

Abstract
A central question in Alzheimer’s disease concerns the mechanism by which β-amyloid contributes to neuropathology, and in particular whether intracellular versus extracellular β-amyloid plays a critical role. Alzheimer transgenic mouse studies demonstrate brain dysfunction, as β-amyloid levels rise, months before the appearance of β-amyloid plaques. We have now used immunoelectron microscopy to determine the subcellular site of neuronal β-amyloid in normal and Alzheimer brains, and in brains from Alzheimer transgenic mice. We report that β-amyloid 42 localized predominantly to multivesicular bodies of neurons in normal mouse, rat, and human brain. In transgenic mice and human Alzheimer brain, intraneuronal β-amyloid 42 increased with aging and β-amyloid 42 accumulated in multivesicular bodies within presynaptic and especially postsynaptic compartments. This accumulation was associated with abnormal synaptic morphology, before β-amyloid plaque pathology, suggesting that intracellular accumulation of β-amyloid plays a crucial role in Alzheimer’s disease.
β-amyloid (Aβ) plays a central role in the pathogenesis of Alzheimer’s disease (AD). 1 Increasing reports suggest that the Aβ42 form of Aβ can accumulate within neurons with aging and that intraneuronal Aβ may be directly involved in the pathogenesis of AD. 2-4 AD transgenic mouse studies indicate that synaptic, behavioral, and physiological functions begin to decline 5-7 as brain Aβ levels rise 8,9 before the appearance of Aβ plaques. These studies suggest that intracellular Aβ accumulation may be important in AD.
Although Aβ40 is the predominantly secreted Aβ species in tissue culture, the longer Aβ42 is more important in AD. Aβ42 is increased in familial AD (FAD) because of mutations in the amyloid precursor protein (APP) or presenilins 1 and 2, and it accumulates as the first Aβ species in Down’s syndrome and AD. 1 Cell biological studies indicate that Aβ generation occurs in the Golgi apparatus (Golgi), especially the trans-Golgi network, and to a lesser extent in the endoplasmic reticulum (ER). The ER appears to be more selective for Aβ42 than Aβ40 generation. 3,4 Moreover, the ER pool of Aβ42 appears to be secreted less efficiently than that from the Golgi. Aβ peptide generation also occurs in the endocytic pathway after internalization from the plasma membrane. 10,11
AD pathogenesis has traditionally been characterized by extracellular, aggregated, and plaque-associated Aβ. However, the extent to which extracellular Aβ causes cell death in AD is unclear. An increasing number of reports demonstrate behavioral, synaptic, physiological, and oxidative abnormalities months before the appearance of Aβ plaques in FAD transgenic mice that develop AD-like plaque pathology. 5,6,9,12 In Tg2576 mice, which express the human APP Swedish 670/671 FAD mutation, 13 brain levels of Aβ rise sharply as measured by enzyme-linked immunosorbent assay (ELISA) at 6 to 10 months, 8 before development of plaques, which appear at ∼11 to 13 months. 14 Preplaque increases in soluble Aβ42, but not Aβ40, have been reported in Down’s syndrome brain. 15 Studies of postmortem human brain show that soluble Aβ correlates better than insoluble Aβ or extracellular Aβ plaques with cognitive dysfunction in patients with AD. 16 Thus, soluble Aβ42 protofibrils may play a critical pathogenic role. 1
The location of the soluble Aβ42 protofibrils and how they cause toxicity are of growing interest. Aβ protofibrils are preferentially generated within cells rather than extracellularly. 17 Neuronal NT2 cells contain abundant intracellular Aβ42, which increases dramatically with time (aging) in culture. 18 Transgenic mice expressing a FAD mutant presenilin 1 were reported to develop intraneuronal Aβ42 accumulation and neurodegeneration. 19 Intracellular Aβ accumulation has been associated with pathology in inclusion body myositis 20 and mucopolysaccharidosis. 21 Thus, cumulative indirect evidence suggests that there may be a pathological role for neuronal Aβ accumulation in AD. Using three different well-characterized pairs of polyclonal antibodies against Aβ40 or Aβ42, we previously reported that intraneuronal Aβ42, as viewed by light microscopy, accumulates especially within AD-susceptible pyramidal neurons in postmortem tissue of patients with clinically defined early cognitive impairment. 2 Intraneuronal Aβ42, but not Aβ40, accumulation with AD pathology now has been reported by light microscopy by several other groups. 22-26 In the present study, the subcellular localization of Aβ within neurons was examined using immunogold electron microscopy (EM) to localize where brain Aβ increases occur before the development of plaques, in a mouse model of AD-like Aβ pathology. We provide evidence implicating intraneuronal Aβ42 accumulation in synaptic pathology both in AD transgenic mouse and human AD brains.
Materials and Methods
Antibodies
Monoclonal antibodies to the C-terminus of Aβ40 and Aβ42 were MBC40 and MBC42, respectively, generated by Dr. Haruyasu Yamaguchi, Gunma University, Gunma, Japan. MBC42 does not recognize Aβ40 or Aβ43. 27 The specificities of MBC40 and MBC42 antibodies were determined by immunoprecipitation of human APP–transfected neuroblastoma (N2a) cells, synthetic Aβ40 or Aβ42 with MBC40 or MBC42 antibody, followed by Western blotting with 6E10 monoclonal antibody (Signet Laboratories, Dedham, MA) as previously described. 28 Aβ42 immuno-EM results were confirmed using a commercially available polyclonal anti-Aβ42 antibody (Chemicon Inc., Temecula, CA). The C-terminus of full-length APP was recognized using antibody 369. 29
Animals
Well-established Tg2576 mice (n = 22) with the human APP Swedish 670/671 mutation of varying ages (four, 2 months; three, 3 months; two, 4 months; one, 8 months; three, 9 months; four, 10 months; two, 11 to 12 months; two, 16 to 17 months; one, 20 months) were used in this study. APP knockout mice and wild-type littermates (n = 4, each; two 6-month-old and two 12-month-old mice, each) were generously provided by Dr. Hui Zheng, Baylor College of Medicine, Houston, TX. Sprague-Dawley rats (300 to 325 g) were obtained from Taconic Farms (Germantown, NY). All methods were approved by the Weill Medical College of Cornell University Institutional Animal Care and Use Committee and conformed to National Institute of Health guidelines.
Human Brain Tissue
Human cortical brain tissue was obtained from the Department of Pathology, Weill Medical College of Cornell University as a result of neurosurgical procedures unrelated to this study. Neurosurgical AD cortical tissue was obtained (n = 2, ages 54 and 62 years). Relatively normal cortical brain tissue was obtained from emergency aneurysm clipping and benign tumor resection (n = 2, ages 44 and 54 years).
Pre-Embedding Immunogold Electron Microscopy
Mice and rats were anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and perfused via the ascending aorta with 3.75% acrolein (Polyscience, Warrington, PA) and 2% paraformaldehyde in 0.1 mol/L of phosphate buffer (pH 7.4). Human brain biopsy tissue was rapidly immersion-fixed with 1.875% acrolein and 2% paraformaldehyde in 0.1 mol/L of phosphate buffer. Brain tissue was cut (40-μm thick) on a vibrating microtome and treated with 1% sodium borohydride as previously described. 30
Some sections were processed for EM using immunoperoxidase, as described previously, 30 that revealed an analogous staining pattern (predominantly MVBs) with MBC42 antibody, but the subcellular localization was not as discreet as with immunogold. For immunogold labeling, free-floating sections were labeled with either MBC42, MBC40, 369, or Chemicon Aβ42 antibodies by the immunogold-silver procedure of Chan and colleagues. 31 For the processing, the tissue was incubated in goat anti-mouse (for MBC40 and MBC42) or goat anti-rabbit (for 369 and Chemicon anti-Aβ42 antibody) IgG conjugated to 1-nm gold particles (Amersham, Arlington Heights, IL) in 0.01% gelatin and 0.08% bovine serum albumin in phosphate-buffered saline (PBS). The conjugated gold particles were enhanced by treatment with silver solution (IntenSE, Amersham).
Dual immuno-EM localization was performed as previously described 30 now using polyclonal rabbit anti-APP (369) and monoclonal anti-Aβ42 (MBC42). Sections were incubated with both 369 and MBC42, and then processed first for immunoperoxidase localization of APP and then with the immunogold-silver method for localization of Aβ42. Aβ42 antibody concentrations were 1:50, Aβ40 antibody concentrations were 1:20, and APP antibody concentrations were 1:500. Sections were fixed in 2% osmium tetroxide in phosphate buffer, embedded in EMBed 812, sectioned (65- to 76-nm thick) and counterstained with uranyl acetate and Reynolds’ lead citrate. 30 Final preparations were examined with a Philips CM10 electron microscope. Morphological terminology is consistent with Peters and colleagues. 32 Quantification was done by counting gold particles from random images taken at the same magnification from analogous cerebral cortical regions with neuronal soma (layer 5) or processes of either 2- or 10-month-old Tg2576 mice. Student’s t-test was used for statistical analyses. Final illustrations were generated from scanned photographic prints (for Figure 1E ▶ ; Figure 3, B and C ▶ ; Figure 4A ▶ ; and Figure 5, A, B, and C ▶ ) or from a high-resolution digital imaging charge-coupled device camera system (Advanced Microscopy Techniques Corp., Danvers, MA) and processed on a Power Macintosh 8500/120 using Adobe Photoshop 6.0 (Adobe System, Mountain View, CA) and Quark X-Press 3.32 (Quark, Denver, CO).
Figure 1.
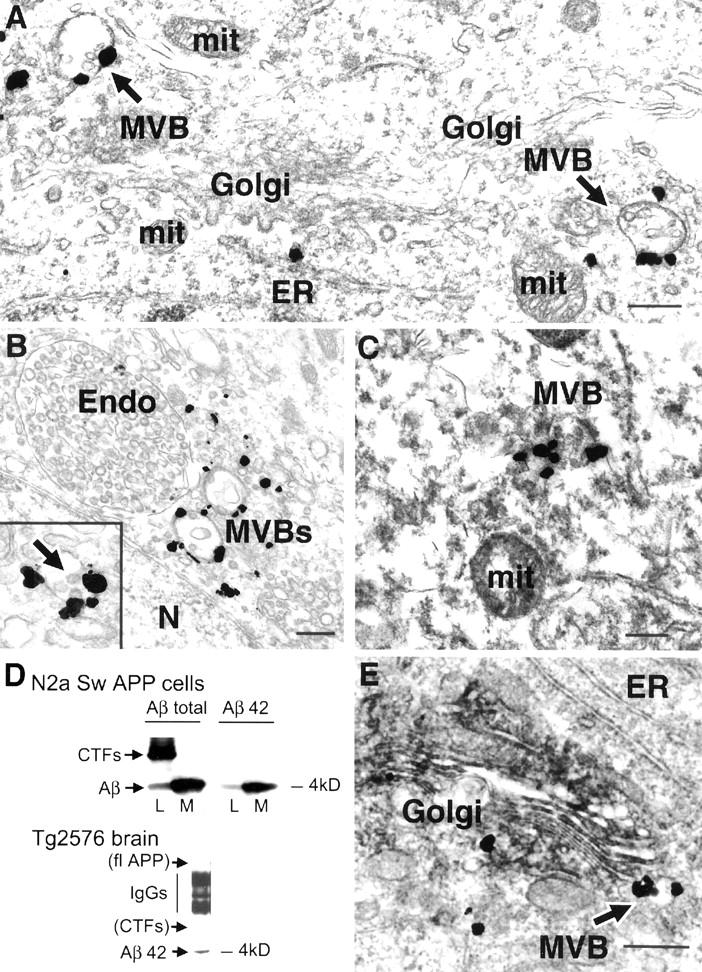
Ultrastructural localization of Aβ42 in neurons of normal mouse, rat, and human brain. A: Prominent Aβ42 immunogold labeling in MVBs (arrows) within a neuronal perikaryon in hippocampus (CA1) of a normal 11-month-old mouse. Note also an isolated gold particle in the ER. B: Perikaryon in CA1 region of hippocampus of a normal young rat; note Aβ42 immunogold labeling of two MVBs adjacent to a larger unlabelled endosome. Occasional gold particles also stained small clear vesicles and at times, gold particles are found in poorly identifiable locations. Inset demonstrates an example of MVB Aβ42 staining in another hippocampal perikaryon of rat brain; this MVB contains several intravesicular vesicles (arrow) that are characteristic of MVBs. C: Aβ42 immunoreactivity in a MVB of a normal human perikaryon derived from a rapidly fixed specimen of cerebral cortex from a 44-year-old with surgical resection in the setting of an acute aneurysm. D: Monoclonal anti-Aβ42 antibody MBC42 does not cross-react with APP CTFs in APP Swedish-transfected N2a cells (top) or Tg2576 mouse brain (bottom). For N2a cells, total Aβ (left) or Aβ42 (right) are shown in lysate (L) and conditioned media (M). Only the antibody directed at the mid-portion of total Aβ (4G8) but not anti-Aβ42 (MBC42) reacts against APP CTFs. The lack of MBC42 staining of CTFs is also demonstrated by this immunoprecipitation/Western blot of 12-month-old Tg2576 mouse brain, where Aβ42 is evident but not CTFs or full-length APP (flAPP). IgG bands represent nonspecific immunoglobulin bands observed as a results of the IP/Western blot. E: Dual-labeling immuno-EM showing full-length APP and APP CTFs (using C-terminal APP antibody 369) with dark immunoperoxidase reaction product primarily in Golgi, and immunogold labeling of Aβ42 (using monoclonal antibody MBC42) demonstrating labeling of an MVB (arrow) close to Golgi in this neuron from mouse cerebral cortex. Abbreviations: MVB, multivesicular body; Golgi, Golgi apparatus; ER, endoplasmic reticulum; mit, mitochondrion; Endo, endosome; N, nucleus. Scale bars: 300 nm (A–C); 500 nm (E).
Figure 3.

Neuronal Aβ42 immunoreactivity increases with aging in the brains of Tg2576 mice containing the APP Swedish 670/671 FAD mutation. A: Immunoprecipitation and Western blot of 4-kd human Aβ in equivalent brain tissue (parietal cortex) of Tg2576 mice at 3, 9, and 16 months. Note the total Aβ increases in preplaque mouse brains between 3 and 9 months. B and C: Aβ42 immunogold MVB labeling (arrows) of representative dendrites from young (2 months; B) and older preplaque (10 months; C) Tg2576 mice. Abbreviations: Den, dendrite; MVB, multivesicular body. D: Bar graph indicating the number of gold particles per MVB in neuronal soma (left) or dendrites (right) in 2-month-old (open bars) and 10-month-old (filled bars) Tg2576 mice (asterisk denotes statistical significance). E: Frequency distribution of total number of MVBs containing various numbers of gold particles in distal dendritic processes of young (2 to 3 months, open circles) and old (10 months, filled circles) Tg2576 mice. Scale bars, 500 nm (A–C).
Figure 4.
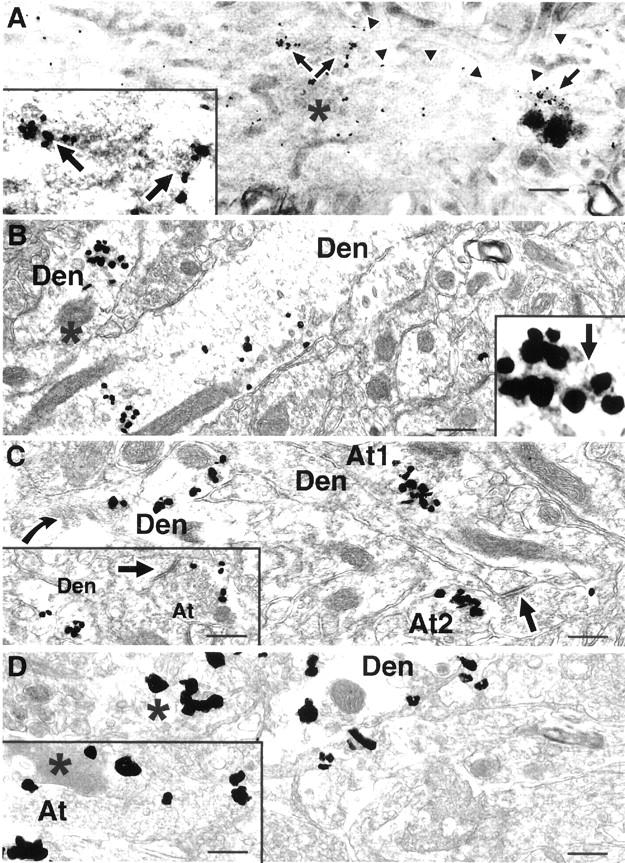
Aβ42 accumulates within neuronal processes in Tg2576 mice before plaque formation. A: A dendritic process with significant MVB-associated Aβ42 shows several apparent morphological abnormalities in the cortex of a preplaque 10-month-old mouse (arrowheads indicate outer membrane of this dendrite). Increased Aβ42 immunogold labeling is seen within this dendrite, which, based on its relative darkness (asterisk), appears to be degenerating. Unusual images of Aβ42 MVBs associated with lysosomes (such as the MVB under the single arrow at the far right) and abnormally dark Aβ42 immunogold MVBs (see two MVBs in inset and MVB under the single arrow) are observed. Inset is higher magnification of two darker appearing Aβ42 immunogold-associated MVBs above the paired arrows. B: In a 17-month-old mouse (B to D), a distended dendritic profile traversing the image with Aβ42 immunogold, and absence of normal cytoskeletal organelles. A second dendrite with Aβ42 is seen at the left with an abnormal dense body (asterisk) close to multiple MVB-associated gold particles; inset is a higher magnification of this MVB, in which characteristic small intravesicular vesicles can be seen (arrow). C: Axon terminal (At1) with substantial Aβ42 immunogold and some electron-dense components suggestive of degeneration. Another axon terminal (At2) with Aβ42 accumulation close to a synapse (arrow) with a dendritic spine. At left is a dendrite with abnormally stacked membranes (curved arrow) and Aβ42 accumulation. Inset shows increase in Aβ42 immunogold within dendritic and axonal compartments close to another synaptic density (arrow). D: At left, is a poorly defined process (asterisk) with Aβ42 accumulation also containing clusters of abnormal, darker membranes. Inset shows degenerating dark profiles (asterisk) seemingly within an axon terminal that contains clusters of Aβ42 gold labeling. Abbreviations: Den, dendrite; At, axon terminal. Scale bars, 300 nm.
Figure 5.
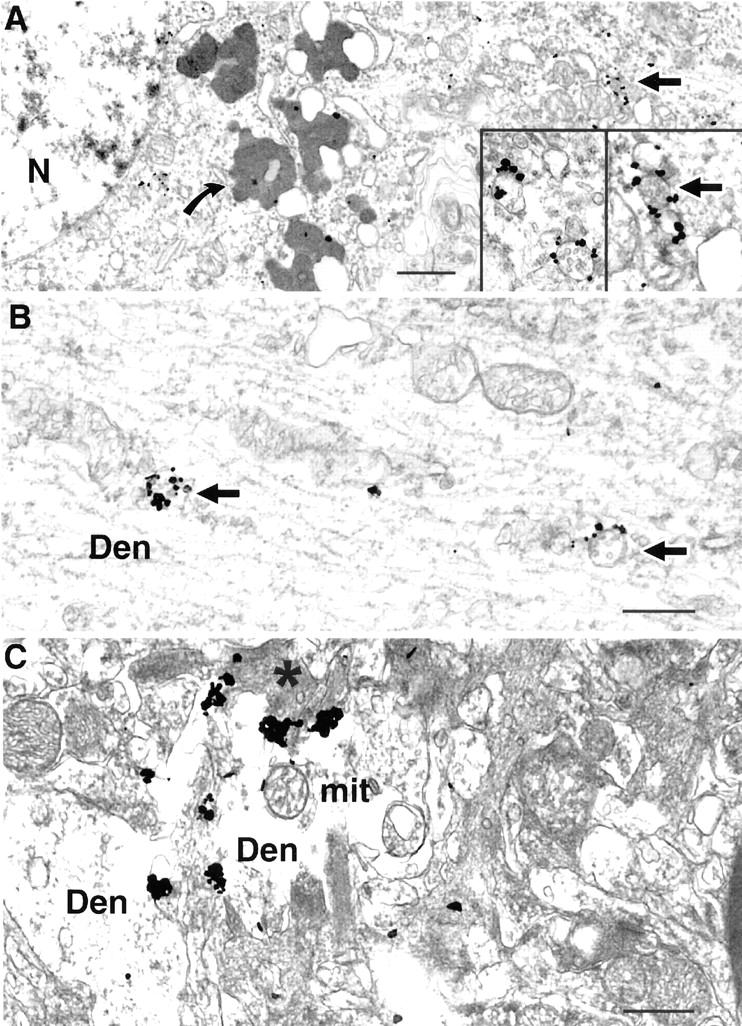
Aβ42 accumulates within neuronal soma and processes in human AD brain. A: Human AD cortical brain specimen showing substantial amounts of Aβ42 labeling within MVBs of an abnormal appearing neuronal soma (arrow); curved arrow indicates lipofuscin devoid of specific immunogold labeling. Nucleus has abnormal appearing accumulation of heterochromatin near the outer membrane. Insets represent higher power views of Aβ42 immunogold labeling of MVBs in neuronal soma. B: Multiple Aβ42 gold particles are associated with two MVBs in a dendrite. C: Multiple aggregated Aβ42 immunogold particles are found within a disrupted, swollen dendrite containing electron-dense material (asterisk), indicative of degeneration. Abbreviations: Den, dendrite; mit, mitochondrion; N, nucleus. Scale bars: 1 μm (A); 500 nm (B and C).
Immunoprecipitation/Western Blot for Aβ
Equal amounts (25 mg) of brain tissue snap-frozen in liquid nitrogen derived from analogous cortical regions from brains of Tg2576 mice sacrificed at different ages (3, 9, and 16 months) or N2a cells stably transfected with Swedish 670/671 FAD mutation human APP (generously provided by Drs. Gopal Thinakaran and Sangram Sisodia, University of Chicago, Chicago, IL) were assayed for determination of Aβ levels as previously described for cell lysates 28 with slight modifications. Samples from frontal and parietal cortex were analyzed for Aβ. Brain samples or N2a cells were lysed in 6% or 4% sodium dodecyl sulfate, respectively, vortexed, and heated at 95°C for several minutes, followed by sonication. Samples were spun at 15,000 × g, and supernatants adjusted to 190 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 8.3, 6 mmol/L ethylenediaminetetraacetic acid, and 2.5% Triton X-100 and incubated overnight with antibody 4G8 (Signet Laboratories), followed by addition of secondary rabbit anti-mouse antibody (Cappell) for 1 hour and then addition of protein A-Sepharose beads (Pharmacia) for 2 hours (all at 4°C). Samples were run on 10 to 20% Tris-Tricine sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels followed by electrophoretic transfer to polyvinylidene-difluoride membrane (Millipore, Bedford, MA). Polyvinylidene-difluoride membranes were boiled in PBS for 5 minutes, followed by Western blotting with 6E10 for human Aβ and visualization after enhanced chemiluminescence on Kodak X-OMAT AR5 film (Eastman-Kodak, Rochester, NY).
Results
Intraneuronal Aβ42 Resides on MVBs
In agreement with a previous report, 27 standard fixation of normal rat brain for EM with paraformaldehyde/glutaraldehyde revealed primarily isolated Aβ42 immunolabeling at the plasma membrane (data not shown). In contrast, fixation with a mixture of acrolein and paraformaldehyde used in the present study, which allows for more optimal antigen detection by immuno-EM, 33 revealed marked intraneuronal Aβ, especially Aβ42. In normal mouse (Figure 1A) ▶ and rat (Figure 1B) ▶ brain, Aβ42 was most prominent on multivesicular bodies (MVBs), especially of pyramidal neurons in the cerebral cortex and hippocampus, which are particularly prone to AD neuropathology. Intraneuronal Aβ42 was less pronounced in neurons known to be relatively resistant to AD, such as in the basal ganglia. More than half of Aβ42 gold particles labeling identifiable organelles within neurons in the brain of normal mouse or rat were found on MVBs. Moreover, rapidly fixed brain tissue from patients undergoing brain surgery for noninvasive neurological disease (n = 2, ages 44 and 54) also revealed Aβ42 especially on MVBs within neurons (Figure 1C) ▶ . In contrast to Aβ42, neuronal Aβ40 was not convincingly evident in normal mouse, rat, or human brain.
The monoclonal anti-Aβ42 antibody 27 did not show cross-reactivity to full-length APP or APP C-terminal fragments (CTFs) by Western analysis of either APP-transfected neuroblastoma (N2a) cells or Tg2576 mouse brain (Figure 1D) ▶ . Peptide competition abolished Aβ42 detection both by Western blot and by immunohistochemistry. Lack of cross-reactivity with APP also can be deduced from the fact that full-length APP/CTFs localized predominantly to the Golgi, although a minor amount was also detected in MVBs, of neuronal cell preparations (using antibody 369 directed at the C-terminus of APP). 34 Golgi predominance of full-length APP staining by immuno-EM with antibody 369 was now observed within neurons of the brain, and immuno-EM double labeling was used to demonstrate the differential staining of full-length APP/CTFs and Aβ42 (Figure 1E) ▶ . We confirmed our MBC42 Aβ42 immuno-EM labeling with the widely used polyclonal Aβ42 antibody from Chemicon, which gave analogous Aβ42 immunoreactivity (data not shown). In addition to the predominant site of Aβ42 on the outer membrane of small MVBs (∼60% of identifiable labeling in young 2- to 3-month-old mouse neuronal soma), Aβ42 immunogold particles were observed on very small vesicles (∼35% of identifiable labeling). Furthermore, immunogold labeling also was observed on ER (∼5% of identifiable labeling; Figure 1A ▶ ), similar to that previously observed in cultured neurons infected transiently with Semliki Forest virus containing human APP. 35 Aβ42 was only occasionally seen at the plasma membrane, and in contrast to full-length APP, only a minor amount of Aβ42 was observed on the Golgi apparatus compared with its predominance on MVBs (Figure 1) ▶ .
APP Knockout Mice Lack Intraneuronal MVB Aβ42
To further confirm the specificity of the Aβ immunoreactivity, well-characterized APP knockout mice (n = 4) 36 were compared with wild-type mice (n = 4) of equal age (two 6-month-old and two 12-month-old littermates) that were perfused and processed for immunogold EM in parallel. APP knockout mice did not demonstrate the Aβ42 on MVBs observed in normal wild-type mice (compare Figure 2, A and B ▶ ).
Figure 2.

Absence of intraneuronal Aβ42 in APP knockout mice. A: Immunogold labeling of several MVBs (arrows) in a neuronal perikaryon from a wild-type mouse brain. B: Absence of immunogold labeling of MVBs in a neuron from an APP knockout mouse. A and B were CA1 hippocampal sections from 12-month-old littermates processed in parallel. Insets in A and B represent lower power views to emphasize the lack of Aβ42 immunoreactivity in several neurons of APP knockout mouse brain; Aβ42 gold particles within several neuronal perikarya from wild-type mouse are evident (inset in A) compared with lack of gold particles in several neurons of a knockout mouse (inset in B) processed in parallel. Arrows indicate MVBs. Abbreviations: MVBs, multivesicular bodies; N, nucleus. Scale bars, 1 μm.
Intraneuronal Aβ42 Increases with Aging
Aβ was examined by immuno-EM in Tg2576 transgenic mice. Two independent previous Aβ ELISA studies demonstrated exponential brain Aβ increases occurring before Aβ plaque deposition in Tg2576 mice. 8,9 We used Aβ immunoprecipitation and Western blot to further confirm brain Aβ increases in our Tg2576 mice and observed analogous increases of brain Aβ (Figure 3A) ▶ as reported by ELISA, including preplaque between 3 and 9 months of age. To determine the location of Aβ increases, mice were perfused at different ages ranging from 2 months to 20 months (n = 16) and processed for immunogold EM localization of Aβ42. Relatively less Aβ42 was found in 2- to 4-month-old mice. In contrast, significantly greater amounts of intraneuronal Aβ42 were found in 10-month-old mice before the onset of plaque pathology (Figure 3, B and C) ▶ . The Aβ42 increases were most prominent on MVBs, particularly at nerve terminals (presynaptic and especially postsynaptic compartments). The immunogold labeling of Aβ42 with aging in brain tissue was quantified from representative young (2-month-old) and older (10-month-old) Tg2576 mice. The MVB-associated gold particles in neuronal cell bodies (n = 20 each, from young and old) or processes of cerebral cortex were counted. Neuronal soma from 2-month-old mice had 1.48 ± 0.3 gold particles per MVB whereas neurons from older mice had 2.02 ± 0.4 gold particles per MVB (Figure 3D) ▶ . This increase of MVB-associated gold particles in neuronal soma with aging did not reach statistical significance. Aβ42 increases were most pronounced within neuronal processes (profiles), especially small dendrites (<1.5 μm in diameter) and axon terminals. Thirty images of dendritic profiles were taken each from 2-month-old and from 10-month-old mice at random at the same magnification. The number of gold particles per MVB in processes was 1.50 ± 0.29 and 3.42 ± 0.45 for 2- and 10-month-old mice, respectively (Figure 3D ▶ ; P < 0.033). MVB-associated gold particles were only rarely found in dendrites of 2-month-old mice. In contrast, numerous MVBs labeled with several gold particles were found in 10-month-old mouse brain. No MVBs with three or more gold particles were observed in 2-month-old dendritic profiles, whereas nine MVBs with three or more gold particles were observed in 10-month-old processes (Figure 3E) ▶ . Lack of a specific marker for MVBs precludes biochemical quantification for potential changes in number of MVBs with aging in Tg2576 mice. Immuno-EM is not an optimal method for quantification and counting of MVBs by EM is further confounded by our observation that MVBs in distal processes of 10-month-old Tg2576 mice tended to be larger and contained a greater number of intravesicular vesicles than in processes of 2-month-old mice (compare Figure 3, B and C ▶ ). To reduce potential bias from gold particles, we quantified total MVBs in dendrites based on morphology by EM without immunogold labeling. The number of MVBs almost doubled in dendrites of young 2-month-old compared to older 10-month-old preplaque Tg2576 mice (1.96-fold increase; P < 0.03).
Aβ42 Accumulation within Nerve Terminals Is Associated with Neuropathology
Before the development of AD plaque pathology, we observed accumulation of Aβ42 presynaptically within axon terminals and especially postsynaptically within distal dendrites. At 10 months of age and before Aβ plaques, dendrites were found that displayed multiple gold particles and morphological alterations, such as atypically dark Aβ42 immunogold-associated MVBs, unusual clusters of ER in the vicinity of the Aβ42 immunogold-associated MVBs, and abnormally collapsed, dark membranes consistent with degenerating processes in the vicinity of especially numerous gold particles (Figure 4A) ▶ . At later ages, when plaques were forming, Aβ42-accumulating processes were more numerous and showed strikingly abnormal morphological changes in the absence of an associated Aβ plaque (Figure 4 ▶ ; B, C, and D). Increased intraneuronal Aβ42 immunogold labeling often was affiliated with swollen processes, sometimes within areas lacking normal appearing organelles or including dark structures resembling degeneration (Figure 4 ▶ ; B, C, and D). Aβ42 accumulation could also be seen associated with disrupted appearing MVBs (Figure 4B) ▶ . The presence of abnormal appearing APP-containing neurites within plaques has been known 37 and isolated Aβ fibrils have been noted in such dystrophic neurites, 38 but this is the first study showing that Aβ42 accumulates in synaptic compartments and is associated with morphological changes. Interestingly, in a recent study isolated neurons in Down’s syndrome brain with marked intraneuronal Aβ42 accumulation were reported to display concomitant terminal dUTP nick-end labeling staining indicative of apoptosis. 26 In contrast to Aβ42, Aβ40 increases were less evident in Aβ42-accumulating neurites, but Aβ40 was seen to accumulate in Aβ extracellular plaques and vessel walls (data not shown).
In human AD cortical brain tissue (n = 2, ages 54 and 62), as in the case of the transgenic mice, we observed Aβ42 accumulation especially within neuronal processes and associated abnormal morphological changes (Figure 5) ▶ . Compared with non-AD cortical biopsy tissue, AD cortex had marked increases of Aβ42 immunogold particles both in soma and processes, even including areas not directly associated with Aβ plaques. MVBs with 5 to 20 gold particles, not seen in normal human brain, could be seen in human AD brain (Figure 5B) ▶ . In AD brain, Aβ42 immunogold accumulation was seen in disrupted swollen dendritic profiles displaying degenerative changes (Figure 5C) ▶ . In AD and older plaque bearing Tg2576 mouse brain, astrocytes and microglia in the vicinity of Aβ plaques contained Aβ42; this Aβ42 was found especially within bundles of intermediate filaments in fibrous astrocytes (data not shown).
Discussion
This study demonstrates that MVBs are the major subcellular site of accumulation of Aβ42 within neurons. We provide data supporting a pathogenic role for intraneuronally accumulating Aβ42 because in Tg2576 mice and in AD brain it is directly associated with abnormal cellular morphology. The finding that Aβ42 accumulation and early cellular pathology occurs specifically in distal processes and at synapses could explain observations of early decreases in synaptic markers in preplaque AD transgenic mice. 12 The observations that substantial Aβ accumulates within synaptic compartments, and is associated with cellular pathology, provides a molecular basis for the clinical observation correlating severity of dementia with markers of synaptic loss. 39
Aβ peptides were initially believed to be an abnormal and toxic by-product of APP metabolism, until it was found in the early 1990s that all cells seem to normally secrete Aβ. 1 Aβ was subsequently thought to be generated and rapidly secreted at the plasma membrane, until cell biological studies demonstrated that Aβ peptides can be found intracellularly 3 and even increase in amount with neuronal maturation or time in culture. 3,18 Our observation of Aβ42 associated with MVBs in normal brain raises the possibility that this small hydrophobic peptide may have a normal biological function. Even though APP has been studied intensively since its identification as the precursor to Aβ in the late 1980s, the normal function of APP remains unknown. APP knockout mice were not found to have an obvious abnormal phenotype, 36 but subsequent studies have indicated that APP knockout neurons have decreased neurite extension in tissue culture 40 and deficits in synaptic plasticity. 41 The normal presence and increases with aging of Aβ42, the first Aβ peptide found in early diffuse plaques in Down’s syndrome and AD, within neurons in Tg2576 mice and in AD brain, also in areas not adjacent to plaques, suggests that this intracellular Aβ may be the site of Aβ aggregation and neurotoxicity, subsequently leading to “extracellular” appearing Aβ plaques. We and others reported images of Aβ plaques seemingly originating from within neuronal soma in AD. 2,22 We did not observe such images in Tg2576 mice. We hypothesize that the formation of plaques and lack of neuron cell death seen in APP FAD mutant transgenic mice may reflect Aβ42 accumulation within distant neuronal processes with synaptosis and plaque formation, with lesser Aβ accumulation within soma, thereby avoiding cell death, possibly secondary to the absence of human τ. 42,43
Our study does not provide information on the NH2-terminus of MVB-associated Aβx-42, with AβAsp1 and AβGlu11 being major NH2-terminal Aβ peptides generated by neurons. 46,47 Because the earliest plaques with AD pathology have been reported to be NH2 terminally truncated Aβx-42, 46 we, and others, have hypothesized that NH2-terminally truncated Aβ42, such as AβGlu11-42, may be especially important in AD pathogenesis. This study again suggests, as did our and other previous studies, 22-26 that Aβx-42, and not Aβx-40, is the major intraneuronal Aβ peptide in brain. Tissue culture studies and brain ELISA studies of AD and Tg2576 mouse brain demonstrate considerable levels of both Aβ40 and Aβ42 peptides. Brain Aβ40 may either not be visualized as well by immunohistochemical methods or may mainly reflect that associated with blood vessels and Aβ plaques. In the present study, some Aβ40 immunogold labeling, although less apparent than Aβ42, was observed on neuronal MVBs in brain with aging.
Relatively little is known about the biological function of MVBs, which are defined by their ultrastructural appearance on EM. No clear distinction exists between MVBs and endosomes, except that MVBs tend to be regarded as smaller than, and potentially even be derived from, endosomes. In normal brain, Aβ42 appeared to localize especially to small MVBs, but with aging Aβ42 could also be observed on larger MVB/endosomes. MVBs are generally viewed as part of the endosomal/lysosomal system and both early and, more typically, late endosomes have been described as MVBs. 47,48 Aβ is known to be generated in the endosomal-lysosomal system 10,11 and early lysosomal abnormalities in AD have been described. 49,50 APP metabolism within Niemann-Pick cells demonstrated a pool of accumulating intracellular Aβ42 in endosomes that is differentially regulated from the constitutive Aβ secretory pathway. 51,52 Possibly this pool of Aβ42 represents the MVB-associated pool of Aβ42. The ER has been described as an important site of Aβ42 formation and we observed some Aβ42 in ER of neurons in brain. Indeed, MVBs are often seen in the vicinity of the ER and may represent extensions of smooth ER. 32 Similarly, the Golgi is a major site of Aβ generation in the secretory pathway and MVBs also are Golgi associated (Figure 1E) ▶ . 32 We also observed Aβ42 in very small vesicles that may represent recycling vesicles between MVBs and major intracellular organelles and/or the plasma membrane. With Aβ42 accumulation on MVBs with aging and AD pathology, structures representing disrupted MVBs were observed (Figure 4B) ▶ . Because pre-embedding immuno-EM generally underestimates actual quantity of antigen, such MVB disruption could lead to release of enough Aβ42 to induce neurotoxicity. Specifically Aβ42 treatment of cells was reported to induce leakage of lysosomal contents into the cytoplasm. 53 Recently, it was demonstrated that Aβ1-42, but not Aβ1-40 or Aβ42-1, is exquisitely neurotoxic when introduced into the cytosol of cultured primary neurons. 54 Thus, leakage of Aβ42 from MVBs into the cytosol may be a critical neurotoxic event in AD.
The mechanism whereby intracellular Aβ accumulates, aggregates, and is neurotoxic to neurons remains to be elucidated. Our studies suggest but do not prove that Aβ42 is the toxic entity. It was recently reported that BACE, presenilin 1, and APP are all transported along axons in membrane vesicles that require APP/kinesin-1 interactions and that vesicle preparations from peripheral nerves can generate Aβ. 55 Based on these findings and the fact that these vesicles also carry additional vital cargo proteins, such as the neurotrophin receptor, TrkA, Kamal and colleagues 55 hypothesized that loss of trophic support to distal synapses from aberrant Aβ accumulation or altered APP function could hold up this cargo and lead to neurodegeneration in AD. 55,56 A neuron-specific C-terminal type kinesin superfamily protein (KIFC2) was found to be associated with MVBs by immuno-EM and to be important for dendritic transport of MVBs. 57 Our study supports the scenario that accumulating Aβ42 located on the outer membrane of MVBs could directly associate and disrupt kinesin (and possibly even τ-)-mediated transport of MVBs.
Intracellular aggregation of proteins is increasingly being linked to neurodegenerative diseases, 58 including AD. FAD mutations increase not only secreted, but also intracellular Aβ42. 59,60 Recently, a FAD mutation was discovered that leads not to increased but decreased levels of Aβ secretion, and to increased generation of protofibrils, leading the authors to suggest that intracellular Aβ increases may be especially important for AD. 61 The present study supports the concept that intracellular Aβ is important in the genesis of AD. Previous experiments indicated that radiolabeled Aβ42 can be internalized by neurons, 62 suggesting that one route of intraneuronal Aβ may be via neuronal uptake and subsequent intracellular accumulation. A recent study demonstrated that epidermal growth factor and transforming growth factor-α internalize after binding to the epidermal growth factor receptor initially via clathrin-coated vesicles, but at 30 minutes of chase are found on the outer membrane of MVBs, 63 analogous to the location of Aβ42 on the outside membrane of MVBs. Thus, MVB-associated Aβ42 may be derived from endocytosis at the plasma membrane. Internally generated Aβ might be expected to be prominent at synapses, because APP, and therefore Aβ, is transported down axons 64 and dendrites. 65 Indeed, the Aβ domain of APP appears critical for this sorting of APP for transport. 65 Progressive accumulation and aggregation of intracellular Aβ42 in dendrites/distal processes and presynaptic and postsynaptic compartments could lead to dystrophic neurite swellings, further APP conversion to Aβ42 within these processes, release of this Aβ42 from normal compartments, disruption of distal processes and synapses, and then eventually to dissolution of processes/synapses with the emergence of “extracellular” Aβ plaques. The mechanism(s) by which Aβ42 increases on MVBs, aggregates, and becomes neurotoxic within neurons and their processes, remain(s) to be determined. A better understanding of the biogenesis and biological role(s) of MVBs and a more detailed understanding of the subcellular events involved in Aβ-induced AD neuropathology may be of value in the development of more effective therapies for AD.
Acknowledgments
We thank Dr. Jean Paul Vonsattel, Columbia University, for helpful discussions.
Footnotes
Address reprint requests to Dr. Gunnar K. Gouras, Department of Neurology and Neuroscience, Weill Medical College of Cornell University, 525 East 68th St., New York, NY 10021. E-mail: gkgouras@med.cornell.edu.
Supported by the National Institutes of Health (grants NS02037, AG09464, and HL18974), the Alzheimer’s Association, the American Academy of Neurology Education and Research Foundation, and Paul Beeson Physician Faculty Scholar Award (to G. K. G.).
References
- 1.Selkoe DJ: Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 2001, 81:741-766 [DOI] [PubMed] [Google Scholar]
- 2.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR: Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 2000, 156:15-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson CA, Doms RW, Lee VM: Intracellular APP processing and A beta production in Alzheimer disease. J Neuropathol Exp Neurol 1999, 58:787-794 [DOI] [PubMed] [Google Scholar]
- 4.Hartmann T: Intracellular biology of Alzheimer’s disease amyloid beta peptide. Eur Arch Psychiatry Clin Neurosci 1999, 249:291-298 [DOI] [PubMed] [Google Scholar]
- 5.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K: Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 1998, 4:97-100 [DOI] [PubMed] [Google Scholar]
- 6.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK: Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 1999, 2:271-276 [DOI] [PubMed] [Google Scholar]
- 7.Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F: Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 1999, 274:6483-6492 [DOI] [PubMed] [Google Scholar]
- 8.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG: Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 2001, 21:372-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM: Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci 2001, 21:4183-4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koo EH, Squazzo SL: Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 1994, 269:17386-17389 [PubMed] [Google Scholar]
- 11.Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH: Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta 42. J Biol Chem 1999, 274:18851-18856 [DOI] [PubMed] [Google Scholar]
- 12.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L: Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 1999, 96:3228-3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G: Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274:99-102 [DOI] [PubMed] [Google Scholar]
- 14.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT: APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 1997, 56:965-973 [DOI] [PubMed] [Google Scholar]
- 15.Teller JK, Russo C, DeBusk LM, Angelini G, Zaccheo D, Dagna-Bricarelli F, Scartezzini P, Bertolini S, Mann DM, Tabaton M, Gambetti P: Presence of soluble amyloid beta-peptide precedes amyloid plaque formation in Down’s syndrome. Nat Med 1996, 2:93-95 [DOI] [PubMed] [Google Scholar]
- 16.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL: Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 1999, 46:860-866 [DOI] [PubMed] [Google Scholar]
- 17.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ: The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry 2000, 39:10831-10839 [DOI] [PubMed] [Google Scholar]
- 18.Skovronsky DM, Doms RW, Lee VM: Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol 1998, 141:1031-1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T: Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med 1999, 5:560-564 [DOI] [PubMed] [Google Scholar]
- 20.Askanas V, Engel WK: Inclusion-body myositis: newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuropathol Exp Neurol 2001, 60:1-14 [DOI] [PubMed] [Google Scholar]
- 21.Ginsberg SD, Galvin JE, Lee VM, Rorke LB, Dickson DW, Wolfe JH, Jones MZ, Trojanowski JQ: Accumulation of intracellular amyloid-beta peptide (A beta 1-40) in mucopolysaccharidosis brains. J Neuropathol Exp Neurol 1999, 58:815-824 [DOI] [PubMed] [Google Scholar]
- 22.D’Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH: Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 2001, 38:120-134 [DOI] [PubMed] [Google Scholar]
- 23.Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC: Intraneuronal Abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med 2001, 125:489-492 [DOI] [PubMed] [Google Scholar]
- 24.Mochizuki A, Tamaoka A, Shimohata A, Komatsuzaki Y, Shoji S: Abeta42-positive non-pyramidal neurons around amyloid plaques in Alzheimer’s disease. Lancet 2000, 355:42-43 [DOI] [PubMed] [Google Scholar]
- 25.Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA: Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 2001, 306:116-120 [DOI] [PubMed] [Google Scholar]
- 26.Busciglio J, Pelsman A, Wong C, Pigino G, Yuan ML, Mori H, Yankner BA: Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron 2002, 33:677-688 [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi H, Maat-Schieman ML, van Duinen SG, Prins FA, Neeskens P, Natte R, Roos RA: Amyloid beta protein (Abeta) starts to deposit as plasma membrane-bound form in diffuse plaques of brains from hereditary cerebral hemorrhage with amyloidosis-Dutch type, Alzheimer disease and nondemented aged subjects. J Neuropathol Exp Neurol 2000, 59:723-732 [DOI] [PubMed] [Google Scholar]
- 28.Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, Xu H: Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci 2001, 21:2561-2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P: Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA 1990, 87:6003-6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milner TA, Lee A, Aicher SA, Rosin DL: Hippocampal alpha2a-adrenergic receptors are located predominantly presynaptically but are also found postsynaptically and in selective astrocytes. J Comp Neurol 1998, 395:310-327 [PubMed] [Google Scholar]
- 31.Chan J, Aoki C, Pickel VM: Optimization of differential immunogold-silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods 1990, 33:113-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters A, Palay SL, Webster HD: The Fine Structure of the Nervous System. 1991. Oxford University Press, Oxford
- 33.Leranth C, Pickel VM: Neuroanatomical Tract Tracing Methods 2. 1989:pp 120-170 Plenum, New York
- 34.Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P: Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci 1994, 14:3122-3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K: Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med 1997, 3:1016-1020 [DOI] [PubMed] [Google Scholar]
- 36.Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW: Beta-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 1995, 81:525-531 [DOI] [PubMed] [Google Scholar]
- 37.Perry G, Siedlak S, Mulvihill P, Kancherla M, Mijares M, Kawai M, Gambetti P, Sharma S, Maggiora L, Cornette J: Immunolocalization of the amyloid precursor protein within the senile plaque. Prog Clin Biol Res 1989, 317:1021-1025 [PubMed] [Google Scholar]
- 38.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D: Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer’s disease. J Neurosci 1996, 16:5795-5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R: Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991, 30:572-580 [DOI] [PubMed] [Google Scholar]
- 40.Perez RG, Zheng H, Van der Ploeg LH, Koo EH: The beta-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J Neurosci 1997, 17:9407-9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJS: Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 1999, 38:349-359 [DOI] [PubMed] [Google Scholar]
- 42.Gotz J, Chen F, van Dorpe J, Nitsch RM: Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Abeta 42 fibrils. Science 2001, 293:1491-1495 [DOI] [PubMed] [Google Scholar]
- 43.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E: Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293:1487-1491 [DOI] [PubMed] [Google Scholar]
- 44.Gouras GK, Xu H, Jovanovic JN, Buxbaum JD, Wang R, Greengard P, Relkin NR, Gandy S: Generation and regulation of beta-amyloid peptide variants by neurons. J Neurochem 1998, 71:1920-1925 [DOI] [PubMed] [Google Scholar]
- 45.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC: BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 2001, 4:233-234 [DOI] [PubMed] [Google Scholar]
- 46.Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ: Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 1996, 3:16-32 [DOI] [PubMed] [Google Scholar]
- 47.Gruenberg J, Maxfield FR: Membrane transport in the endocytic pathway. Curr Opin Cell Biol 1995, 7:552-563 [DOI] [PubMed] [Google Scholar]
- 48.Katzmann DJ, Babst M, Emr SD: Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106:145-155 [DOI] [PubMed] [Google Scholar]
- 49.Nixon RA, Cataldo AM, Mathews PM: The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: a review. Neurochem Res 2000, 25:1161-1172 [DOI] [PubMed] [Google Scholar]
- 50.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA: Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 2000, 157:277-286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamazaki T, Chang TY, Haass C, Ihara Y: Accumulation and aggregation of amyloid {beta}-protein in late endosomes of Niemann-Pick type C cells. J Biol Chem 2000, 276:4454-4460 [DOI] [PubMed] [Google Scholar]
- 52.Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T: Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci 2002, 22:1679-1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG: Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res 1998, 52:691-698 [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, McLaughlin R, Goodyer C, LeBlanc A: Selective cytotoxicity of intracellular amyloid {beta} peptide1-42 through p53 and Bax in cultured primary human neurons. J Cell Biol 2002, 156:519-529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS: Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 2001, 414:643-648 [DOI] [PubMed] [Google Scholar]
- 56.Sisodia SS: Biomedicine: a cargo receptor mystery APParently solved? Science 2002, 295:805-807 [DOI] [PubMed] [Google Scholar]
- 57.Saito N, Okada Y, Noda Y, Kinoshita Y, Kondo S, Hirokawa N: KIFC2 is a novel neuron-specific C-terminal type kinesin superfamily motor for dendritic transport of multivesicular body-like organelles. Neuron 1997, 18:425-438 [DOI] [PubMed] [Google Scholar]
- 58.Trojanowski JQ, Lee VM: “Fatal attractions” of proteins. A comprehensive hypothetical mechanism underlying Alzheimer’s disease and other neurodegenerative disorders. Ann NY Acad Sci 2000, 924:62-67 [DOI] [PubMed] [Google Scholar]
- 59.Wild-Bode C, Yamazaki T, Capell A, Leimer U, Steiner H, Ihara Y, Haass C: Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J Biol Chem 1997, 272:16085-16088 [DOI] [PubMed] [Google Scholar]
- 60.Martin BL, Schrader-Fischer G, Busciglio J, Duke M, Paganetti P, Yankner BA: Intracellular accumulation of beta-amyloid in cells expressing the Swedish mutant amyloid precursor protein. J Biol Chem 1995, 270:26727-26730 [DOI] [PubMed] [Google Scholar]
- 61.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L: The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci 2001, 4:887-893 [DOI] [PubMed] [Google Scholar]
- 62.Bahr BA, Hoffman KB, Yang AJ, Hess US, Glabe CG, Lynch G: Amyloid beta protein is internalized selectively by hippocampal field CA1 and causes neurons to accumulate amyloidogenic carboxyterminal fragments of the amyloid precursor protein. J Comp Neurol 1998, 397:139-147 [PubMed] [Google Scholar]
- 63.Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH: Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 2002, 156:843-854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL: Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA 1990, 87:1561-1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tienari PJ, De Strooper B, Ikonen E, Simons M, Weidemann A, Czech C, Hartmann T, Ida N, Multhaup G, Masters CL, Van Leuven F, Beyreuther K, Dotti CG: The beta-amyloid domain is essential for axonal sorting of amyloid precursor protein. EMBO J 1996, 15:5218-5229 [PMC free article] [PubMed] [Google Scholar]