

Abstract
Heart failure and hypertension have each been linked to an induction of oxidative stress transduced by neurohormones, such as angiotensin II and catecholamines. Herein, we hypothesized that aldosterone (ALDO) likewise induces oxidative stress and accounts for a proinflammatory/fibrogenic phenotype that appears at vascular and nonvascular sites of injury found in both right and left ventricles in response to ALDO/salt treatment and that would be sustained with chronic treatment. Uninephrectomized rats received ALDO (0.75 μg/hour) together with 1% dietary NaCl, for 3, 4, or 5 weeks. Other groups received this regimen in combination with an ALDO receptor antagonist, spironolactone (200 mg/kg p.o. daily), or an antioxidant, either pyrrolidine dithiocarbamate (PDTC) (200 mg/kg s.c. daily) or N-acetylcysteine (NAC) (200 mg/kg i.p. daily). Unoperated and untreated age- and gender-matched rats served as controls. We monitored spatial and temporal responses in molecular and cellular events using serial, coronal sections of right and left ventricles. Our studies included: assessment of systolic blood pressure; immunohistochemical detection of NADPH oxidase expression and activity; analysis of redox-sensitive nuclear factor-κB activation; in situ localization of intercellular adhesion molecule-1, monocyte chemoattractant protein-1, and tumor necrosis factor-α mRNA expression; monitoring cell growth and infiltration of macrophages and T cells; and analysis of the appearance and quantity of fibrous tissue accumulation. At week 3 of ALDO/salt treatment and comparable to controls, there was no evidence of oxidative stress or pathological findings in the heart. However, at weeks 4 and 5 of treatment, increased gp91phox and 3-nitrotyrosine expression and persistent activation of RelA were found in endothelial cells and inflammatory cells that appeared in the perivascular space of intramural coronary arteries and at sites of lost cardiomyocytes in both ventricles. Coincident in time and space with these events was increased mRNA expression of intercellular adhesion molecule-1, monocyte chemoattractant protein-1, and tumor necrosis factor-α. Macrophages, lymphocytes, and proliferating endothelial and vascular smooth muscle cells and fibroblast-like cells were seen at each of these sites, together with an accumulation of fibrillar collagen, or fibrosis, as evidenced by a significant increase in ventricular collagen volume fraction. Co-treatment with spironolactone, PDTC, or NAC attenuated these molecular and cellular responses as well as the appearance of fibrosis at vascular and nonvascular sites of injury. Furthermore, elevated systolic blood pressure in ALDO-treated rats was partially suppressed by spironolactone or either antioxidant. Thus, chronic ALDO/salt treatment is accompanied by a time-dependent sustained activation of NADPH oxidase with 3-nitrotyrosine generation and nuclear factor-κB activation expressed by endothelial cells and inflammatory cells. This leads to a proinflammatory/fibrogenic phenotype involving vascular and nonvascular sites of injury found, respectively, in both normotensive and hypertensive right and left ventricles. Spionolactone, PDTC, and NAC each attenuated these responses suggesting ALDO/salt induction of oxidative/nitrosative stress is responsible for the appearance of this proinflammatory phenotype.
Heart failure and hypertension have each been linked to an induction of oxidative stress. 1-6 This includes enhanced generation of reactive oxygen species by the myocardium, 6-8 aorta, 9-13 kidneys, 14 and skeletal musculature. 15 Moreover, biomarkers of free radical activity and lipid peroxidation are elevated in plasma 16-19 and expired air. 20 Oxidative stress, in turn, has been held responsible for the proinflammatory/fibrogenic phenotype 14,21-24 and abnormal vasomotor reactivity of the involved vasculature. 10,12,13,25-27
The pathophysiological basis for the induction of oxidative stress in these settings has been related to an activation of NADPH oxidase, a major determinant of vascular tissue redox state, transduced by circulating neurohormones that include angiotensin (Ang) II 28 and catecholamines. 29 Herein, we hypothesized that aldosterone (ALDO), another member of the renin-angiotensin-aldosterone system, likewise contributes to the induction of oxidative stress and the proinflammatory/fibrogenic phenotype that appears at vascular and nonvascular sites of injury in the rodent heart in response to ALDO/salt administration and that would be sustained with chronic treatment. We and others have shown that this model rapidly suppresses plasma renin activity and circulating angiotensin II while raising plasma levels of this steroid molecule to those found in congestive heart failure or with adrenal adenoma. 30 We compared ALDO-treated rodents to those co-treated with an ALDO receptor antagonist, spironolactone (Spi), or with an antioxidant, either pyrrolidine dithiocarbamate (PDTC) or N-acetylcysteine (NAC), and to untreated/unoperated, age-matched controls.
We monitored the temporal and spatial association between molecular and cellular events associated with this phenotype. This specifically included: immunohistochemical detection of 1) NADPH oxidase, a major source of reactive oxygen species within the vasculature, via expression of its membrane-bound flavoprotein subunit (gp91phox); 2) expression of 3-nitrotyrosine, a stable protein moiety formed by the interaction of superoxide with nitric oxide and resultant peroxynitrite reaction with tyrosine residues, which is indicative of nitrosative stress; and 3) activation of redox-sensitive transcription factor nuclear factor-κB (NF-κB) and specifically its RelA subunit. By in situ hybridization we localized mRNA expression of genes regulated by NF-κB. These included intercellular adhesion molecule (ICAM)-1; monocyte chemoattractant protein (MCP)-1; and a proinflammatory cytokine, tumor necrosis factor (TNF)-α. Cellular responses were examined by immunohistochemical detection of macrophages, T lymphocytes, and cell growth by BrdU labeling. Finally, the histochemical detection of fibrosis was monitored by picrosirius red labeling and collagen volume fraction at vascular and nonvascular sites of cardiac injury was determined by videodensitometry.
Materials and Methods
Animal Model
Eight-week-old male Sprague-Dawley rats (Harlan, Indianapolis, IN) were used in this study. Five animal groups were studied (n = 8 in each group): 1) untreated and unoperated rats served as controls; 2) uninephrectomized rats on 1% NaCl/0.4% KCl diet received ALDO (0.75 μg/hour) by implanted minipump for 3, 4, or 5 weeks; 3) uninephrectomized rats on the same salt diet and dose of ALDO also received an ALDO receptor antagonist, Spi (200 mg/kg/day), 31 given by gavage for 4 weeks; 4) uninephrectomized rats on the same salt diet received ALDO plus an antioxidant, PDTC (200 mg/kg s.c. daily), 32 also considered an inhibitor of NF-κB, for 4 weeks; and 5) uninephrectomized rats on the same salt diet received ALDO plus an antioxidant, NAC (200 mg/kg i.p. daily) for 4 weeks. Animals were sacrificed at weeks 3, 4, and 5. The dose of ALDO selected raises its plasma levels to those seen in humans with congestive heart failure. 33 Rats received BrdU (25 mg/rat, i.p.) for 3 days before sacrifice. Hearts were removed, rinsed in cold saline solution, and frozen in isopentane with dry ice for in situ hybridization, immunohistochemistry, and histochemistry. The study was approved by this Institution’s Animal Care and Use Committee.
NADPH Oxidase and 3-Nitrotyrosine Expression and NF-κB Activation
Coronal cryostat sections (6 μm) were prepared, air-dried, fixed in 10% buffered formalin for 5 minutes, and washed in phosphate-buffered saline (PBS) for 10 minutes. Sections were then incubated with primary antibody against gp91phox at a dilution of 1:100; 34 3-nitrotyrosine at a dilution of 1:100 (Upstate Biotech, Lake Placid, NY); or anti-RelA (Chemicon, Temecula, CA) at a dilution of 1:100 in PBS containing 1% bovine serum albumin for 60 minutes. Sections were then washed in PBS for 10 minutes and incubated with IgG-peroxidase-conjugated secondary antibody (Sigma, St. Louis, MO) with a dilution of 1:150, washed in PBS for 10 minutes, incubated with 0.5 mg/ml diaminobenzidine tetrahydrochloride 2-hydrate and 0.05% H2O2 for 10 minutes, and again washed in PBS. Negative control sections were incubated with secondary antibody alone, stained with hematoxylin, dehydrated, mounted, and viewed by light microscopy. 21
Gene Expression of Proinflammatory Mediators
The localization and optical density of mRNA levels of ICAM-1, MCP-1, and TNF-α were detected by quantitative in situ hybridization. Coronal cryostat sections (16 μm) were fixed in 4% formaldehyde for 10 minutes, washed with phosphate buffer (pH 7.4), and incubated in 0.25% acetic anhydride in 0.1 mol/L TE-HCl for 10 minutes. Sections were then hybridized (overnight at 45°C) with a random primed 35S-dATP-labeled ICAM-1, MCP-1, or TNF-α cDNA-probe (American Type Culture Collection, Rockville, MD). Sections were washed, dried, and subsequently exposed to Kodak Biomax X-ray film (Eastman-Kodak, Rochester, NY). After exposure, film was developed and sections stained with hematoxylin and eosin (H&E). Quantification of mRNA density in the heart was performed using a computer image analysis system (NIH Image, 1.60) as previously reported. 35 Sections stained with H&E were overlapped with radiographical film and examined under light microscopy to determine the distribution of detected mRNA.
Inflammatory Cell Responses
Cell proliferation and monocyte and lymphocyte infiltration were detected by immunohistochemical BrdU, ED-1, and T-cell labeling, respectively. BrdU is incorporated into DNA during the S-phase of the cell cycle. Cryostat sections (6 μm) were incubated with primary BrdU antibody (Sigma, St. Louis, MO) at a dilution of 1:1000; anti-ED-1 antibody (Harlan Bioproducts for Science, Indianapolis, IN) at a dilution of 1:140 or anti-T-cell antibody (ABCAM, Cambridge, UK) at a dilution of 1:200 in PBS containing 1% bovine serum albumin for 30 minutes. Sections were then incubated with IgG-peroxidase-conjugated secondary antibody (Sigma) with dilution of 1:150. Negative control sections were incubated with secondary antibody alone. 21
Cardiac Morphology
Cryostat sections (6 μm) of heart were prepared to determine morphology by H&E and fibrillar collagen accumulation by collagen-specific picrosirius red staining and observed by light microscopy as previously reported. 33 Collagen volume fraction of each section (total three sections/heart) was determined using a computer image analyzing system (NIH image, 1.60) as previously reported. 35
Systolic Blood Pressure
Systolic blood pressure was measured at sacrifice representing week 3, 4, or 5 of treatment and in controls by tail cuff method, as previously reported. 36
Statistical Analysis
Statistical analysis of in situ hybridization and systolic blood pressure findings was performed using analysis of variance. Values are expressed as mean ± SEM with P < 0.01 considered significant. Multiple group comparisons among controls and each group were made by Scheffé’s F-test.
Results
NADPH Oxidase Expression and 3-Nitrotyrosine Staining
NADPH oxidase catalyzes the one-electron reduction of molecular oxygen to superoxide anion, which can react with nitric oxide to form short-lived peroxynitrite. Peroxynitrite forms stable 3-nitrotyrosine-conjugated protein moieties. 37 To investigate the role of oxidative stress associated with ALDO/salt treatment, we analyzed NADPH oxidase expression and activity in ALDO-treated rat hearts. Expression of NADPH oxidase was determined by immunohistochemical detection of the heme-bearing subunit (gp91phox), and 3-nitrotyrosine staining was monitored as a biomarker of oxidative/nitrosative stress. In ALDO-treated rats, gp91phox labeling was first observed within the perivascular space and at microscopic sites of repair during week 4 (Figure 1A) ▶ and week 5 of ALDO treatment. Cells expressing gp91phox at these sites were primarily inflammatory and endothelial cells. Oxidase activation, as detected by 3-nitrotyrosine staining, was not seen at week 3 of treatment or in the normal myocardium of untreated controls (not shown). However, 3-nitrotyrosine staining was observed in inflammatory and endothelial cells in the perivascular space and microscopic sites of injury (Figure 1B) ▶ in the heart at week 4 and 5 of ALDO/salt treatment.
Figure 1.
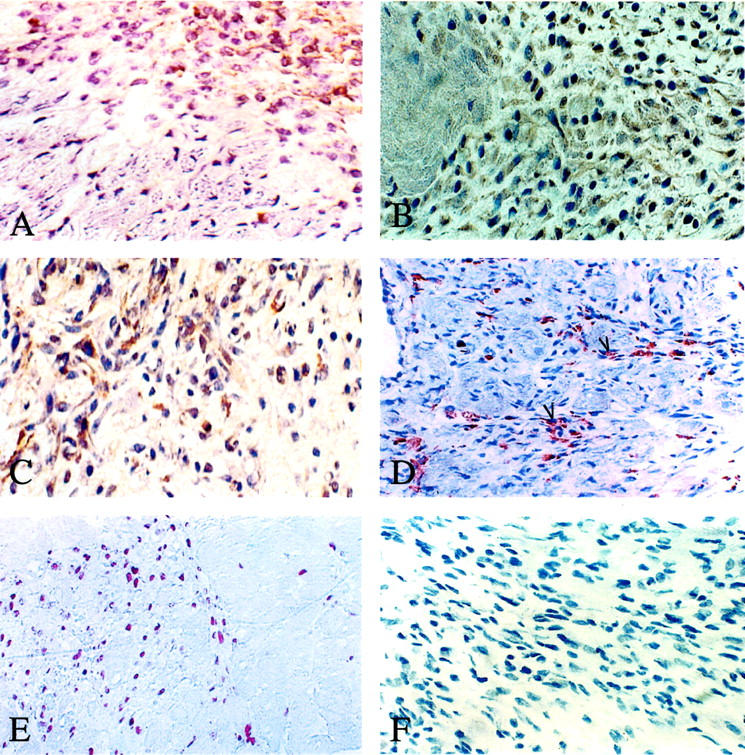
Expression of gp91phox and 3-nitrotyrosine as detected by immunolabeling, NF-κB activation by RelA labeling, macrophage infiltration by ED-1 labeling, and cell proliferation by BrdU labeling are presented at sites of cardiac repair induced by ALDO treatment. Positive staining appears as brown. A: In rats receiving 4 weeks of ALDO treatment, NADPH oxidase was expressed by inflammatory cells within perivascular space (not shown) and sites of microscopic injury. B: 3-Nitrotyrosine staining was present in inflammatory cells at these sites. C: Activated NF-κB was observed in vascular endothelial cells (not shown) and inflammatory cells found in the injured myocardium of both ventricles. D: Numerous ED-1-positive macrophages appeared within the perivascular space and sites of scarring. E: Abundant BrdU-positive cells were likewise seen at these sites. F: Negative control for gp91phox and 3-nitrotyrosine. Original magnifications: ×360 (A–C, F); ×280 (D, E).
NF-κB Activation
The NF-κB heterodimer normally remains in an inactive form bound to its inhibitory protein subunit IκB. On stimulation, phosphorylated IκB is degraded allowing NF-κB to translocate from the cytosol into the cell nucleus, where it binds to promoter regions to initiate transcription of specific genes that encode host defense responses. 38 The presence of activated NF-κB (ie, its p65 subunit or RelA) can be detected by immunohistochemistry. In the present study, cardiac RelA labeling was negative in control rats and rats with 3 weeks of ALDO/salt treatment (not shown); however, it was positive at sites of repair in both right and left ventricles after 4 and 5 weeks treatment (Figure 1) ▶ , where it was found primarily in vascular endothelial cells and inflammatory cells (Figure 1C) ▶ at vascular and nonvascular sites of inflammation (see below).
Gene Expression of Proinflammatory Mediators
As detected by quantitative in situ hybridization (Figure 2) ▶ , low optical density ICAM-1, MCP-1, and TNF-α mRNA expression was observed in both right and left ventricles of control rats (Figure 2; A, D, G) ▶ and those who received 3 weeks of ALDO/salt treatment (not shown). At 4 weeks of ALDO treatment, gene expression of ICAM-1 (Figure 2B) ▶ , MCP-1 (Figure 2E) ▶ , and TNF-α (Figure 2H) ▶ were markedly increased within the perivascular space of intramural coronary arteries and at sites of microscopic scarring found in both ventricles. The mRNA expression of these proinflammatory mediators remained elevated at these sites at week 5 of continued ALDO/salt treatment (not shown). This contrasts to ICAM-1, MCP-1, and TNF-α gene expression in noninjured myocardium that remained no different from control hearts. Quantitative optical density of cardiac ICAM-1, MCP-1, and TNF-α gene expression is shown in Figures 3, 4, and 5 ▶ ▶ ▶ , respectively.
Figure 2.
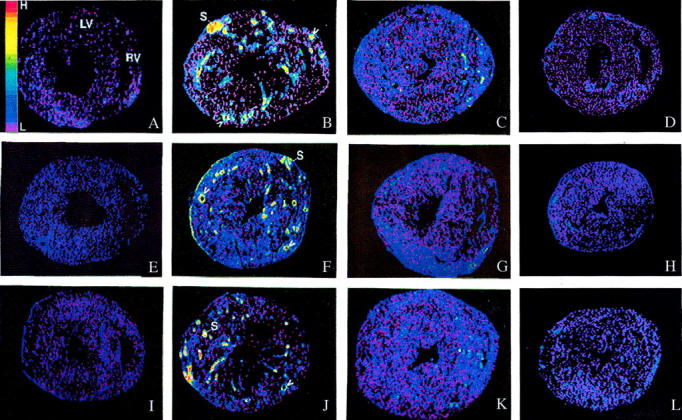
Transcription of ICAM-1, MCP-1, and TNF-α in the rat heart by in situ hybridization. In the normal rat heart, low levels of ICAM-1 (A), MCP-1 (E), and TNF-α (I) gene expression were present in both right (RV) and left (LV) ventricles. At 4 weeks of ALDO/salt treatment, gene expression of ICAM-1 (B), MCP-1 (F), and TNF-α (J) were increased within the perivascular space of intramural coronary arteries (arrowheads) and sites of myocardial scarring (S) in both ventricles. These responses were markedly attenuated by co-treatment with PDTC (C, G, and K, respectively) or with Spi (D, H, and L). NAC was similarly protective (not shown). High (H) and low (L) optical densities are color coded and presented at the left side of A.
Figure 3.

Quantitative optical density of ICAM-1 mRNA in the control rat hearts and those hearts obtained from ALDO-, ALDO + Spi-, ALDO + PDTC-, and ALDO + NAC-treated rats. Compared to controls, ICAM-1 mRNA was markedly increased in both ventricles in ALDO-treated rats for 4 weeks, which was prevented by co-treatment with either Spi, PDTC, or NAC. *, P < 0.01 compared to controls; †, P < 0.01 compared to ALDO-treated rats.
Figure 4.

Quantitative optical density of MCP-1 mRNA in control hearts and those hearts obtained from ALDO-, ALDO + Spi-, ALDO + PDTC-, and ALDO + NAC-treated rats for 4 weeks. Compared to controls, MCP-1 mRNA was markedly increased in ALDO-treated rats, which was prevented by co-treatment with either Spi, PDTC, or NAC. *, P < 0.01 compared to controls; †, P < 0.01 compared to ALDO-treated rats.
Figure 5.

Quantitative optical density of TNF-α mRNA in control rat hearts and those hearts obtained ALDO-, ALDO + Spi-, ALDO + PDTC-, and ALDO + acetylcysteine-treated rats. Compared to controls, TNF-α mRNA was markedly increased in ALDO-treated rats, which was prevented by co-treatment with either Spi, PDTC, or NAC. *, P < 0.01 compared to controls; †, P < 0.01 compared to ALDO-treated rats.
Monocyte/Macrophage and Lymphocyte Infiltration, Cell Proliferation, and Fibrosis
At sites of repair, infiltrating monocytes differentiate into macrophages and express ED-1. By immunohistochemistry, macrophages and lymphocytes were not observed in the heart of untreated controls or those with 3 weeks ALDO treatment (not shown). However, at weeks 4 and 5 of treatment, macrophages (Figure 1D) ▶ and T cells (not shown) were observed within the perivascular space of intramyocardial coronary arteries and at sites of myocardial injury.
Vascular endothelial cell, smooth muscle cell, and fibroblast-like cell proliferation are a feature of repair tissue, which contributes to angiogenesis and collagen formation. By immunohistochemical detection of BrdU labeling, abundant proliferating cells were observed within the perivascular space and at microscopic sites of repair at week 4 (Figure 1E) ▶ and week 5 of ALDO/salt treatment, but not at week 3.
Histochemical detection of fibrillar collagen by picrosirius red staining revealed an accumulation of collagen within the perivascular space of intramural coronary arteries and at sites of scarring that replaced lost myocytes during week 4 of ALDO/salt treatment, which resulted in increased collagen volume fraction (Figure 6) ▶ . This perivascular fibrosis and microscopic scarring, which was not seen at week 3 of treatment or in untreated controls, became more evident at week 5 of continued ALDO treatment.
Figure 6.

Collagen volume fraction (%), a marker of fibrosis, in control rat hearts and those hearts obtained from ALDO-, ALDO + Spi-, ALDO + PDTC-, and ALDO + NAC-treated rats. Compared to controls, collagen volume fraction was markedly increased in the right and left ventricles of ALDO-treated rats, which was prevented by co-treatment with either Spi, PDTC, or NAC. *, P < 0.01 compared to controls; †, P < 0.01 compared to ALDO-treated rats.
Response to Spironolactone and Antioxidants
Four weeks of Spi co-treatment abrogated the ALDO/salt-induced activation of NADPH oxidase and NF-κB. Transcription of ICAM-1, MCP-1, and TNF-α; inflammatory cell response; and the appearance of fibrosis in the heart were also significantly attenuated by Spi co-treatment. Quantitative data for gene expression of proinflammatory mediators and cardiac fibrosis are presented in Figures 2 to 6 ▶ ▶ ▶ , respectively.
Co-treatment with either PDTC or NAC also prevented ALDO/salt-induced NADPH oxidase and NF-κB activation (not shown). Cardiac gene expression of ICAM-1, MCP-1, and TNF-α in ALDO-treated animals was also primarily attenuated. These data are shown in Figures 2 to 5 ▶ ▶ ▶ , respectively. Inflammatory cell infiltration and cell proliferation in ALDO-treated rats were abrogated by co-treatment with either PDTC or NAC (not shown). Collagen volume fraction of the ventricles was markedly attenuated in rats receiving ALDO plus PDTC or ALDO plus NAC (Figure 6) ▶ .
Compared to controls, systolic blood pressure in ALDO-treated rats was elevated at each time point (Table 1) ▶ and confirms previous findings. 39 For the dose selected and as previously reported, 39 Spi treatment normalized blood pressure, whereas PDTC and NAC partially suppressed systolic blood pressure (Table 1) ▶ .
Table 1.
Systolic Blood Pressure in Rats Treated with ALDO
| Groups | Controls | ALDO | +Spi | +PDTC | +NAC |
|---|---|---|---|---|---|
| 3 weeks | 115 ± 11 | 143 ± 13* | 120 ± 9† | 123 ± 11† | 126 ± 14† |
| 4 weeks | 118 ± 10 | 182 ± 19* | 132 ± 14† | 140 ± 12*† | 149 ± 12*† |
| 5 weeks | 121 ± 13 | 189 ± 16* | 131 ± 10† | 144 ± 12*† | 145 ± 13*† |
*P < 0.01 compared to control group.
†P < 0.01 compared to ALDO group.
Discussion
Herein we addressed a role for elevations in circulating ALDO (inappropriate for dietary salt intake) in contributing to a proinflammatory/fibrogenic cardiac phenotype that appears within both the normotensive right and hypertensive left ventricles of the rat heart. This study yielded several major findings.
First is the coupling of an inflammatory response to ALDO-/salt-mediated induction of oxidative stress. Superoxide generation is related to an NADH/NADPH oxidase present in inflammatory cells and in endothelial cells and adventitial fibroblasts of vascular tissue. 9,10,40-43 NADPH-oxidase expression, as assessed by gp91phox labeling and suggestive of enhanced superoxide production, was elevated at sites of vascular and nonvascular repair seen in both the left and right ventricles in rats treated with ALDO/salt for 4 and 5 weeks. Furthermore, staining of 3-nitrotyrosine at sites of cardiac repair provided indirect evidence of superoxide generation. Co-treatment with Spi, an ALDO receptor antagonist, or two different antioxidants, PDTC or NAC, each abrogated this response at vascular and nonvascular sites of cardiac injury. In a deoxycorticosterone acetate/salt model, another model of chronic mineralocorticoid excess antioxidant treatment proved renoprotective by attenuating NF-κB activation and inflammatory cell invasion in renal parenchyma. 44
What induces oxidative stress in rats treated with ALDO/salt remains unknown. Elevated arterial pressure has been held responsible for enhanced oxygen radical production by aortic tissue in rats with acquired or genetic hypertension. 11-13,25,45 However, normotensive tissues (eg, pulmonary artery and right ventricle) have not been examined and the hypertension induced by systemic administration of norepinephrine was not associated with oxidative stress. 12 Superoxide production has been observed in arterioles and postcapillary venules of Dahl salt-sensitive hypertensive rats and rats receiving a pressor dose of angiotensin II, 11,22 in which elevations in intraluminal pressure do not reach these segments of the vasculature. Activated immune cells, not blood pressure, have been incriminated in initiating oxidative stress in Dahl salt-sensitive hypertensive rats. 46,47 Therefore, the role of hemodynamic factors in these models remains uncertain. An elevation in arterial pressure is expected in the ALDO/salt model. This confounding variable raises the question of whether hemodynamic factors are involved in promoting the proinflammatory vascular phenotype in response to ALDO/salt treatment. A series of studies conducted throughout the past decade favors the importance of the circulating hormone (plus salt). The adverse structural remodeling of the coronary vasculature appears throughout the heart when circulating levels of ALDO are chronically increased, from either endogenous or exogenous sources. This includes the normotensive right atria and ventricle and left atrium. 33,48 Arterial hypertension induced by infrarenal abdominal aortic banding is not accompanied by a remodeling of intramyocardial coronary arteries. 33 Neither treatment with ALDO plus dietary salt deprivation nor high dietary salt treatment alone leads to this vascular remodeling. 49 Furthermore, treatment with an mineralocorticoid receptor antagonist has proven cardioprotective. Given systemically, in either nondepressor or depressor dosages, together with ALDO/salt, such an agent prevents vascular remodeling independent of an elevation in arterial pressure. 49 In the present study, we selected a 200-mg/kg/day dose of Spi, which our previous studies have demonstrated is a depressor dose. 49 When a mineralocorticoid receptor antagonist is infused into a cerebral ventricle, it prevents the appearance of hypertension that accompanies the systemic administration of ALDO/salt, but not cardiac fibrosis. 50,51 We therefore would conclude that the signal that initiates an up-regulation of oxidative stress in the ALDO/salt model remains unknown, but is not arterial hypertension. We might hypothesize a Na+-dependent effect of ALDO that promotes Mg2+ efflux from lymphocytes 52 with subsequent [Mg2+]i deficiency and reduced (Mg2+-dependent) Na,K-ATPase activity leading to an elevation in [Na+]i and [Ca2+]i. Intracellular Ca2+ loading is a major determinant to the induction of oxidative stress in peripheral blood mononuclear cells, as is Mg2+ deficiency. 53 We are currently exploring this hypothesis.
Our second major finding demonstrated rats treated with ALDO/salt for 4 and 5 weeks (but not 3 weeks) was accompanied by activation of NADPH oxidase and redox-sensitive NF-κB, a central mediator of an inflammatory response and several proinflammatory mediators, which it regulates. Included with NF-κB activation is an increased gene expression of an adhesion molecule, a chemokine, and a proinflammatory cytokine (ICAM-1, MCP-1, and TNF-α, respectively). NF-κB activation appeared at week 4 of ALDO/salt treatment and was localized to endothelial cells of intramural coronary arteries and arterioles and infiltrating inflammatory cells. Activation of this redox-sensitive transcription factor remained elevated at week 5 of ALDO/salt administration indicating it was not desensitized or down-regulated. An activation of oxidative stress leading to inflammatory responses has been proposed as contributing to the progressive nature of heart failure. 5 Elevations in circulating levels of proinflammatory cytokines (eg, TNF-α and interleukin-6), together with elevated plasma levels of ALDO, are recognized features of heart failure of diverse etiological origins. 54-63 These cytokines adversely influence myocardial and cardiac myocyte contractility 62 and contribute to a catabolic state that besets diverse organs. 15,55,64
Lastly, this study has demonstrated that the ALDO/salt-induced proinflammatory phenotype is a necessary requisite to the accumulation of fibrous tissue at vascular and nonvascular sites of injury in the heart. Spi, PDTC, and NAC each prevented the appearance of this phenotype, including the expression of a cascade of effector proteins that contribute to the homing and transmigration of circulating monocytes/macrophages and T cells into injured vascular and nonvascular sites of cardiac tissue. The necessary role of inflammatory cells in promoting subsequent fibrosis has previously been reported by Nicoletti and co-workers 65,66 in rats with genetic hypertension.
The importance of ALDO in the pathophysiology of salt and water retention in congestive heart failure is well established. 67 In a recently reported, controlled clinical trial (RALES) conducted in 19 countries on 5 continents, using either Spi or placebo, in combination with ACE inhibitor and loop diuretic, a favorable impact in reducing all-cause mortality and cardiac mortality was observed in patients with severe heart failure. 68 Findings of the present study may offer an additional perspective to the favorable response to Spi. The cardioprotective response we observed in response to either Spi or an antioxidant implicates oxidative stress in response to inappropriate elevations in circulating ALDO. Superoxide production reduces nitric oxide bioactivity while reducing the expression of nitric oxide synthase. 26,69 Aldosteronism therefore predisposes to endothelial dysfunction, which has been observed in patients with heart failure and individuals with ALDO-producing adrenal adenoma and that was reversed by either Spi or surgical resection of the tumor. 30,70 Moreover, and as reaffirmed in our study, systemic treatment with an ALDO receptor antagonist prevented cardiac remodeling by fibrillar collagen. 49-51 In a substudy to the RALES trial, Zannad and colleagues 71 found a reduction in elevated serological levels of markers of collagen synthesis in those individuals randomized to Spi, which accounted for the observed survival benefit not seen in those receiving placebo.
In summary, chronic ALDO/salt treatment is accompanied by a proinflammatory/fibrogenic phenotype involving the intramural coronary circulation and myocardium of both right and left ventricles and which is based on induction of oxidative/nitrosative stress (NADPH oxidase and 3-nitrotyrosine protein expression) at these vascular and nonvascular sites of injury. Activation of redox-sensitive NF-κB, which transactivates the expression of proinflammatory mediators, appears in endothelial cells of the involved coronary vasculature and is persistent during continued ALDO/salt treatment. The proinflammatory cardiac phenotype is requisite to the appearance of fibrosis at these sites. Spi, PDTC, NAC each attenuate these responses suggesting ALDO-induced oxidative/nitrosative stress is involved in triggering this phenotype through a signaling pathway that remains to be identified.
Acknowledgments
We thank Yuanjian Chen for skillful technical assistance and Richard A. Parkinson, MEd., for editorial support.
Footnotes
Address reprint requests to Yao Sun, M.D., Ph.D., University of Tennessee Health Science Center, Division of Cardiovascular Diseases, 956 Court Ave., Rm B310, Memphis, TN 38163. E-mail: yasun@utmem.edu.
Supported, in part, by the National Heart, Lung, and Blood Institute (R01-HL67888 to Y. S., R01-HL6229 to K. T. W., and R01-HL66575).
References
- 1.Weglicki WB, Kramer JH, Mak IT: The role of antioxidant drugs in oxidative injury of cardiovascular tissue. Heart Fail Rev 1999, 4:183-192 [Google Scholar]
- 2.Ball AM, Sole MJ: Oxidative stress and the pathogenesis of heart failure. Cardiol Clin 1998, 16:665-675 [DOI] [PubMed] [Google Scholar]
- 3.Keith ME, Jeejeebhoy KN, Langer A, Kurian R, Barr A, O’Kelly B, Sole MJ: A controlled clinical trial of vitamin E supplementation in patients with congestive heart failure. Am J Clin Nutr 2001, 73:219-224 [DOI] [PubMed] [Google Scholar]
- 4.Sole MJ, Jeejeebhoy KN: Conditioned nutritional requirements and the pathogenesis and treatment of myocardial failure. Curr Opin Clin Nutr Metab Care 2000, 3:417-424 [DOI] [PubMed] [Google Scholar]
- 5.Singal PK, Khaper N, Farahmand F, Bello-Klein A: Oxidative stress in congestive heart failure. Curr Cardiol Rep 2000, 2:206-211 [DOI] [PubMed] [Google Scholar]
- 6.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P: Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res 2001, 89:279-286 [DOI] [PubMed] [Google Scholar]
- 7.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A: Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res 1999, 85:357-363 [DOI] [PubMed] [Google Scholar]
- 8.Nath R, Kumar D, Li T, Singal PK: Metallothioneins, oxidative stress and the cardiovascular system. Toxicology 2000, 155:17-26 [DOI] [PubMed] [Google Scholar]
- 9.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG: Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 1996, 97:1916-1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang HD, Pagano PJ, Du Y, Cayatte AJ, Quinn MT, Brecher P, Cohen RA: Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circ Res 1998, 82:810-818 [DOI] [PubMed] [Google Scholar]
- 11.Swei A, Lacy F, DeLano FA, Schmid-Schonbein GW: Oxidative stress in the Dahl hypertensive rat. Hypertension 1997, 30:1628-1633 [DOI] [PubMed] [Google Scholar]
- 12.Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG: Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 1997, 95:588-593 [DOI] [PubMed] [Google Scholar]
- 13.Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA: Superoxide anion production is increased in a model of genetic hypertension: role of the endothelium. Hypertension 1999, 33:1353-1358 [DOI] [PubMed] [Google Scholar]
- 14.Haugen EN, Croatt AJ, Nath KA: Angiotensin II induces renal oxidant stress in vivo and heme oxygenase-1 in vivo and in vitro. Kidney Int 2000, 58:144-152 [DOI] [PubMed] [Google Scholar]
- 15.Tsutsui H, Ide T, Hayashidani S, Suematsu N, Shiomi T, Wen J, Nakamura K, Ichikawa K, Utsumi H, Takeshita A: Enhanced generation of reactive oxygen species in the limb skeletal muscles from a murine infarct model of heart failure. Circulation 2001, 104:134-136 [DOI] [PubMed] [Google Scholar]
- 16.McMurray J, McLay J, Chopra M, Bridges A, Belch JJ: Evidence for enhanced free radical activity in chronic congestive heart failure secondary to coronary artery disease. Am J Cardiol 1990, 65:1261-1262 [DOI] [PubMed] [Google Scholar]
- 17.Belch JJ, Bridges AB, Scott N, Chopra M: Oxygen free radicals and congestive heart failure. Br Heart J 1991, 65:245-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz-Velez CR, Garcia-Castineiras S, Mendoza-Ramos E, Hernandez-Lopez E: Increased malondialdehyde in peripheral blood of patients with congestive heart failure. Am Heart J 1996, 131:146-152 [DOI] [PubMed] [Google Scholar]
- 19.Keith M, Geranmayegan A, Sole MJ, Kurian R, Robinson A, Omran AS, Jeejeebhoy KN: Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol 1998, 31:1352-1356 [DOI] [PubMed] [Google Scholar]
- 20.Sobotka PA, Brottman MD, Weitz Z, Birnbaum AJ, Skosey JL, Zarling EJ: Elevated breath pentane in heart failure reduced by free radical scavenger. Free Radic Biol Med 1993, 14:643-647 [DOI] [PubMed] [Google Scholar]
- 21.Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC: NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 2000, 35:193-201 [DOI] [PubMed] [Google Scholar]
- 22.Piqueras L, Kubes P, Alvarez A, O’Connor E, Issekutz AC, Esplugues JV, Sanz MJ: Angiotensin II induces leukocyte-endothelial cell interactions in vivo via AT1 and AT2 receptor-mediated P-selectin upregulation. Circulation 2000, 102:2118-2123 [DOI] [PubMed] [Google Scholar]
- 23.Mervaala EM, Müller DN, Park JK, Schmidt F, Löhn M, Breu V, Dragun D, Ganten D, Haller H, Luft FC: Monocyte infiltration and adhesion molecules in a rat model of high human renin hypertension. Hypertension 1999, 33:389-395 [DOI] [PubMed] [Google Scholar]
- 24.Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Pritzl P, Floege J, Schwartz SM: Renal injury from angiotensin II-mediated hypertension. Hypertension 1992, 19:464-474 [DOI] [PubMed] [Google Scholar]
- 25.Somers MJ, Mavromatis K, Galis ZS, Harrison DG: Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation 2000, 101:1722-1728 [DOI] [PubMed] [Google Scholar]
- 26.Gryglewski RJ, Palmer RM, Moncada S: Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320:454-456 [DOI] [PubMed] [Google Scholar]
- 27.Schnackenberg CG, Wilcox CS: Two-week administration of tempol attenuates both hypertension and renal excretion of 8-isoprostaglandin f2α. Hypertension 1999, 33:424-428 [DOI] [PubMed] [Google Scholar]
- 28.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M: Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 2000, 20:2175-2183 [DOI] [PubMed] [Google Scholar]
- 29.Singal PK, Beamish RE, Dhalla NS: Potential oxidative pathways of catecholamines in the formation of lipid peroxides and genesis of heart disease. Adv Exp Med Biol 1983, 161:391-401 [DOI] [PubMed] [Google Scholar]
- 30.Taddei S, Virdis A, Mattei P, Salvetti A: Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 1993, 21:929-933 [DOI] [PubMed] [Google Scholar]
- 31.Theuer J, Dechend R, Muller DN, Park JK, Fiebeler A, Barta P, Ganten D, Haller H, Dietz R, Luft FC: Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc Disord 2002, 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi K, Takahata H, Kitagawa N, Kitange G, Kaminogo M, Shibata S: N-acetylcysteine inhibited nuclear factor-κB expression and the intimal hyperplasia in rat carotid arterial injury. Neurol Res 2001, 23:731-738 [DOI] [PubMed] [Google Scholar]
- 33.Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT: Remodeling of the rat right and left ventricle in experimental hypertension. Circ Res 1990, 67:1355-1364 [DOI] [PubMed] [Google Scholar]
- 34.Burritt JB, Quinn MT, Jutila MA, Bond CW, Jesaitis AJ: Topological mapping of neutrophil cytochrome b epitopes with phage-display libraries. J Biol Chem 1995, 270:16974-16980 [DOI] [PubMed] [Google Scholar]
- 35.Sun Y, Ratajska A, Weber KT: Inhibition of angiotensin-converting enzyme and attenuation of myocardial fibrosis by lisinopril in rats receiving angiotensin II. J Lab Clin Med 1995, 126:95-101 [PubMed] [Google Scholar]
- 36.Hilgers KF, Hartner A, Porst M, Veelken R, Mann JF: Angiotensin II type 1 receptor blockade prevents lethal malignant hypertension: relation to kidney inflammation. Circulation 2001, 104:1436-1440 [DOI] [PubMed] [Google Scholar]
- 37.Beckman JS, Koppenol WH: Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 1996, 271:C1424-C1437 [DOI] [PubMed] [Google Scholar]
- 38.May MJ, Ghosh S: Signal transduction through NF-κB. Immunol Today 1998, 19:80-88 [DOI] [PubMed] [Google Scholar]
- 39.Brilla CG, Matsubara LS, Weber KT: Anti-aldosterone treatment and the prevention of myocardial fibrosis in primary and secondary hyperaldosteronism. J Mol Cell Cardiol 1993, 25:563-575 [DOI] [PubMed] [Google Scholar]
- 40.Pagano PJ, Ito Y, Tornheim K, Gallop PM, Tauber AI, Cohen RA: An NADPH oxidase superoxide-generating system in the rabbit aorta. Am J Physiol 1995, 268:H2274-H2280 [DOI] [PubMed] [Google Scholar]
- 41.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT: Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci USA 1997, 94:14483-14488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohazzab KM, Kaminski PM, Wolin MS: NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol 1994, 266:H2568-H2572 [DOI] [PubMed] [Google Scholar]
- 43.Jones SA, O’Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT: Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol 1996, 271:H1626-H1634 [DOI] [PubMed] [Google Scholar]
- 44.Beswick RA, Zhang H, Marable D, Catravas JD, Hill WD, Webb RC: Long-term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension 2001, 37:781-786 [DOI] [PubMed] [Google Scholar]
- 45.Gonick HC, Ding Y, Bondy SC, Ni Z, Vaziri ND: Lead-induced hypertension: interplay of nitric oxide and reactive oxygen species. Hypertension 1997, 30:1487-1492 [DOI] [PubMed] [Google Scholar]
- 46.Shen K, DeLano FA, Zweifach BW, Schmid-Schonbein GW: Circulating leukocyte counts, activation, and degranulation in Dahl hypertensive rats. Circ Res 1995, 76:276-283 [DOI] [PubMed] [Google Scholar]
- 47.Swei A, Lacy F, Delano FA, Parks DA, Schmid-Schonbein GW: A mechanism of oxygen free radical production in the Dahl hypertensive rat. Microcirculation 1999, 6:179-187 [PubMed] [Google Scholar]
- 48.Sun Y, Ramires FJA, Weber KT: Fibrosis of atria and great vessels in response to angiotensin II or aldosterone infusion. Cardiovasc Res 1997, 35:138-147 [DOI] [PubMed] [Google Scholar]
- 49.Brilla CG, Weber KT: Mineralocorticoid excess, dietary sodium and myocardial fibrosis. J Lab Clin Med 1992, 120:893-901 [PubMed] [Google Scholar]
- 50.Young M, Fullerton M, Dilley R, Funder J: Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest 1994, 93:2578-2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young M, Head G, Funder J: Determinants of cardiac fibrosis in experimental hypermineralocorticoid states. Am J Physiol 1995, 269:E657-E662 [DOI] [PubMed] [Google Scholar]
- 52.Delva P, Pastori C, Degan M, Montesi G, Brazzarola P, Lechi A: Intralymphocyte free magnesium in patients with primary aldosteronism: aldosterone and lymphocyte magnesium homeostasis. Hypertension 2000, 35:113-117 [DOI] [PubMed] [Google Scholar]
- 53.Weglicki WB, Mak IT, Stafford RE, Dickens BF, Cassidy MM, Phillips TM: Neurogenic peptides and the cardiomyopathy of magnesium-deficiency: effects of substance P-receptor inhibition. Mol Cell Biochem 1994, 130:103-109 [DOI] [PubMed] [Google Scholar]
- 54.Mann DL: Tumor necrosis factor and viral myocarditis: the fine line between innate and inappropriate immune responses in the heart. Circulation 2001, 103:626-629 [DOI] [PubMed] [Google Scholar]
- 55.Anker SD, Chua TP, Ponikowski P, Harrington D, Swan JW, Kox WJ, Poole-Wilson PA, Coats AJS: Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation 1997, 96:526-534 [DOI] [PubMed] [Google Scholar]
- 56.Munger MA, Johnson B, Amber IJ, Callahan KS, Gilbert EM: Circulating concentrations of proinflammatory cytokines in mild or moderate heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 1996, 77:723-727 [DOI] [PubMed] [Google Scholar]
- 57.Testa M, Yeh M, Lee P, Fanelli R, Loperfido F, Berman JW, LeJemtel TH: Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol 1996, 28:964-971 [DOI] [PubMed] [Google Scholar]
- 58.Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL: Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol 1996, 27:1201-1206 [DOI] [PubMed] [Google Scholar]
- 59.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL: Cytokines and cytokine receptors in advanced heart failure. An analysis of the cytokine database from the Vesnarinone Trial (VEST). Circulation 2001, 103:2055-2059 [DOI] [PubMed] [Google Scholar]
- 60.Francis GS, Benedict C, Johnstone DE, Kirlin PC, Nicklas J, Liang C, Kubo SH, Rudin-Toretsky E, Yusuf S: Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure: a substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 1990, 82:1724-1729 [DOI] [PubMed] [Google Scholar]
- 61.Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL: Cellular basis for the negative inotropic effects of tumor necrosis factor-α in the adult mammalian heart. J Clin Invest 1993, 92:2303-2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bozkurt B, Kribbs SB, Clubb FJ, Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL: Pathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in rats. Circulation 1998, 97:1382-1391 [DOI] [PubMed] [Google Scholar]
- 63.Feldman AM, Combes A, Wagner D, Kadakomi T, Kubota T, Li YY, McTiernan C: The role of tumor necrosis factor in the pathophysiology of heart failure. J Am Coll Cardiol 2000, 35:537-544 [DOI] [PubMed] [Google Scholar]
- 64.Anker SD, Clark AL, Teixeira MM, Hellewell PG, Coats AJS: Loss of bone mineral in patients with cachexia due to chronic heart failure. Am J Cardiol 1999, 83:612-615 [DOI] [PubMed] [Google Scholar]
- 65.Nicoletti A, Heudes D, Mandet C, Hinglais N, Bariety J, Michel J-B: Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res 1996, 32:1096-1107 [DOI] [PubMed] [Google Scholar]
- 66.Nicoletti A, Mandet C, Challah M, Bariety J, Michel JB: Mediators of perivascular inflammation in the left ventricle of renovascular hypertensive rats. Cardiovasc Res 1996, 31:585-595 [PubMed] [Google Scholar]
- 67.Weber KT: Aldosterone in congestive heart failure. N Engl J Med 2001, 345:1689-1697 [DOI] [PubMed] [Google Scholar]
- 68.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes W: The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999, 341:709-717 [DOI] [PubMed] [Google Scholar]
- 69.Peterson TE, Poppa V, Ueba H, Wu A, Yan C, Berk BC: Opposing effects of reactive oxygen species and cholesterol on endothelial nitric oxide synthase and endothelial cell caveolae. Circ Res 1999, 85:29-37 [DOI] [PubMed] [Google Scholar]
- 70.Farquharson CAJ, Struthers AD: Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin I/angiotensin II conversion in patients with chronic heart failure. Circulation 2000, 101:594-597 [DOI] [PubMed] [Google Scholar]
- 71.Zannad F, Alla F, Dousset B, Perez A, Pitt B: Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators Circ 2000, 102:2700-2706 [DOI] [PubMed] [Google Scholar]