

Abstract
Cisplatin is an anticancer drug that has enjoyed remarkable success against testicular tumors, but dose limiting side-effects have limited its application against a broader range of cancers. Previous studies have shown that high-mobility group (HMG) domain proteins such as HMG1 sensitize cells to cisplatin by shielding its major DNA adducts from nucleotide excision repair. Estrogen treatment increases HMG1 mRNA levels in breast cancer MCF-7 cells. Herein, we describe that treatment of human cancer cells having steroid hormone receptors with the appropriate hormone, estrogen and/or progesterone, significantly increases the potency of cisplatin and its analogue carboplatin by causing the overexpression of HMG1. These findings suggest that the proper combination of these drugs, which are already approved by the Food and Drug Administration, could have potential benefit in treating tumors such as ovarian or breast that carry the hormone receptors.
Cisplatin [cis-diamminedichloroplatinum(II) also known as cis-DDP] is a widely used antitumor drug for the treatment of testicular, breast, ovarian, lung, head, and neck tumors (1–5). DNA is the primary cytotoxic target of cisplatin in vivo (6). The major cisplatin-DNA adducts are 1,2-intrastrand d(GpG) and d(ApG) crosslinks, and the adducts significantly bend and distort normal B DNA (7, 8). This structural distortion in DNA affects DNA replication and transcription (9–13) and is recognized by a variety of structure-specific DNA-binding proteins, such as DNA repair and high-mobility group (HMG) domain proteins (13).
HMG domain proteins are architectural proteins that facilitate cellular functions requiring chromosomal DNA (14). HMG domain proteins bind specifically to the major cisplatin-DNA adducts, forming a stable platinum-DNA-protein ternary complex (15, 16). There is evidence implicating the involvement of the platinum-DNA-protein complex in mediating cisplatin cytotoxicity by blocking nucleotide excision repair of the DNA damage, a process termed repair shielding. In yeast, interruption of the HMG domain protein Ixr1 caused a 2- to 6-fold desensitization to cisplatin compared with that in wild-type cells (17, 18). In vitro experiments revealed that a variety of HMG domain proteins, including HMG1, tsHMG, and SRY, blocked removal of cisplatin intrastrand d(GpG) adducts when added in a nucleotide excision repair assay (19–21). Depleting HMG1 and HMG2 from cell extracts by immunoprecipitation enhanced excision repair of cisplatin-modified DNA (22). Furthermore, introducing HMG2 by transfection enhanced the cisplatin sensitivity of a lung adenocarcinoma cell line (23). Until the present work, however, there was, to our knowledge, no evidence that overexpression of an HMG domain protein via a natural signal transduction pathway in human cancer cells could increase their sensitivity to cisplatin.
HMG1 is a structure-specific HMG domain protein with little or no sequence specificity. It is abundant in all tissues and species (24). HMG1 binds preferentially to cruciform DNA, cisplatin-modified DNA, and other distorted structures (14). It has a multitude of functions including association with chromosomes and interaction with Oct, Hox, p53 protein, and some components of the basal transcriptional machinery (25–29). HMG1 also functions as an architectural protein to facilitate the binding of steroid hormone receptors, such as estrogen receptors (ER), progesterone receptors (PR), androgen receptors, and glucocorticoid receptors to their cognate DNA binding sites (30). Binding of ER to estrogen-responsive elements and PR to progesterone-responsive elements induces DNA bending (31) and, accordingly, increases HMG1 affinity. The binding of HMG1 further alters the structure of the target DNA and facilitates formation of a more stable receptor-DNA complex (32–34). The transcriptional activity of these steroid hormones is enhanced in mammalian cells that are expressing HMG1 transiently (30, 32). Mice deficient in HMG1 die of hypoglycemia, implicating a role for HMG1 in glucocorticoid-dependent gene regulatory pathways (35).
The rationale for the discovery described herein was derived from previous work reporting that the mRNA level of chromosomal architectural protein HMG1 is up-regulated when human MCF-7 breast cancer cells are treated with estrogen (36). We corroborate such overexpression at the protein level. Because of the inverse relationship between the levels of HMG domain proteins and the ability of cells to repair cisplatin-DNA adducts both in vivo and in vitro, described above, we predicted that steroid hormone treatment would sensitize these cells to cisplatin. The present results confirm this prediction and suggest that a combination of cisplatin or carboplatin with estrogen and/or progesterone, all of which have been approved by the Food and Drug Administration, may be of clinical significance.
Materials and Methods
Cell Culture.
Cells were grown in DMEM (GIBCO/BRL) containing 10% (vol/vol) heat-inactivated FBS (GIBCO/BRL), 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C under a 5% CO2(g) atmosphere.
Clonogenic Assays.
Cells were seeded on 6-well plates (Corning) at a density of 400 cells per well. After 24 h, a fresh stock of steroid hormone, estrogen or progesterone, was prepared in N,N-dimethylformamide and added to the plates at a final hormone concentration described below for each experiment. Control plates were treated with the same volume of N,N-dimethylformamide without hormone. For cotreatment, the hormone was added at the same time as cisplatin. For pretreatment, the hormone was added 2 h, 4 h, or 24 h before cisplatin. After 4 h of cisplatin treatment, the cells were washed with PBS, and fresh medium was added. After 10 days, the cell colonies were stained with a 1% methylene blue (Fluka)/50% (vol/vol) ethanol solution and were then counted. Each point is an average of three independent determinations ± 1 SD (Eq. 1) of the cell count.
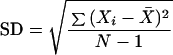 |
1 |
Immunofluorescence.
The cells were grown to 70% confluence on 12-mm glass coverslips in 6-well plates and subsequently treated with estrogen or progesterone for 0–24 h. After hormone treatment, the cells were permeabilized with 25% (vol/vol) acetic acid in methanol for 10 min at room temperature and washed with PBS. The permeabilized cells were incubated for 30 min at 37°C with 1:100 dilution (for MCF-7 cells) or 1:500 dilution (for Evsa-T cells) of anti-HMG1 polyclonal antibody (PharMingen), washed, and incubated subsequently with 1:200 dilution of goat anti-rabbit IgG conjugated to fluorescein (Biosource International, Camarillo, CA) for 30 min at 37°C. Finally, the cells were visualized with a fluorescent light microscope (Zeiss Axiophot).
Flow Cytometry.
About 106 cells were collected, fixed in 70% (vol/vol) ethanol, and then incubated in 1 ml of PBS containing 50 μg/ml propidium iodide (Sigma) and 250 μg/ml RNase A (Roche Molecular Biochemicals) at 37°C for 30 min to stain the DNA and eliminate RNA. For each sample, 100,000 cells were analyzed in a fluorescence-activated cell sorter (FACScan, Becton Dickinson) for the cell-cycle profile by using cell quest and modifit software.
Analysis of Platinum-DNA Adducts Levels.
About 107 cells were collected and then lysed in 2 ml of 100 mM Tris⋅HCl, pH 8.5/5 mM EDTA/0.2% SDS/200 mM NaCl/100 μg/ml proteinase K. The samples were gently agitated overnight at 55°C in the incubator. The samples were extracted twice with 2 ml of phenol/chloroform/isoamyl alcohol and followed by two extractions with chloroform/isoamyl alcohol. Genomic DNA was precipitated with 2 ml of isopropanol, and the DNA was removed and dissolved in 10 mM Tris⋅HCl (pH 7.5) buffer. The DNA was treated with RNase and quantitated by UV-visible spectroscopy. The amount of platinum on the DNA was determined by using a Perkin–Elmer HGA-800 AAnalyst 300 atomic absorption spectrometer.
Results
HMG1 Overexpression in MCF-7 and Evsa-T Cells.
For MCF-7 cells, a mammalian breast cancer cell line, a single 10−7 M dose of estrogen increases HMG1 mRNA level by 1.5- to 2.5-fold (36). The exact mechanism of this up-regulation is not known, although HMG1 overexpression may be necessary to facilitate transcriptional activation of hormone-responsive genes. We find by immunofluorescence that the elevated level of HMG1 mRNA transcript corresponds to higher protein levels (Fig. 1 A–C). After MCF-7 cells were treated with 10−7 M estrogen for 0.5 h, the fluorescence signal from labeled nuclear HMG1 was much stronger than in the untreated cells (Fig. 1B). MCF-7 cells contain both ER and PR and thus also respond to progesterone treatment (10−6 M), up-regulating HMG1 within 0.5 h (Fig. 1C). Evsa-T is a breast cancer cell line that has PR but not ER. Estrogen treatment had no effect on HMG1 levels in Evsa-T cells (data not shown). Progesterone treatment in Evsa-T cells raised HMG1 protein levels as detected by immunofluorescence but required a 4-h treatment (Fig. 1, compare E and F) in contrast to MCF-7 cells. The kinetics of HMG1 overexpression has also been confirmed by Western analysis. The amount of HMG1 peaked at 1.4-fold within 2 h of estrogen treatment in MCF-7 cells and leveled off by 24 h; the amount of HMG1 increased by about 2-fold after a 4-h progesterone treatment in Evsa-T cells (data not shown).
Figure 1.

HMG1 levels in hormone-treated MCF-7 and Evsa-T cells detected by immunofluorescence. (A) MCF-7 controls. (B) MCF-7 treated with 10−7 M estrogen (0.5 h). (C) MCF-7 treated with 10−6 M progesterone (0.5 h). (D) Evsa-T controls. (E) Evsa-T treated with 10−6 M progesterone (0.5 h). (F) Evsa-T treated with 10−6 M progesterone (4 h). (Bar = 50 μm.)
Sensitization of Cells to Cisplatin by Estrogen or Progesterone.
To assess further the involvement of HMG domain proteins in mediating cisplatin cytotoxicity, we investigated the effects of elevated HMG1 levels on the sensitivity of cells to the drug. As initially anticipated from the repair shielding hypothesis, we found that elevated HMG1 expression levels were indeed paralleled by increased sensitivity toward cisplatin. In MCF-7 cells, estrogen or progesterone treatment increased cisplatin sensitivity about 2-fold, according to the cisplatin concentrations where only 50% of the cells are viable (Fig. 2A). A combination of estrogen and progesterone sensitized the cells to cisplatin by a factor of 4 (Fig. 2A); this additive effect suggests that they independently up-regulate HMG1. In Evsa-T cells, progesterone treatment induced 1.5-fold sensitization toward cisplatin, whereas estrogen treatment had no effect because of the absence of ER (Fig. 2B). In addition, the cisplatin sensitivity of HeLa cells, an ER-negative cervical cancer cell line, did not change after estrogen treatment (Fig. 2C).
Figure 2.

Cell survival assays. (A) The effects of estrogen and progesterone cotreatment on cisplatin sensitivity of MCF-7 cells. MCF-7 cells were cotreated with 2 × 10−7 M estrogen, 2 × 10−7 M progesterone, or both hormones with cisplatin for 4 h. (B) The effects of hormones on cisplatin sensitivity of Evsa-T cells. Evsa-T cells were pretreated with 10−7 M estrogen or 10−6 M progesterone for 2 h before a 4-h cisplatin treatment. (C) The effects of 10−7 M estrogen on cisplatin sensitivity of HeLa cells.
The timing of hormone treatment plays an important role in the degree of sensitization of cells to cisplatin. Cotreatment of estrogen and cisplatin caused 2-fold sensitization in MCF-7 cells; pretreatment with estrogen for 24 h did not cause any sensitization (data not shown). In Evsa-T cells, the effect of sensitization by progesterone was not observed with cotreatment (data not shown) but was 2-fold with a 2-h pretreatment of progesterone (Fig. 2B). Carboplatin is an analogue of cisplatin that can form similar bifunctional DNA adducts, because the products of aquation are the same for the two compounds (37). Estrogen had no effect on the carboplatin sensitivity of cells treated simultaneously with the two reagents, but a 24-h pretreatment of carboplatin followed by a 4-h estrogen treatment increased the sensitivity by 2-fold (Fig. 3).
Figure 3.

The effects of estrogen on the carboplatin sensitivity of MCF-7 cells. MCF-7 cells were pretreated with carboplatin for 24 h before the addition of 10−7 M estrogen for 4 h.
Effects of Hormones on Cell Proliferation and Sensitivity to Other Cytotoxic Agents.
Estrogen induces general cell proliferation (38) and regulates human mammary epithelial cell morphogenesis (39, 40). We have considered the possibility that the sensitization of cells toward cisplatin may be a consequence of hormone-induced cell proliferation. Accordingly, we investigated whether the effects of hormone treatment conditions used in the cell survival assays, 4-h exposure to estrogen or progesterone, affected cell growth. FACS analysis revealed no change in cell-cycle profile after hormone treatment (Table 1). Cell proliferation rate, determined in cell counting assays, was similarly unaffected by the transient hormone treatments (data not shown).
Table 1.
Cell-cycle analysis of MCF-7 cells treated with steroid hormones
| Cell treatment conditions | Percentage
|
||
|---|---|---|---|
| G0–G1 | S | G2–M | |
| N,N-dimethylformamide 4 h, wash, wait 24 h | 57 | 35 | 8 |
| Estrogen 4 h, wash, wait 24 h | 56 | 35 | 9 |
| Progesterone 4 h, wash, wait 24 h | 54 | 37 | 9 |
trans-Diamminedichloroplatinum(II) (trans-DDP) is a clinically inactive isomer of cisplatin that forms DNA adducts not recognized by HMG1 (15). The trans-DDP sensitivity of MCF-7 cells was unaffected by estrogen treatment (data not shown). Calicheamicin is another cytotoxic agent that causes double-stranded DNA cleavage (41) in a manner that does not involve HMG1. The sensitivity of MCF-7 cells toward calicheamicin was also unaffected by estrogen treatment (data not shown). We conclude that the hormone treatment did not sensitize the cells to all cytotoxic agents but only to cisplatin or carboplatin via a pathway that involves HMG1, presumably by repair shielding.
Platinum-DNA Adduct Levels.
It is possible that DNA platination levels could be elevated as a consequence of hormone treatment, as suggested by reports that active promoter sites are preferentially platinated by the drug (42, 43). We have investigated the bound-platinum levels on genomic DNA in cells treated with steroid hormones by platinum atomic absorption spectroscopy. MCF-7 cells were treated with a range of cisplatin concentrations (0–100 μM) for 4 h and then immediately harvested, and genomic DNA was extracted to evaluate the initial platination levels. The platinum signals were not detected with the atomic absorption spectrometer in samples treated with 10 μM, 20 μM, or 50 μM cisplatin. For samples treated with 100 μM of cisplatin, the rb values, defined as the number of platinum atoms per nucleotide, were 3.48 × 10−4 for control cells and 3.33 × 10−4 for estrogen-treated cells. The results indicate that the platinum adduct levels on genomic DNA are comparable in control cells and cells treated with estrogen.
Discussion
HMG1 already exists at a high level, between 10,000 and 100,000 copies per cell (24, 44), and an excess amount of the protein is toxic (45), which may explain why only a moderate level of overexpression of HMG1 was observed in the present study. Why then does the moderate up-regulation observed in this study increase drug sensitivity? One possibility is that endogenous HMG1 proteins are already involved in complexes with chromatin and transcription factors. HMG1 transiently expressed as a scaffold to facilitate ER- or PR-mediated transcription may be more readily available to bind to cisplatin-DNA intrastrand crosslinks. The 2-fold difference in cisplatin sensitivity is in good accord with the approximately 2-fold increase in HMG1 protein levels.
The timing of the hormone treatments reflects the kinetics of HMG1 up-regulation and cisplatin aquation chemistry. Cotreatment of estrogen and cisplatin is more effective in sensitizing MCF-7 cells than pretreatment for 24 h (data not shown). HMG1 protein levels increase within 0.5–2 h after estrogen treatment in MCF-7 cells, whereas cisplatin may take a few hours to enter the cell, undergo aquation, and bind to DNA (46). By the time platinum-DNA damage occurs, HMG1 levels are high and can block nucleotide excision repair efficiently. When cells are pretreated with estrogen for 24 h, the amount of HMG1 has leveled off at the time that platinum-DNA adducts form, and consequently, the sensitization effect is less. On the other hand, pretreatment of estrogen for 24 h sensitizes MCF-7 cells toward carboplatin more than cotreatment. This observation is readily explained by the difference in the rate of DNA binding for the two drugs. To obtain the same degree of DNA damage, it is necessary to use a 7.5-fold longer incubation time for carboplatin than for cisplatin (47–49), owing to the slow rate of aquation of the former. This difference may account for the required preincubation period. In contrast to MCF-7 cells, HMG1 levels increase in Evsa-T cells only after 4 h of progesterone treatment. The effect of sensitization by progesterone is not observed with cotreatment but is 2-fold greater with 2-h pretreatment of progesterone.
Steroid hormones can induce higher transcriptional activities of cell-cycle control genes (39, 40) and could trigger imbalance in the cells and subsequent cisplatin sensitization. The transient steroid hormone treatment does not change the cell growth and proliferation characteristics of MCF-7 cells, according to FACScan analysis and cell-counting assays. Progesterone does not cause cell proliferation but still evokes the same sensitivity of cells to cisplatin. In addition, the sensitivity toward other cytotoxic agents, such as trans-DDP and calicheamicin, is not affected by hormone treatments. The above evidence argues against higher transcriptional activity being the main reason for the enhanced platinum-induced toxicity of treated cells.
Another hypothesis that active promoter sites are preferentially platinated by cisplatin was also investigated. Such sites should be equally accessible to cisplatin and trans-DDP, however, and the trans-DDP sensitivity was not influenced by hormone treatment. In addition, platinum atomic absorption data show similar levels of platinum-DNA adducts on genomic DNA from estrogen-treated and control cells.
In summary, all available evidence supports the hypothesis that hormone receptors are essential for HMG1 up-regulation and subsequent cisplatin/carboplatin sensitization in the cells. Cisplatin produces many side effects including nephrotoxicity, neurotoxicity, and emesis (50). Our study shows that the potency of cisplatin can be increased through hormone treatment. From a clinical perspective, estrogen and/or progesterone treatment should allow the currently applied cisplatin regimens to show increased cytotoxicity toward cancer cells. Even a factor of 2 could be quite important in medical applications. In addition, the enhanced potency of the drug may be sufficient to overcome some acquired and intrinsic resistance. Many breast and ovarian tumors express high levels of ER, PR, or both (51, 52), and these cancers would be good candidates for cisplatin/carboplatin treatment in conjunction with hormone therapy. Because estrogen has been implicated in the etiology of breast cancer owing to its proliferative properties, progesterone may be preferred for patients with breast cancer. For patients with ovarian and cervical cancer, carboplatin is the standard chemotherapeutic agent because of its diminished nephrotoxicity compared with that of cisplatin (37). Even though pathways other than repair shielding by HMG1 could be responsible for the observed sensitization, this work establishes the potential for treating patients with ovarian cancer with estrogen/progesterone in combination with carboplatin in clinical trials.
Acknowledgments
We thank J. M. Essigmann for access to tissue culture facilities and the Massachusetts Institute of Technology Center for Cancer Research for providing the FACS facilities. We greatly appreciate help received from R. Hynes and J. Trevithick in conjunction with the immunofluorescence study. The HeLa cell line was a gift from T. de Lange (Rockefeller University, New York), and the Evsa-T cell line was from G. Leclercq (Institut Jules Bordet, Brussels). This work was supported by a grant from the National Cancer Institute (to S.J.L.), by the Howard Hughes Medical Institute (to Q.H.), and by the Ludwig Cancer Research Fellowship (to C.H.L.).
Abbreviations
- ER
estrogen receptor
- PR
progesterone receptor
- trans-DDP
trans-diamminedichloroplatinum(II)
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.100108697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.100108697
References
- 1.Loehrer P J, Einhorn L H. Ann Intern Med. 1984;100:704–713. doi: 10.7326/0003-4819-100-5-704. [DOI] [PubMed] [Google Scholar]
- 2.Morris M, Eifel P J, Lu J, Grigsby P W, Levenback C, Stevens R E, Rotman M, Gershenson D M, Mutche D G. N Engl J Med. 1999;340:1137–1143. doi: 10.1056/NEJM199904153401501. [DOI] [PubMed] [Google Scholar]
- 3.Rose P G, Bundy B N, Watkin E B, Thigpen J T, Deppe G, Maiman M A, Clarke-Pearson D L, Insalaco S. N Engl J Med. 1999;340:1144–1153. doi: 10.1056/NEJM199904153401502. [DOI] [PubMed] [Google Scholar]
- 4.Keys H M, Bundy B N, Stehman F B, Muderspach L I, Chafe W E, Suggs C L, Walker J L, Gersell D. N Engl J Med. 1999;340:1154–1161. doi: 10.1056/NEJM199904153401503. [DOI] [PubMed] [Google Scholar]
- 5.Thomas G M. N Engl J Med. 1999;340:1198–1200. doi: 10.1056/NEJM199904153401509. [DOI] [PubMed] [Google Scholar]
- 6.Lepre C A, Lippard S J. Nucleic Acids Mol Biol. 1990;4:9–38. [Google Scholar]
- 7.Yang D, Wang A J. Prog Biophys Mol Biol. 1996;66:81–111. doi: 10.1016/s0079-6107(96)00017-x. [DOI] [PubMed] [Google Scholar]
- 8.Gelasco A, Lippard S J. In: Topics in Biological Inorganic Chemistry. Clarke M J, Sadler P J, editors. Vol. 1. Berlin: Springer; 1998. pp. 1–43. [Google Scholar]
- 9.Pinto A L, Lippard S J. Proc Natl Acad Sci USA. 1985;82:4616–4619. doi: 10.1073/pnas.82.14.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciccarelli R B, Solomon M J, Varshavsky A, Lippard S J. Biochemistry. 1985;24:7533–7540. doi: 10.1021/bi00347a005. [DOI] [PubMed] [Google Scholar]
- 11.Comess K M, Burstyn J N, Essigmann J M, Lippard S J. Biochemistry. 1992;31:3975–3990. doi: 10.1021/bi00131a013. [DOI] [PubMed] [Google Scholar]
- 12.Mello J A, Lippard S J, Essigmann J M. Biochemistry. 1995;34:14783–14791. doi: 10.1021/bi00045a020. [DOI] [PubMed] [Google Scholar]
- 13.Jamieson E R, Lippard S J. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 14.Bustin M, Reeves R. Prog Nucleic Acid Res Mol Biol. 1996;54:35–97. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 15.McA'Nulty M M, Lippard S J. In: Nucleic Acids and Molecular Biology. Eckstein F, Lilley D M J, editors. Vol. 9. Berlin: Springer; 1995. pp. 264–284. [Google Scholar]
- 16.Whitehead J P, Lippard S J. In: Metal Ions in Biological Systems. Sigel A, Sigel H, editors. Vol. 32. New York: Dekker; 1996. pp. 687–725. [PubMed] [Google Scholar]
- 17.Brown S J, Kellett P J, Lippard S J. Science. 1993;261:603–605. doi: 10.1126/science.8342024. [DOI] [PubMed] [Google Scholar]
- 18.McA'Nulty M M, Lippard S J. Mutat Res. 1996;362:75–86. doi: 10.1016/0921-8777(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 19.Huang J, Zamble D B, Reardon J T, Lippard S J, Sancar A. Proc Natl Acad Sci USA. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zamble D B, Mu D, Reardon J T, Sancar A, Lippard S J. Biochemistry. 1996;35:10004–10013. doi: 10.1021/bi960453+. [DOI] [PubMed] [Google Scholar]
- 21.Trimmer E E, Zamble D B, Lippard S J, Essigmann J M. Biochemistry. 1997;37:352–362. doi: 10.1021/bi971675q. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Liu X, Glassman A B, Keating M J, Stros M, Plunkett W, Yang L. Cancer Res. 1997;57:1487–1494. [PubMed] [Google Scholar]
- 23.Arioka H, Nishio K, Ishida T, Fukumoto H, Fukuoka K, Nomoto T, Kurokawa H, Yokote H, Abe S, Saijo N. Jpn J Cancer Res (GANN) 1999;90:108–115. doi: 10.1111/j.1349-7006.1999.tb00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zlatanova J, van Holde K. BioEssays. 1998;20:584–588. doi: 10.1002/(SICI)1521-1878(199807)20:7<584::AID-BIES10>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 25.Landsman D, Bustin M. BioEssays. 1993;15:539–546. doi: 10.1002/bies.950150807. [DOI] [PubMed] [Google Scholar]
- 26.Bianchi M E, Beltrame M. Am J Hum Genet. 1998;63:1573–1577. doi: 10.1086/302170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falciola L, Spada F, Calogero S, Langst G, Voit R, Grummt I, Bianchi M E. J Cell Biol. 1997;137:19–26. doi: 10.1083/jcb.137.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grosschedl R, Giese K, Pagel J. Trends Genet. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 29.Grosschedl R. Curr Opin Cell Biol. 1995;7:362–370. doi: 10.1016/0955-0674(95)80091-3. [DOI] [PubMed] [Google Scholar]
- 30.Boonyaratanakornkit V, Melvin V, Prendergast P, Altmann M, Ronfani L, Bianchi M E, Taraseviciene L, Nordeen S K, Allegretto E A, Edwards D P. Mol Cell Biol. 1998;18:4471–4487. doi: 10.1128/mcb.18.8.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nardulli A M, Greene G L, Shapiro D J. Mol Endocrinol. 1993;2:331–340. doi: 10.1210/mend.7.3.8483477. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C C, Krieg S, Shapiro D J. Mol Endocrinol. 1999;13:632–643. doi: 10.1210/mend.13.4.0264. [DOI] [PubMed] [Google Scholar]
- 33.Onate S A, Prendergast P, Wagner J P, Nissen M, Reeves R, Pettijohn D E, Edwards D P. Mol Cell Biol. 1994;14:3376–3391. doi: 10.1128/mcb.14.5.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melvin V S, Edwards D P. Steroids. 1999;64:576–586. doi: 10.1016/s0039-128x(99)00036-7. [DOI] [PubMed] [Google Scholar]
- 35.Calogero S, Grassi F, Aguzzi A, Voigtlander T, Ferrier P, Ferrari S, Bianchi M E. Nat Genet. 1999;22:276–280. doi: 10.1038/10338. [DOI] [PubMed] [Google Scholar]
- 36.Chau K Y, Lam H Y P, Lee K L D. Exp Cell Res. 1998;241:269–272. doi: 10.1006/excr.1998.4052. [DOI] [PubMed] [Google Scholar]
- 37.Fichtinger-Schepman A M J, van Dijk-Knijnenburg H C M, van der Velde-Visser S D, Berends F, Baan R A. Carcinogenesis. 1995;16:2447–2453. doi: 10.1093/carcin/16.10.2447. [DOI] [PubMed] [Google Scholar]
- 38.Lippman M, Bolan G, Huff K. Cancer Res. 1976;36:4595–4601. [PubMed] [Google Scholar]
- 39.Ciocca D R, Fanelli M A. Trends Endocrinol Metab. 1997;8:313–321. doi: 10.1016/s1043-2760(97)00122-7. [DOI] [PubMed] [Google Scholar]
- 40.Diaz-Perez M J, Zannis-Hadjopoulos M, Price G B, Wainer I W. J Cell Biochem. 1998;70:323–329. doi: 10.1002/(sici)1097-4644(19980901)70:3<323::aid-jcb5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 41.Kuduvalli P N, Townsend C A, Tullius T D. Biochemistry. 1995;34:3899–3906. doi: 10.1021/bi00012a005. [DOI] [PubMed] [Google Scholar]
- 42.Haghighi A, Lebedeva S, Gherset R A. Biochemistry. 1999;38:12432–12438. doi: 10.1021/bi991079r. [DOI] [PubMed] [Google Scholar]
- 43.Caliaro M J, Vitaux P, Lochon I, Nehme A, Valette A, Canal P, Bugat R, Jozan S. Br J Cancer. 1997;75:333–340. doi: 10.1038/bjc.1997.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J D. Molecular Biology of the Cell. New York: Garland; 1989. pp. 496–497. [Google Scholar]
- 45.Wang H, Bloom O, Zhang M, Vishnubhakat J M, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 46.Bancroft D, Lepre C A, Lippard S J. J Am Chem Soc. 1990;112:6860–6871. [Google Scholar]
- 47.Los G, Verdegaal E, Noteborn H P J M, Ruevekamp M, Graeff A D, Meesters E W, Huinink D T B, McVie J G. Biochem Pharmacol. 1991;42:357–363. doi: 10.1016/0006-2952(91)90723-i. [DOI] [PubMed] [Google Scholar]
- 48.Knox R J, Friedlos F, Lydall D A, Roberts J J. Cancer Res. 1986;46:1972–1979. [PubMed] [Google Scholar]
- 49.Hongo A, Seki S, Akiyama K, Kudo T. Int J Biochem. 1994;26:1009–1016. doi: 10.1016/0020-711x(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 50.Van Hoff D D, Schilsky R, Reichert C M, Reddick R L, Rozenscweig M, Young R C, Muggia F M. Cancer Treat Rep. 1979;63:1527–1531. [PubMed] [Google Scholar]
- 51.Ferno M, Johansson B U, Olsson N H, Ryden S, Sellberg G. Acta Oncol. 1990;29:129–135. doi: 10.3109/02841869009126532. [DOI] [PubMed] [Google Scholar]
- 52.Slotman B J, Rao B R. Anticancer Res. 1988;8:417–434. [PubMed] [Google Scholar]