

Abstract
Gastric fundic gland polyps (FGPs) occur in two distinct clinicopathological scenarios: sporadic and familial adenomatous polyposis (FAP) associated. FAP-associated FGPs arise through somatic second hit alterations of the adenomatous polyposis coli (APC) gene and frequently demonstrate epithelial dysplasia (Am J Pathol 2000, 157:747–754). Sporadic FGPs, in contrast, tend to contain β-catenin gene mutations and only infrequently show dysplasia (Am J Pathol 2001, 158:1005–1010). However, sporadic FGPs with dysplasia have not been previously investigated. We studied 13 sporadic FGPs with surface/foveolar low-grade dysplasia or changes indefinite for dysplasia for alterations in the APC/β-catenin pathway, using chromosome 5q allelic loss assays and direct DNA sequencing of the mutation cluster region in exon 15 of APC and the phosphorylation region in exon 3 of β-catenin. In addition, to evaluate for possible additional genetic alterations in FGPs, all cases were evaluated for microsatellite instability using fluorescent-based amplification of a standard panel of five microsatellite markers. Alterations in APC were present in seven (53.8%) FGPs, including two cases with bi-allelic APC inactivation (truncating intragenic mutation plus 5q allelic loss), two cases with APC mutation only, and three cases with 5q allelic loss only. In contrast, only two (15.4%) FGPs contained stabilizing β-catenin mutations. All 13 FGPs were microsatellite stable. These results indicate that sporadic FGPs with dysplasia/indefinite for dysplasia are molecularly similar to FAP-associated FGPs, and are dissimilar to the more common sporadic nondysplastic FGPs. Mutations in APC and β-catenin, despite occurring in the same genetic pathway, show differing biological properties, a phenomenon that has previously been demonstrated in colorectal neoplasms. The lack of microsatellite instability in FGPs in this study and of K-ras mutations in a previous study suggests that secondary genetic alterations are rare in both dysplastic and nondysplastic FGPs.
Fundic gland polyps (FGPs) constitute the most common gastric polyps. 1 FGPs arise in two distinct clinicopathological scenarios: sporadic and syndromic. Sporadic FGPs are encountered in 0.9 to 1.9% of patients who undergo upper endoscopic evaluation, and are most frequent in middle-aged women. 2-4 Syndromic FGPs are found in patients with familial adenomatous polyposis (FAP), a setting in which the polyps are frequently multiple and can occur even at young ages. 3-6 Histopathologically, in both settings, these distinctive corpus- or fundic-based polyps are composed of cystically dilated oxyntic glands containing attenuated parietal cells, chief cells, and mucous neck cells (Figure 1) ▶ . Most commonly, the surface epithelium of FGPs closely resembles the normal gastric surface epithelium, with a single layer of nondysplastic gastric mucin cells, each containing an apical cap of neutral mucin (Figure 1) ▶ .
Figure 1.
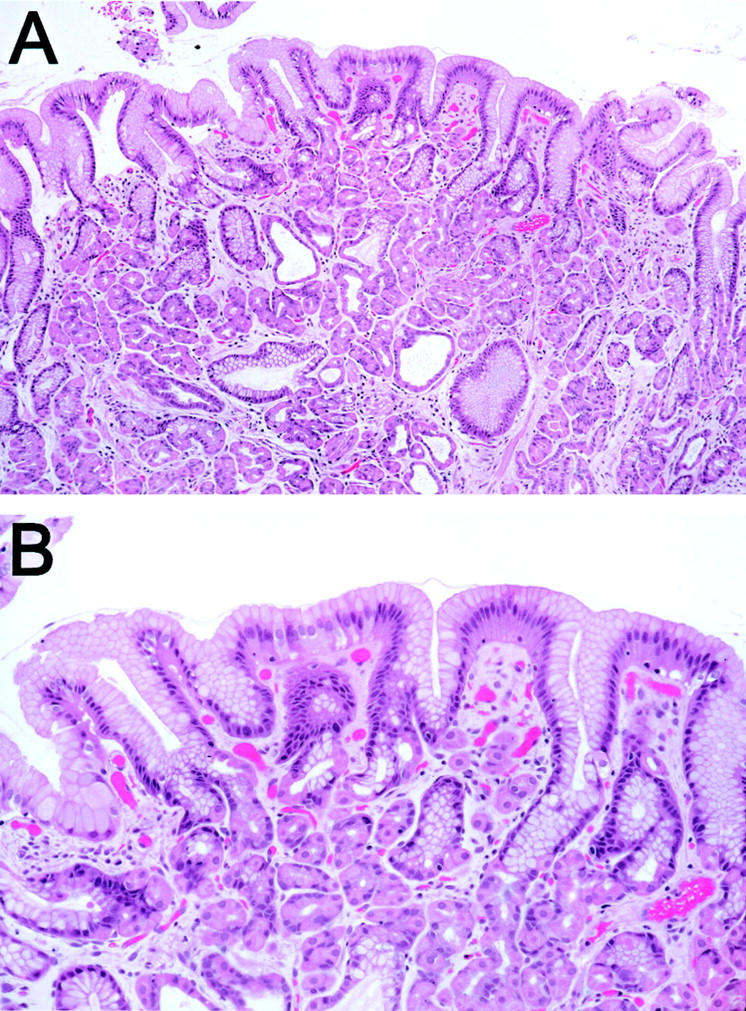
Histological appearance of a nondysplastic FGP. A: At low power, the FGP shows characteristic, cystically dilated, and budded oxyntic glands. B: At higher power, the surface and foveolar epithelium overlying the dilated glandular compartment is composed of nondysplastic gastric mucin cells, with small, basally oriented nuclei and abundant apical mucin.
In some FGPs, dysplasia of the foveolar epithelium is encountered. Dysplasia in FGPs is characterized by crowding of surface and neck gastric mucin cells, which display enlarged hyperchromatic nuclei and loss of cytoplasmic mucin. 7,8 However, the underlying cystically dilated oxyntic glands in these polyps do not demonstrate epithelial dysplasia. 7,8 The frequency of dysplasia in FGPs is closely associated with the setting in which they arise. Dysplasia in FAP-associated FGPs is common, whereas it is rare in sporadic FGPs. In a large study, Wu and colleagues 7 found low-grade dysplasia in 25% of FAP-associated FGPs, but in only 3% of sporadic FGPs; furthermore, immunohistochemical markers of epithelial proliferation were dysregulated in both those FGPs with dysplasia and those considered indefinite for dysplasia.
We have recently studied the genetic alterations in FGPs and have found evidence for dysregulation of the adenomatous polyposis coli (APC)/β-catenin pathway similar to that previously demonstrated in colorectal adenomas. 8,9 Interestingly, these studies and others have suggested that divergent genetic alterations are present in FAP-associated and sporadic FGPs. FAP-associated FGPs, with their attendant risk of epithelial dysplasia, arise through second-hit inactivating APC gene mutations or chromosome 5q allelic loss, 9,10 whereas sporadic FGPs that are negative for dysplasia instead show a high rate of activating β-catenin mutations. 8,11,12 However, sporadic FGPs with dysplasia have not yet been evaluated. In this study we examine the role of the APC/β-catenin pathway and microsatellite instability in a series of sporadic FGPs with epithelial dysplasia or cellular changes indefinite for dysplasia.
Materials and Methods
Specimen Selection and Histological Evaluation
The study population consisted of 13 sporadic FGPs (designated SD1 to SD13) that demonstrated either low-grade epithelial dysplasia (eight cases) or surface/foveolar changes indefinite for dysplasia (five cases). Cases were accrued both by prospective evaluation of FGPs seen at routine sign-out between March 2001 to March 2002, as well as by a retrospective search of the computerized Surgical Pathology files at The Johns Hopkins Hospital for FGPs in non-FAP patients that contained “dysplasia” or “indefinite for dysplasia” in the diagnostic report between January 1995 to March 2002. On rereview of the formalin-fixed, hematoxylin and eosin (H&E)-stained histological sections, FGPs were graded as either low-grade dysplasia or indefinite for dysplasia according to previously published criteria. 7 Briefly, FGPs with low-grade dysplasia showed nuclear enlargement, hyperchromatism, crowding, and decreased intracellular mucin content in the surface/foveolar epithelium (Figure 2) ▶ . FGPs graded as indefinite for dysplasia had milder cytological changes that were considered insufficient for a diagnosis of low-grade dysplasia (Figure 2) ▶ .
Figure 2.
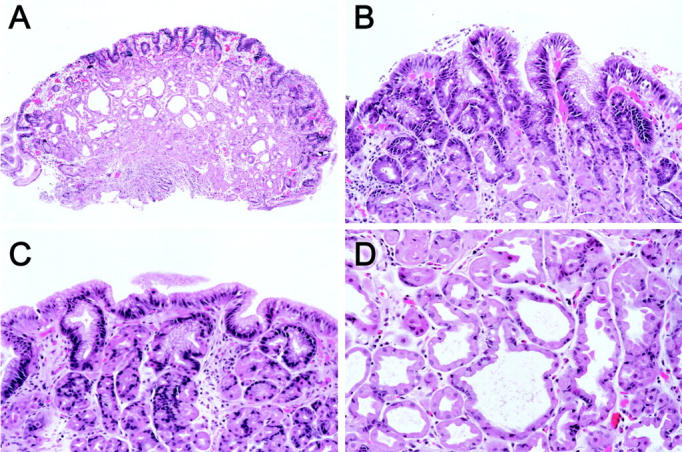
Histological features of sporadic FGPs with dysplasia. A: At low power, the characteristic architecture of a FGP is seen. However, the surface appears hyperchromatic in comparison with the FGP in Figure 1 ▶ . B: Low-grade dysplasia of the surface/foveolar epithelium. The nuclei are enlarged, hyperchromatic, and crowded, and there is decreased cytoplasmic mucin content. C: Epithelial changes indefinite for dysplasia. The cytological features are intermediate between those of the nondysplastic FGP in Figure 1 ▶ and the FGP with low-grade dysplasia in B. D: The attenuated parietal cells and chief cells lining the dilated oxyntic glands do not demonstrate dysplasia.
DNA Extraction
Microdissection of H&E-stained slides for DNA extraction was performed from formalin-fixed, paraffin-embedded specimens using a 27½-guage tuberculin needle. In one FGP (SD1), the surface/foveolar epithelium and the underlying dilated oxyntic glands were separately microdissected and sequenced. We did not separately microdissect the glandular and surface/foveolar compartments in the remaining 12 FGPs, because we have previously shown that there is excellent correlation between the genetic status of the two compartments. 8,9 Normal control DNA was microdissected from nonpolypoid gastric mucosa (10 cases), duodenum (two cases), and liver (one case). Genomic DNA was extracted as described previously. 13
Mutation Analysis of the β-Catenin and APC Genes
Direct DNA sequencing of DNA was used to evaluate for mutations in the β-catenin and APC genes. For β-catenin, genomic DNA was amplified by polymerase chain reaction (PCR) using the primer pair: 5′-ATGGAACCAGACAGAAAAGC-3′ (forward) and 5′-GCTACTTGTTCTGAGTGAAG-3′ (reverse). These amplified a 200-bp fragment in exon 3 of the β-catenin gene that encompasses the region for glycogen synthase kinase-3β (GSK-3β) phosphorylation and subsequent ubiquitin-mediated degradation of the β-catenin protein. PCR reactions were performed under standard conditions in a 50-μl volume containing 2 μl of genomic DNA, 10 mmol/L dNTP mix, 2.25 U AmpliTaq Gold (Applied Biosystems, Foster City, CA), 0.125 U PFU DNA Polymerase (Stratagene, La Jolla, CA), and 20 pmol of forward and reverse primers. PCR conditions consisted of an initial denaturation at 95°C for 10 minutes, 40 cycles of 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes, and a final extension at 72°C for 10 minutes on a GeneAmp PCR System 9700 (Applied Biosystems). For APC, four sets of oligonucleotide primers (A1: 5′-CAGACTTATTGTGTAGAAGA-3′ and A2: 5′-CTCCTGAAGAAAATTCAACA-3′ for codons 1260 to 1359; B1: 5′-AGGGTTCTAGTTTATCTTCA-3′ and B2: 5′-TCTGCTTGGTGGCATGGTTT-3′ for codons 1339 to 1436; C1: 5′-GGCATTATAAGCCCCAGTGA-3′ and C2: 5′-AAATGGCTCATCGAGGCTCA-3′ for codons 1417 to 1516; D1: 5′-ACTCCAGATGGATTTTCTTG-3′ and D2: 5′-GGCTGGCTTTTTTGCTTTAC-3′ for codons 1497 to 1596) were used to amplify the mutation cluster region in exon 15 of the APC gene. 14 PCR reactions were performed in a reaction mixture as described above. PCR conditions for APC consisted of an initial denaturation step of 95°C for 10 minutes, 40 cycles (94°C for 1 minute, 55°C for 1 minute, and 68°C for 1.5 minutes for APC-B, -C, and -D primer pairs and 94°C for 1 minute, 52°C for 1 minute, and 68°C for 1.5 minutes for APC-A), followed by a final extension at 72°C for 10 minutes.
PCR products were purified using shrimp alkaline phosphatase and exonuclease I (Amersham, Buckinghamshire, UK). The purified PCR products were sequenced on an ABI Prism 3700 DNA Analyzer (Applied Biosystems) using the ABI Prism Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems). The same primers were used for both amplification and sequencing. The resulting sequencing data were analyzed using GeneScan Analysis software (Applied Biosystems) in accordance with the manufacturer’s protocol. All mutations were verified in both forward and reverse directions.
Allelic Loss on Chromosome 5q
Allelic loss on 5q was evaluated by microsatellite assays using three microsatellite markers (D5S82, D5S299, and D5S346). Assays were performed by fluorescent-labeled PCR amplification using fluorescent dye-labeled forward primer and unlabeled reverse primer. The forward primer was end-labeled with 6-FAM (Applied Biosystems). PCR reactions were performed in 15-μl reaction volumes that contained 40 ng of genomic DNA, 9 μl of ABI Prism True Allele PCR Premix (Applied Biosystems), 5 pmol of 6-FAM-labeled forward primer, and 10 pmol of unlabeled reverse primer. The following cycling conditions were used: denaturation at 95°C for 6 minutes; 45 cycles of 94°C for 45 seconds, 55°C for 45 seconds, and 72°C for 1 minute; and a final extension at 72°C for 30 minutes. The resulting PCR products were diluted with 30 μl of H2O, and a 1.0-μl aliquot of each diluted fluorescent-labeled PCR product was combined with 12 μl of formamide and 0.5 μl of GeneScan 400HD (ROX) size standard (Applied Biosystems). The samples were then capillary electrophoresed on an ABI 3700 DNA Analyzer and analyzed using GeneScan Analysis software. Allelic loss was considered to be present when there was at least a 50% reduction in the height of a heterozygous peak as compared with normal control DNA in at least one informative microsatellite marker.
Microsatellite Instability Analysis
MSI testing was performed using the five microsatellite loci (D2S123, D5S346, D17S250, Bat-25, and Bat-26) recommended by the 1997 National Cancer Institute (NCI)-sponsored consensus conference. 15 Assays were performed as described above for 5q allelic loss. Chromatograms were interpreted according to the criteria described in detail by Berg and colleagues. 16 As per NCI criteria, high-level microsatellite instability (MSI-H) is defined as shifting in at least two of five microsatellite loci, low-level microsatellite instability (MSI-L) as shifting in only one locus, and microsatellite stable (MSS) as stability in all five loci. 15
Statistical Analysis
Fisher’s exact test was used to compare gene mutation frequencies in the sporadic FGPs in the current study with those of previously published FAP-associated FGPs 8 and sporadic nondysplastic FGPs. 9,11 Two-tailed P values of <0.05 were considered statistically significant.
Results
Clinical Findings
The 13 FGPs with dysplasia/indefinite for dysplasia were present in 12 patients, including 8 (67%) women and 4 (33%) men. Mean patient age at the time of upper endoscopy was 61 years (range, 37 to 71 years). None of the patients had a clinical history of FAP and none was enrolled in the Johns Hopkins Familial Colorectal Cancer Registry. Review of previous and subsequent surgical pathology specimens in all cases revealed that none of the patients had extraintestinal manifestations of FAP such as desmoid tumors. Only four patients had undergone colonoscopy and biopsy at The Johns Hopkins Hospital; of these four patients, two were negative for colorectal adenomas, one had multifocal polypoid and flat dysplasias in the setting of ileocolonic Crohn’s disease, and one had several colorectal adenomas. None had duodenal or gastric adenomas.
Single FGPs were biopsied in six patients and several FGPs in six patients; however, none of the patients had fundic gland polyposis (>10 FGPs). 11 In one patient with several FGPs, one polyp showed low-grade dysplasia, one polyp was indefinite for dysplasia, and the others were nondysplastic. In the other five patients with more than one FGP, only one of the FGPs demonstrated dysplasia/indefinite for dysplasia whereas the other FGPs were nondysplastic. As has been described in previous reports, the dysplastic changes in all FGPs were limited to the surface/foveolar epithelium and were not seen in the attenuated parietal cells and chief cells lining the dilated oxyntic glands (Figure 2) ▶ . 7,8
APC and β-Catenin Alterations
Alterations in APC were detected in 7 of 13 (53.8%) FGPs (four demonstrating low-grade dysplasia and three indefinite for dysplasia). These included two FGPs with bi-allelic APC inactivation (intragenic mutation coupled with 5q allelic loss) (Figure 3) ▶ , two FGPs with APC gene mutation only, and three FGPs with 5q allelic loss only. The four intragenic APC mutations would all be predicted to result in protein truncation. Three were frameshift mutations resulting from insertion (two cases) or deletion (one case) of a base (A) into a 6-base poly(A) tract spanning codons 1554 to 1556, and the other was a stop codon mutation (TTA→TAA, codon 1564). This latter mutation occurred in the one FGP in which we separately microdissected the surface/foveolar and dilated glandular compartments; as expected, the mutation was present in both compartments.
Figure 3.

Bi-allelic APC inactivation in a sporadic FGP graded as indefinite for dysplasia (case SD2). A: DNA sequencing chromatogram showing deletion of base (A) from a 6-base poly(A) tract spanning codons 1554 to 1556. B: Allelic loss on chromosome 5q is also present (shown here after amplification with the microsatellite marker D5S299). The ratio of smaller allele (arrow) to larger allele (arrowhead) is inverted in the FGP as compared to the patient’s normal tissue because of loss of smaller allele. Allelic loss can also be appreciated on the sequencing chromatogram in A.
β-catenin exon 3 mutations were identified in only 2 of 13 (15.4%) FGPs (both indefinite for dysplasia). Both were 1-bp missense mutations affecting serine/threonine residues for GSK-3β phosphorylation: codon 33, TCT→TGT (one case) (Figure 4) ▶ and codon 37, TCT→TTT (one case). Both mutations would therefore be expected to interfere with normal phosphorylation of β-catenin protein by GSK-3β and subsequent ubiquitin-mediated protein degradation.
Figure 4.

β-catenin missense mutation in a sporadic FGP graded as indefinite for dysplasia. DNA sequencing chromatogram shows a codon 33 TCT (serine)→TGT (cysteine) mutation in case SD7. This mutation would be expected to result in stabilization of β-catenin protein because of loss of a serine phosphorylation site. A mixture of the mutant and wild-type is present, reflective of the dominant nature of β-catenin mutations.
With the exception of one case, β-catenin mutations and APC alterations were mutually exclusive. In this case (SD7, a polyp graded as indefinite for dysplasia), the FGP contained a codon 33 β-catenin mutation as well as 5q allelic loss with the D5S299 marker (D5S82 and D5S346 did not show loss of heterozygosity). If the 5q allelic loss in this FGP is disregarded as a nonspecific genetic abnormality, then overall six (46.2%) FGPs demonstrated APC alterations, two FGPs (15.4%) demonstrated β-catenin mutations, and five (38.5%) did not show identifiable genetic alterations (Table 1) ▶ .
Table 1.
Sporadic Fundic Gland Polyps with Epithelial Dysplasia/Indefinite for Dysplasia
| Polyp designation | Age/sex | Epithelial dysplasia | β-catenin mutation | APC mutation | 5q allelic loss | MSI |
|---|---|---|---|---|---|---|
| SD1 | 49 /F | Low-grade | Wild-type | 1564X | + | Stable |
| SD2 | 52 /M | Indefinite | Wild-type | 1554-1556FS | + | Stable |
| SD3 | 67 /M | Low-grade | Wild-type | 1554-1556FS | − | Stable |
| SD4 | 59 /F | Indefinite | Wild-type | 1554-1556FS | − | Stable |
| SD5 | 71 /M | Low-grade | Wild-type | Wild-type | + | Stable |
| SD6 | 48 /F | Low-grade | Wild-type | Wild-type | + | Stable |
| SD7 | 69 /F | Indefinite | S33C | Wild-type | + | Stable |
| SD8 | 50 /F | Indefinite | S37F | Wild-type | − | Stable |
| SD9 | 59 /F | Low-grade | Wild-type | Wild-type | − | Stable |
| SD10 | 61 /F | Low-grade | Wild-type | Wild-type | − | Stable |
| SD11 | 46 /M | Indefinite | Wild-type | Wild-type | − | Stable |
| SD12 | 37 /F | Low-grade | Wild-type | Wild-type | − | Stable |
| SD13 | 62 /F | Low-grade | Wild-type | Wild-type | − | Stable |
Fundic gland polyps SD4 and SD9 were separate fundic gland polyps from the same patient. Locations of somatic mutations in β-catenin and APC are shown by codon.
FS, frameshift mutation; MSI, microsatellite instability.
Microsatellite Instability
All 13 FGPs in this study were microsatellite stable (Table 1) ▶ .
Discussion
We have identified APC alterations in 7 of 13 (53.8%) of sporadic FGPs with epithelial dysplasia or surface/foveolar changes indefinite for dysplasia, including 2 FGPs with bi-allelic APC inactivation (truncating APC mutations coupled with 5q allelic loss), 2 FGPs with truncating APC gene mutations only, and 3 FGPs with 5q allelic loss only. In contrast, in a previous study of 13 sporadic FGPs that were negative for dysplasia, we found only one FGP with 5q allelic loss (7.7%), and no APC gene mutations (53.8% versus 7.7%, P = 0.030). 8 However, this is nearly identical to the frequency of APC alterations previously demonstrated in a series of 41 FAP-associated FGPs (51%, P > 0.999). 8 In addition, the types of APC alterations found in FAP-associated FGPs and sporadic FGPs with dysplasia/indefinite for dysplasia are quite similar; frameshift mutations in a 6-base poly(A) tract spanning codons 1554 to 1556 in exon 15 are the most common alteration in both FAP-associated FGPs and dysplastic/indefinite sporadic FGPs, whereas exon 15 stop codon mutations are less frequent. 8,10
Three previous studies of sporadic FGPs have shown high prevalences of activating β-catenin gene mutations of 91%, 9 76%, 11 and 64.4%. 12 In particular, the two studies of sporadic FGPs performed in our laboratory looked exclusively at nondysplastic polyps, and demonstrated exon 3 missense mutations or in-frame deletion mutation in 52 of 57 and 47 of 62 FGPs, respectively (total = 99 of 119, 83.2%). 9,11 These mutations would be predicted to result in β-catenin protein stabilization. In contrast, only 2 of 13 (15.4%) sporadic FGPs with dysplasia/indefinite for dysplasia in the current study contained β-catenin gene mutations (P < 0.00001). These results suggest that whereas nondysplastic sporadic FGPs and FAP-associated FGPs arise through different arms of an altered APC/β-catenin pathway, sporadic FGPs with dysplasia/indefinite for dysplasia are molecularly more similar to FAP-associated FGPs.
The role of the APC/β-catenin pathway has been best studied in gastrointestinal adenomas. Among patients with FAP, most of the numerous colorectal and duodenal adenomas (and less frequent gastric adenomas) can be demonstrated to harbor bi-allelic inactivation of the APC gene on chromosome 5q21-22 (germline APC mutation coupled with a somatic, second-hit APC alteration). 17-19 A major function of the APC tumor-suppressor protein involves regulation of the level of cellular β-catenin protein. β-catenin functions as both as a submembranous component of the cadherin-mediated cell-cell adhesion system as well as a downstream transcriptional activator in the Wnt signaling pathway. 20,21 APC, in cooperation with AXIN and GSK-3β, promotes phosphorylation of serine/threonine residues encoded in exon 3 of the β-catenin gene. 20,22,23 Phosphorylation targets β-catenin for ubiquitin-mediated degradation. Increased levels of cytoplasmic β-catenin can result either from truncating APC gene mutations or from stabilizing β-catenin gene mutations. Increased cytoplasmic β-catenin subsequently translocates to the nucleus where, as part of a β-catenin/Tcf/Lef complex, it can stimulate transcription of cell-cycle regulatory genes including cyclin D1 and c-myc. 24-26
In addition to FAP-associated adenomas, a majority of sporadic gastrointestinal adenomas and adenocarcinomas demonstrate bi-allelic APC inactivation in the form of truncating mutations and/or 5q allelic loss. 17,27 Approximately half of colorectal adenomas without detectable APC alterations instead harbor β-catenin mutations. 27 Interestingly, however, there is some evidence for different biological behavior of APC and β-catenin gene mutations in colorectal neoplasms. β-catenin mutations are found in 12.5% of small (<1 cm) adenomas, 28 but are only rarely (1 to 2%) found in large adenomas 28 or invasive adenocarcinomas, 28,29 suggesting that colonic adenomas with β-catenin mutations are less likely to progress to carcinoma than are those with APC mutations.
Our findings in this current study as well as in previous studies suggest that the functional results of APC versus β-catenin mutations may be different in gastric FGPs as well. FAP-associated FGPs, which are associated with second-hit APC mutations, frequently show epithelial dysplasia, 7,30-32 and neoplastic progression of FAP-associated FGPs in the form of large dysplastic polyps 33 or invasive adenocarcinoma 32,34,35 has been reported, albeit rarely. Sporadic FGPs, with their predominance of β-catenin mutations, only infrequently show dysplasia 7 and have never been reported to progress to adenocarcinoma, even among patients with numerous polyps or fundic gland polyposis. Here we show that dysplasia/cellular changes indefinite for dysplasia in sporadic FGPs is also more likely to be associated with APC alterations than with β-catenin mutations. This distinction is not absolute; we could find no discernable differences (either in the histological appearance, size, or presence of other genetic alterations such as microsatellite instability) between the FGPs indefinite for dysplasia that harbored APC alterations and those that instead harbored β-catenin mutations. However, the high frequency of both APC alterations (in FAP-associated FGPs and dysplastic sporadic FGPs) and β-catenin mutations (in nondysplastic sporadic FGPs) underscores the fact that FGPs are neoplastic lesions harboring clonal genetic alterations similar to those found in gastrointestinal adenomas.
Among colorectal adenomas, it has been demonstrated that alterations in the APC/β-catenin pathway are sufficient for adenoma initiation and early growth. 36 No additional genetic alterations (including K-ras mutations, microsatellite instability, or allelic loss at chromosome 1p) are required for growth up to a size of ∼1 cm in diameter. 36 Likewise, APC and β-catenin mutations are the only high-frequency genetic alterations found in gastric FGPs to date. 8,9,11,12 Although a single large FAP-associated FGP was reported to contain an activating codon 12 K-ras mutation, 33 we previously found only one K-ras mutation among 39 FAP-associated FGPs, and none among 13 sporadic FGPs. 8 In this study, we have additionally shown that microsatellite instability was not present in any of 13 sporadic FGPs. One explanation for this relative lack of secondary genetic alterations among FGPs is that the molecular progression of neoplastic alterations in FGPs could differ from those typically found in colorectal carcinogenesis. However, these results also raise the possibility that alterations in the APC/β-catenin pathway may be the sole genetic alteration in most FGPs.
Although FGPs were once considered to represent hamartomatous or hyperplastic/functional lesions 2,37,38 that were poorly linked to proton pump inhibitor ingestion, 39-44 the available genetic evidence supports that FGPs are neoplastic growths with clonal molecular alterations similar to those of colorectal and duodenal adenomas. Interestingly, this study and previous studies of FGPs highlight several unanswered questions with regard to the morphology and biological behavior of FGPs. First, all polyps of the colorectum and small bowel that harbor mutations in the APC/β-catenin pathway also concomitantly demonstrate epithelial dysplasia, a phenomenon that does not hold true for most FGPs. The data from the current study suggest that FGPs with dysplasia are more likely to harbor APC alterations than β-catenin mutations; however, overall the majority of FAP-associated FGPs with APC mutations are not dysplastic, and only rare FGPs with β-catenin mutations are dysplastic. Furthermore, we have previously demonstrated that identical mutations in APC or β-catenin are present in both the surface/foveolar epithelium and the dilated oxyntic glands, despite the fact that the glandular compartment virtually never shows epithelial dysplasia. 8,9 This was again borne out in the current study, in which the one FGP in which the dysplastic surface and nondysplastic glandular epithelia were separately analyzed showed identical APC stop codon mutations in both compartments. Finally, the almost uniformly benign behavior of FGPs as compared to colorectal adenomas, despite the presence of APC/β-catenin alterations in both types of lesions, is intriguing. It is clear that for colorectal adenomas, it is the accumulation of additional genetic alterations that is associated with neoplastic progression. The results of this study showing no evidence for microsatellite instability in dysplastic FGPs, and a previous study showing only rare K-ras mutations in dysplastic and nondysplastic FGPs, 8 would suggest that these additional genetic alterations tend not to occur in the gastric milieu.
Footnotes
Address reprint requests to Tsung-Teh Wu, M.D., Ph.D., University of Texas M.D. Anderson Cancer Center, Department of Pathology, Box 85, Room G1.3595C, 1515 Holcombe Blvd., Houston, TX 77030-4095. E-mail: twu@mdanderson.org.
References
- 1.Stolte M, Sticht T, Eidt S, Ebert D, Finkenzeller G: Frequency, location, and age and sex distribution of various types of gastric polyp. Endoscopy 1994, 26:659-665 [DOI] [PubMed] [Google Scholar]
- 2.Sipponen P, Laxen F, Seppala K: Cystic ‘hamartomatous’ gastric polyps: a disorder of oxyntic glands. Histopathology 1983, 7:729-737 [DOI] [PubMed] [Google Scholar]
- 3.Marcial MA, Villafana M, Hernandez-Denton J, Colon-Pagan JR: Fundic gland polyps: prevalence and clinicopathologic features. Am J Gastroenterol 1993, 88:1711-1713 [PubMed] [Google Scholar]
- 4.Kinoshita Y, Tojo M, Yano T, Kitajima N, Itoh T, Nishiyama K, Inatome T, Fukuzaki H, Watanabe M, Chiba T: Incidence of fundic gland polyps in patients without familial adenomatous polyposis. Gastrointest Endosc 1993, 39:161-163 [DOI] [PubMed] [Google Scholar]
- 5.Iida M, Yao T, Itoh H, Watanabe H, Kohrogi N, Shigelmatsu A, Iwashita A, Fujishima M: Natural history of fundic gland polyposis in patients with familial adenomatosis coli/Gardner’s syndrome. Gastroenterology 1985, 89:1021-1025 [DOI] [PubMed] [Google Scholar]
- 6.Odze RD, Marcial MA, Antonioli D: Gastric fundic gland polyps: a morphologic study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 1996, 27:896-903 [DOI] [PubMed] [Google Scholar]
- 7.Wu TT, Kornacki S, Rashid A, Yardley JH, Hamilton SR: Dysplasia and dysregulation of proliferation in foveolar and surface epithelia of fundic gland polyps from patients with familial adenomatous polyposis. Am J Surg Pathol 1998, 22:293-298 [DOI] [PubMed] [Google Scholar]
- 8.Abraham SC, Nobukawa B, Giardiello FM, Hamilton SR, Wu TT: Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic APC gene alterations. Am J Pathol 2000, 157:747-754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abraham SC, Nobukawa B, Giardiello FM, Hamilton SR, Wu TT: Sporadic fundic gland polyps: common gastric polyps arising through activating mutations in the β-catenin gene. Am J Pathol 2001, 158:1005-1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toyooka M, Konishi M, Kikuchi-Yanoshita R, Iwama T, Miyaki M: Somatic mutations of the adenomatous polyposis coli gene in gastroduodenal tumors from patients with familial adenomatous polyposis. Cancer Res 1995, 55:3165-3170 [PubMed] [Google Scholar]
- 11.Torbenson M, Lee J-Y, Cruz-Correa M, Ravich W, Rastgar K, Abraham SC, Wu T-T: Sporadic fundic gland polyposis: a clinical, histological, and molecular analysis. Mod Pathol 2002, 15:718-723 [DOI] [PubMed] [Google Scholar]
- 12.Sekine S, Shibata T, Yamauchi Y, Nakanishi Y, Shimoda T, Sakamoto M, Hirohashi S: β-catenin mutations in sporadic fundic gland polyps. Virchows Arch 2002, 440:381-386 [DOI] [PubMed] [Google Scholar]
- 13.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 14.Yashima K, Nakamori S, Murakami Y, Yamaguchi A, Hayashi K, Ishikawa O, Konishi Y, Sekiya T: Mutations of the adenomatous polyposis coli gene in the mutation cluster region: comparison of human pancreatic and colorectal cancers. Int J Cancer 1994, 59:43-47 [DOI] [PubMed] [Google Scholar]
- 15.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S: A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998, 58:5248-5257 [PubMed] [Google Scholar]
- 16.Berg KD, Glaser CL, Thompson RE, Hamilton SR, Griffin CA, Eshleman JR: Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J Mol Diagn 2000, 2:20-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 18.Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, Muraoka M, Takahashi H, Amada Y, Fukayama M, Maeda Y, Iwama T, Mishima Y, Mori T, Koike M: Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res 1994, 54:3011-3020 [PubMed] [Google Scholar]
- 19.Toyooka M, Konishi M, Kikuchi-Yanoshita R, Iwama T, Miyaki M: Somatic mutations of the adenomatous polyposis coli gene in gastroduodenal tumors from patients with familial adenomatous polyposis. Cancer Res 1995, 55:3165-3170 [PubMed] [Google Scholar]
- 20.Barth AI, Nathke IS, Nelson WJ: Cadherins, catenins, and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol 1997, 9:683-690 [DOI] [PubMed] [Google Scholar]
- 21.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W: Functional interaction of β-catenin with the transcriptional factor LEF-1. Nature 1996, 382:638-642 [DOI] [PubMed] [Google Scholar]
- 22.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P: Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 1996, 272:1023-1026 [DOI] [PubMed] [Google Scholar]
- 23.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P: Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA 1995, 92:3046-3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tetsu O, McCormick F: β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398:422-426 [DOI] [PubMed] [Google Scholar]
- 25.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C: Target genes of β-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 1999, 96:1603-1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW: Identification of c-myc as a target of the APC pathway. Science 1998, 281:1509-1512 [DOI] [PubMed] [Google Scholar]
- 27.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW: Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998, 58:1130-1134 [PubMed] [Google Scholar]
- 28.Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML: β-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res 1999, 59:1442-1444 [PubMed] [Google Scholar]
- 29.Kitaeva MN, Grogan L, Williams JP, Dimond E, Nakahara K, Hausner P, DeNobile JW, Soballe PW, Kirsch IR: Mutations in β-catenin are uncommon in colorectal cancer occurring in occasional replication error-positive tumors. Cancer Res 1997, 57:4478-4481 [PubMed] [Google Scholar]
- 30.Domizio P, Talbot IC, Spigelman AD, Williams CB, Phillips RKS: Upper gastrointestinal pathology in familial adenomatous polyposis: results from a prospective study of 102 patients. J Clin Pathol 1990, 43:738-743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertoni G, Sassatelli R, Nigrisoli E, Pennazio M, Tansini P, Arrigoni A, Rossini FP, Ponz de Leon M, Bedogni G: Dysplastic changes in gastric fundic gland polyps of patients with familial adenomatous polyposis. Ital J Gastroenterol Hepatol 1999, 31:192-197 [PubMed] [Google Scholar]
- 32.Zwick A, Munir M, Ryan CK, Gian J, Burt RW, Leppert M, Spiro L, Chey WY: Gastric adenocarcinoma and dysplasia in fundic gland polyps of a patient with attenuated adenomatous polyposis coli. Gastroenterology 1997, 113:659-663 [DOI] [PubMed] [Google Scholar]
- 33.Odze RD, Quinn PS, Terrault NA, Vivona AA, Ward MA, Cohen Z, Gallinger S: Advanced gastroduodenal polyposis with ras mutations in a patient with familial adenomatous polyposis. Hum Pathol 1993, 24:442-448 [DOI] [PubMed] [Google Scholar]
- 34.Coffey RJ, Knight CD, Van Heerden JA, Weiland LH: Gastric adenocarcinoma complicating Gardner’s syndrome in a North American woman. Gastroenterology 1985, 88:1263-1266 [DOI] [PubMed] [Google Scholar]
- 35.Hofgartner WT, Thorp M, Ramus MW, Delorefice G, Chey WY, Ryan CK, Takahashi GW, Lobitz JR: Gastric adenocarcinoma associated with fundic gland polyps in a patient with attenuated familial adenomatous polyposis. Am J Gastroenterol 1999, 94:2275-2281 [DOI] [PubMed] [Google Scholar]
- 36.Lamlum H, Papadopoulou A, Ilyas M, Rowan A, Gillet C, Hanby A, Talbot I, Bodmer W, Tomlinson I: APC mutations are sufficient for the growth of early colorectal adenomas. Proc Natl Acad Sci USA 2000, 97:2225-2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iida M, Yao T, Watanabe H, Itoh H, Iwashita A: Fundic gland polyposis in patients without familial adenomatosis coli: its incidence and clinical features. Gastroenterology 1984, 86:1437-1442 [PubMed] [Google Scholar]
- 38.Odze R: Gastric fundic gland polyps: are they preneoplastic lesions? Gastroenterology 1998, 114:422-423 [DOI] [PubMed] [Google Scholar]
- 39.Kazantsev GB, Schwesinger WH, Heim-Hall J: Spontaneous resolution of multiple fundic gland polyps after cessation of treatment with lansoprazole and Nissen fundoplication: a case report. Gastrointest Endosc 2002, 55:600-602 [DOI] [PubMed] [Google Scholar]
- 40.Vieth M, Stolte M: Fundic gland polyps are not induced by proton pump inhibitor therapy. Am J Clin Pathol 2001, 116:716-720 [DOI] [PubMed] [Google Scholar]
- 41.Declich P, Tavani E, Porcellati M, Bellone S, Grassini R: Long-term omeprazole treatment and fundic gland polyps: a very authoritative proof against a link? Am J Gastroenterol 2001, 96:1650. [DOI] [PubMed] [Google Scholar]
- 42.Graham JR: Omeprazole and gastric polyposis in humans. Gastroenterology 1993, 104:1584. [DOI] [PubMed] [Google Scholar]
- 43.Choudhry U, Boyce HW, Jr, Coppola D: Proton pump inhibitor-associated gastric polyps: a retrospective analysis of their frequency, and endoscopic, histologic, and ultrastructural characteristics. Am J Clin Pathol 1998, 110:615-621 [DOI] [PubMed] [Google Scholar]
- 44.el-Zimaity HM, Jackson FW, Graham DY: Fundic gland polyps developing during omeprazole therapy. Am J Gastroenterol 1997, 92:1858-1860 [PubMed] [Google Scholar]