic cells. J Immunol 1999, 162:3859-3864 [PubMed] [Google Scholar]
- 16.Dehmel B, Mervaala E, Lippoldt A, Gross V, Bohlender J, Ganten D, Luft FC: Pressure-natriuresis and -diuresis in transgenic rats harboring both human renin and human angiotensinogen genes. J Am Soc Nephrol 1998, 9:2212-2222 [DOI] [PubMed] [Google Scholar]
- 17.Mervaala E, Dehmel B, Gross V, Lippoldt A, Bohlender J, Milia AF, Ganten D, Luft FC: Angiotensin-converting enzyme inhibition and AT1 receptor blockade modify the pressure-natriuresis relationship by additive mechanisms in rats with human renin and angiotensinogen genes. J Am Soc Nephrol 1999, 10:1669-1680 [DOI] [PubMed] [Google Scholar]
- 18.Bacher M, Metz CN, Calandra T, Mayer K, Chesney J, Lohoff M, Gemsa D, Donnelly T, Bucala R: An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA 1996, 93:7849-7854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloom BR, Bennett B: Relation of the migration inhibitory factor (MIF) to delayed-type hypersensitivity reactions. Ann NY Acad Sci 1970, 169:258-265 [DOI] [PubMed] [Google Scholar]
- 20.Lin SG, Yu XY, Chen YX, Huang XR, Metz C, Bucala R, Lau CP, Lan HY: De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circ Res 2000, 87:1202-1208 [DOI] [PubMed] [Google Scholar]
- 21.Roger T, David J, Glauser MP, Calandra T: MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 2001, 414:920-924 [DOI] [PubMed] [Google Scholar]
- 22.Burger-Kentischer A, Goebel H, Seiler R, Fraedrich G, Schaefer HE, Dimmeler S, Kleemann R, Bernhagen J, Ihling C: Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation 2002, 105:1561-1566 [DOI] [PubMed] [Google Scholar]
- 23.Niimi R, Nakamura A, Yanagawa Y: Suppression of endotoxin-induced renal tumor necrosis factor-alpha and interleukin-6 mRNA by renin-angiotensin system inhibitors. Jpn J Pharmacol 2002, 88:139-145 [DOI] [PubMed] [Google Scholar]
- 24.Brasier AR, Li J, Wimbish KA: Tumor necrosis factor activates angiotensinogen gene expression by the Rel A transactivator. Hypertension 1996, 27:1009-1017 [DOI] [PubMed] [Google Scholar]
- 25.Brasier AR, Jamaluddin M, Han Y, Patterson C, Runge MS: Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-kappaB (NF-kappaB) transcription factor. Mol Cell Biochem 2000, 212:155-169 [PubMed] [Google Scholar]
- 26.Klahr S, Morrissey JJ: The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int 2000, 57(Suppl)(75):S7-S14 [PubMed] [Google Scholar]
- 27.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK: p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem 1996, 271:23317-23321 [DOI] [PubMed] [Google Scholar]
- 28.Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kubler W, Kreuzer J: Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol 2000, 20:940-948 [DOI] [PubMed] [Google Scholar]
- 29.Touyz RM, Schiffrin EL: Ang II-stimulated superoxide production is mediated via phospholipase D in human vascular smooth muscle cells. Hypertension 1999, 34:976-824 [DOI] [PubMed] [Google Scholar]
- 30.Mervaala EM, Cheng ZJ, Tikkanen I, Lapatto R, Nurminen K, Vapaatalo H, Muller DN, Fiebeler A, Ganten U, Ganten D, Luft FC: Endothelial dysfunction and xanthine oxidoreductase activity in rats with human renin and angiotensinogen genes. Hypertension 2001, 37:414-418 [DOI] [PubMed] [Google Scholar]
- 31.Li N, Karin M: Is NF-kappaB the sensor of oxidative stress? EMBO J 1999, 13:1137-1143 [PubMed] [Google Scholar]
- 32.Karin M, Ben-Neriah Y: Phosphorylation meets ubiquitination: the control of NF-κ B activity. Annu Rev Immunol 2000, 18:621-663 [DOI] [PubMed] [Google Scholar]
- 33.Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM: Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest 1999, 104:1693-1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luft FC: Proinflammatory effects of angiotensin II and endothelin: targets for progression of cardiovascular and renal diseases. Curr Opin Nephrol Hypertens 2002, 11:59-66 [DOI] [PubMed] [Google Scholar]
- 35.Mervaala E, Muller DN, Park JK, Dechend R, Schmidt F, Fiebeler A, Bieringer M, Breu V, Ganten D, Haller H, Luft FC: Cyclosporin A protects against angiotensin II-induced end-organ damage in double transgenic rats harboring human renin and angiotensinogen genes. Hypertension 2000, 35:360-366 [DOI] [PubMed] [Google Scholar]
- 36.Webster JC, Oakley RH, Jewell CM, Cidlowski JA: Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci USA 2001, 98:6865-6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adcock IM, Brown CR, Barnes PJ: Tumour necrosis factor alpha causes retention of activated glucocorticoid receptor within the cytoplasm of A549 cells. Biochem Biophys Res Commun 1996, 225:545-550 [DOI] [PubMed] [Google Scholar]
- 38.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS, Jr: Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science 1995, 270:283-286 [DOI] [PubMed] [Google Scholar]
- 39.Brostjan C, Anrather J, Csizmadia V, Stroka D, Soares M, Bach FH, Winkler H: Glucocorticoid-mediated repression of NFkappaB activity in endothelial cells does not involve induction of IkappaBalpha synthesis. J Biol Chem 1996, 271:19612-19616 [DOI] [PubMed] [Google Scholar]
- 40.Oberholzer A, Oberholzer C, Moldawer LL: Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med 2002, 30(Suppl 1):S58-S63 [PubMed] [Google Scholar]
- 41.Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH: CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99:2468-2476 [DOI] [PubMed] [Google Scholar]
- 42.Gilliet M, Liu YJ: Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med 2002, 195:695-704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nieto E, Escudero E, Navarro E, Yanez-Mo M, Martin A, Perez De Lema G, Sanchez-Madrid F, Mampaso F: Effects of mycophenolate mofetil in mercury-induced autoimmune nephritis. J Am Soc Nephrol 2002, 13:937-945 [DOI] [PubMed] [Google Scholar]
- 44.Fujihara CK, De Lourdes Noronha I, Malheiros, Antunes GR, de Oliveira IB, Zatz R: Combined mycophenolate mofetil and losartan therapy arrests established injury in the remnant kidney. J Am Soc Nephrol 2000, 11:283-290 [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ: Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 2001, 59:2222-2232 [DOI] [PubMed] [Google Scholar]
- 46.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC: Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 1999, 103:945-952 [DOI] [PMC free article] [PubMed] [Google Scholar]