

Abstract
Apoptosis mediated by Fas/Fas ligand (FasL) interaction has been implicated in human disease processes, including pulmonary disorders. However, the role of the Fas/FasL system in acute lung injury (ALI) and in the acute respiratory distress syndrome (ARDS) is poorly defined. Accordingly, we investigated both the soluble and cellular expression of the Fas/FasL system in patients with ALI or ARDS. The major findings are summarized as follows. First, the soluble expression of the Fas/FasL system was assessed in undiluted pulmonary edema fluid and simultaneous plasma. Pulmonary edema fluid obtained from patients with ALI or ARDS (n = 51) had significantly higher concentrations of both soluble Fas (27 ng/ml; median; P < 0.05) and soluble FasL (0.125 ng/ml; P < 0.05) compared to control patients with hydrostatic pulmonary edema (n = 40; soluble Fas, 12 ng/ml; soluble FasL, 0.080 ng/ml). In addition, the concentrations of both soluble Fas and soluble FasL were significantly higher in the pulmonary edema fluid of the patients with ALI or ARDS compared to simultaneous plasma samples (soluble Fas, 16 ng/ml; soluble FasL, 0.058 ng/ml; P < 0.05), indicating local release in the lung. Higher soluble Fas concentrations were associated with worse clinical outcomes. Second, cellular expression of the Fas/FasL system was assessed by semiquantitative immunofluorescence microscopy in lung tissue obtained at autopsy from a different set of patients. Both Fas and FasL were immunolocalized to a greater extent in the patients who died with ALI or ARDS (n = 10) than in the patients who died without pulmonary disease (n = 10). Both proteins were co-expressed by epithelial cells that lined the alveolar walls, as well as by inflammatory cells and sloughed epithelial cells that were located in the air spaces. Semiquantitative immunohistochemistry showed that markers of apoptosis (terminal dUTP nick-end labeling, caspase-3, Bax, and p53) were more prevalent in alveolar wall cells from the patients who died with ALI or ARDS compared to the patients who died without pulmonary disease. These findings indicate that alveolar epithelial injury in humans with ALI or ARDS is in part associated with local up-regulation of the Fas/FasL system and activation of the apoptotic cascade in the epithelial cells that line the alveolar air spaces.
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are characterized by increased protein permeability across the pulmonary microvascular endothelium and alveolar epithelium. 1 The structural manifestation of increased protein permeability is disruption of endothelial and epithelial cells. 2 The mechanisms by which the alveolar epithelium is injured in humans with ALI or ARDS remain uncertain. Recent studies propose that one mechanism may be Fas-dependent epithelial apoptosis. 3,4
The Fas/Fas ligand (FasL) system can initiate apoptosis. Fas (CD95) is a 45-kd type I membrane receptor, a member of the tumor necrosis factor family of cell surface receptors. 5 FasL (CD95L), a 37-kd type II membrane glycoprotein, is also a member of the tumor necrosis factor family of cytokines. 6 Fas and FasL exist in both membrane-bound and soluble forms. Membrane-bound Fas is the natural ligand for soluble FasL. Soluble Fas is produced by alternative splicing of mRNA 7 whereas soluble FasL is generated by cleavage of membrane-bound FasL by metalloproteinases. 8 Apoptosis is induced when membrane-bound or soluble FasL binds to Fas-bearing cells. 9 By contrast, apoptosis is inhibited when soluble Fas binds to either membrane-bound FasL or soluble FasL, thereby preventing FasL from interacting with membrane-bound Fas receptors.
A variety of cell types express FasL protein. Hematopoietic cells, such as lymphocytes, 10-12 monocytes, 13 neutrophils, 14,15 eosinophils, 11 and platelets 16 express FasL protein on their surfaces. Epithelial cells also express Fas and FasL proteins. For example, Fas and FasL proteins have been immunohistochemically localized in situ in airway epithelial cells obtained from resected human lungs. 3 Expression of both Fas and FasL was detected in columnar and basal cells of histologically normal-appearing bronchi.
Circulating soluble FasL is elevated in autoimmune disease, 17 malignancy, 18,19 and congestive heart failure. 20 In pulmonary diseases, soluble FasL is elevated in plasma and/or bronchoalveolar lavage (BAL) fluid from patients with fibrosing pulmonary diseases, 21 hypersensitivity pneumonitis, 22 and bronchiolitis obliterans-organizing pneumonia. 23 Soluble FasL protein was present in BAL fluid of patients with ALI or ARDS before and after the onset of lung injury. 4 That study also reported that the concentration of soluble FasL protein in the BAL fluid correlated both with proapoptotic activity in cultured human distal airway epithelial cells in vitro and decreased patient survival. 4 FasL mRNA also is increased in the cellular component of BAL fluid retrieved from patients with early ARDS. 24 Other in vitro studies have shown that distal lung epithelial cells express Fas on their surface. 25 Together, these studies suggest that the Fas/FasL system may mediate, in part, the injury to distal lung epithelial cells that occurs in patients with ALI or ARDS.
Our study was designed to test two hypotheses regarding the proapoptotic environment in the alveolar air spaces in patients with ALI or ARDS. The first was that soluble FasL would be present in higher concentrations in undiluted pulmonary edema fluid from patients with ALI or ARDS compared to patients with hydrostatic pulmonary edema, and that soluble FasL would be in excess of soluble Fas. Accordingly, the concentration of soluble Fas and soluble FasL was measured in the undiluted pulmonary edema fluid obtained from 51 patients with ALI or ARDS and from 40 control patients with hydrostatic pulmonary edema. The second hypothesis was that Fas and FasL protein would be expressed by distal epithelial cells in the lungs of patients with ALI or ARDS to a greater extent than in patients without ALI or ARDS. To test the second hypothesis, lung tissue was collected at autopsy from a different set of patients who died of ALI or ARDS (n = 10) or without pulmonary disease (n = 10).
Materials and Methods
Soluble Fas/FasL Protein Concentration in Pulmonary Edema Fluid and Plasma
Patient Characteristics
Patients admitted to the adult intensive care units of Moffitt-Long Hospital, University of California, San Francisco, or San Francisco General Hospital between 1981 and 1999 with ALI, ARDS, or hydrostatic pulmonary edema were eligible for inclusion in the study. Additional inclusion criteria were acute respiratory failure requiring mechanical ventilation and aspirable pulmonary edema fluid within 1 hour of tracheal intubation.
Patients with ALI or ARDS were identified by the American European Consensus Conference definitions, which include the acute onset of bilateral infiltrates on chest radiograph, an arterial pO2-to-inspired O2 ratio of ≤300 for ALI and ≤200 for ARDS, and no clinical evidence of elevated left atrial pressure, including a pulmonary arterial wedge pressure of <18 mmHg, if measured. 26 The initial ratio of edema fluid to plasma protein, a measure of alveolar capillary barrier permeability, was required to be >0.65, consistent with increased permeability pulmonary edema. 27 The definition of acute hydrostatic pulmonary edema was based on standard clinical criteria and the presence of a transudative pulmonary edema fluid with an initial ratio of edema fluid to plasma protein <0.65.
Specific criteria used to define hydrostatic pulmonary edema included central venous pressure ≥14 mmHg, pulmonary arterial wedge pressure ≥18 mmHg, or cardiac ejection fraction ≤45% by echocardiogram, radionuclide, or contrast ventriculography, and/or positive physical findings, including a third heart sound and jugular venous distension. Patients who had a mixed cause of pulmonary edema, with elements of both ALI and elevated hydrostatic pressures, were excluded.
This study was approved by the Committee for Human Research at the University of California, San Francisco.
Collection of Pulmonary Edema Fluid
Samples of pulmonary edema fluid were collected as previously described. 28,29 Briefly, a 14-Fr-gauge tracheal suction catheter was advanced through the endotracheal tube into a wedged position in a distal bronchus. Undiluted pulmonary edema fluid was aspirated by gentle suction into a suction trap. A simultaneous plasma sample was obtained by venipuncture or aspiration from an indwelling venous catheter. Pulmonary edema fluid and plasma samples were centrifuged at 3000 × g for 10 minutes. The supernatants were aspirated and stored at −70°C. The initial pulmonary edema fluid sample was obtained within 1 hour of endotracheal intubation in all patients.
Protein Concentration Measurements
The total protein concentration was measured in thawed edema fluid and plasma samples by the biuret method, as previously described. 28,29 If the volume of edema fluid or plasma was insufficient for measurement by the biuret method (<1% of samples), then the total protein concentration was measured by refractometry.
Measurement of Soluble Fas and FasL Protein
Measurements were made in duplicate for thawed edema fluid and plasma samples. Soluble Fas and FasL proteins were measured using commercially available sandwich enzyme-linked immunosorbent assay (ELISA) kits (Fas from Biosource International, Camarillo, CA; FasL from MBL, Nagoya, Japan). To exclude the possibility that substances present in the pulmonary edema fluid or plasma interfered with accurate quantification by the Fas or FasL ELISA antibodies, known concentrations of Fas and FasL proteins were measured by ELISA in the presence of varying volumes of edema fluid and plasma samples. There was no evidence of any interfering substance present in the edema fluid or plasma for either the Fas or FasL ELISA.
Clinical Data Collection
Clinical data were collected from the medical record for each patient, using a standardized data collection form. Demographic data included age, gender, race, and smoking history. Physiological data for the first 24 hours after endotracheal intubation included hemodynamic parameters (pulmonary arterial wedge pressure; central venous pressure; systolic, diastolic, and mean arterial blood pressures; heart rate; cardiac output; systemic vascular resistance; and left ventricular ejection fraction), respiratory and ventilatory parameters [arterial pH, PO2, PCO2, alveolar-arterial PO2 difference, arterial PO2-to-inspiratory O2 fraction (P/F) ratio, tidal volume, minute ventilation, positive end-expiratory pressure, peak inspiratory pressure, and static compliance], and multiorgan system function (measurements of renal, hepatic, central nervous system, gastrointestinal, and hematological function; net fluid balance; and weight). For each patient, the Simplified Acute Physiology Score II (SAPS II) 30 and the Lung Injury Score (LIS) 31 were calculated according to the published algorithms. Organ system failures were defined as previously described. 32 Outcome variables included hospital mortality and the duration of unassisted ventilation as measured by days alive and off of the mechanical ventilator during the 28 days after enrollment.
The primary etiology of ALI or ARDS was determined from a detailed review of the clinical history. Sepsis was defined using standard criteria. 32 Pneumonia was defined as new radiographical infiltrates and positive sputum cultures. Patients with sepsis secondary to pneumonia were classified as having sepsis as the primary etiology of ALI or ARDS. Aspiration of gastric contents was defined as a witnessed aspiration event. The primary etiology of hydrostatic pulmonary edema was also assessed based on a detailed review of the medical chart, and classified as acute exacerbation of chronic congestive heart failure, acute myocardial infarction, or volume overload.
Immunofluorescence Localization of Fas/FasL Proteins in Lung Tissue Sections
Patient Selection
Fresh lung tissue was analyzed from 20 patients admitted to the adult medical intensive care units of the University of Utah Hospital or the LDS Hospital, both in Salt Lake City, between 1995 and 2000, who underwent autopsy procedures for medical reasons. None of these patients had pulmonary edema fluid or plasma collected for analysis. The autopsy tissue, collected within 12 to 14 hours of death, using standard anatomical pathology approaches optimized to preserve tissue architecture and antigen display, was obtained from two groups of 10 patients each. One group died with ALI or ARDS. The other group died without pulmonary disease and served as controls. Diagnostic criteria for ALI or ARDS were as described above (see Patient Characteristics for Soluble Fas/FasL Protein Concentration in Pulmonary Edema Fluid and Plasma). This autopsy protocol was approved by the Institutional Review Board committees at both participating hospitals. The requirement for written informed consent was waived by both committees.
Two to three tissue cubes (>2 × 2 × 2 cm) were obtained from each of the three lobes of the right lung of each patient. The cubes were sliced (3 mm × 2 cm × 2 cm) and the slices were immersed in 10% buffered neutral formalin (VWR, Inc., Media, PA) overnight at 4°C. The slices were placed in 70% ethanol and processed immediately. Paraffin-embedded tissue sections (5 μm) were collected on PLUS slides (VWR, Inc.) and the paraffin was removed from the sections before performing for immunofluorescence microscopy. 33
Tissue Section Preparation for Immunofluorescence Microscopy
The deparaffinized tissue sections were immersed in antigen retrieval solution (S1700; DAKO Corp., Carpinteria, CA) and heated in a water bath for 20 minutes at 90°C. The tissue sections were cooled for 20 minutes at room temperature before endogenous peroxidase activity was quenched by incubating the tissue sections in 3% hydrogen peroxide for 20 minutes at room temperature. Nonspecific binding of avidin and biotin was prevented by treating the tissue sections with avidin/biotin blocking kit (SP-2001; Vector Laboratories, Burlingame, CA). Next, a protein-blocking solution [tyramide signal amplification (TSA) kit NEL701 for Fluorescence System; NEN Life Science Products, Boston, MA] was applied for 30 minutes at room temperature. The tissue sections were washed with 0.01 mol/L of phosphate-buffered saline (PBS) between each of the steps described above. After the tissue sections were treated with the primary and secondary antibodies (described below), the tissue sections were coverslipped, using Vectashield DAPI (4,6-diamidino-2-phenylindole) mount-medium for fluorescence (H-1200, Vector Laboratories). In some instances, we did not use Vectashield DAPI mount; instead, we used nonfluorescent mounting medium.
For each of the immunofluorescence preparations described below, we used one tissue section from each of the 20 patients. Sufficient reagents for each primary/secondary antibody combination were prepared to process all 20 slides together. We processed each set of 20 slides as a batch to ensure that the results were not affected by variations in protocol. The tissue sections were washed three times with 0.01 mol/L of PBS between each of the steps described below.
Single Immunofluorescence Localization of Fas or FasL
One set of 20 tissue sections each was incubated with either 1:250 dilution of anti-Fas mouse monoclonal anti-human antibody or 1:1000 dilution of anti-FasL polyclonal rabbit anti-human antibody (SC-8009 for anti-Fas and SC-834 for anti-FasL, respectively; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) overnight at 4°C. The next morning, we incubated the tissue sections with 1:200 dilution of anti-mouse IgG biotin conjugate or anti-rabbit IgG biotin conjugate, respectively (BA-2000 for anti-mouse or BA-1000 for anti-rabbit, respectively; Vector Laboratories) for 60 minutes at room temperature. The tissue sections were subsequently incubated with 1:200 dilution of streptavidin-horseradish peroxidase conjugate (TSA kit, NEL701) for 60 minutes at room temperature. Fluorochrome amplification was performed by incubating the tissue sections in 1:100 dilution of tyramide-fluorescein isothiocyanate (TSA kit; NEL701) at room temperature for 10 minutes. We counted the number of alveolar lining cells that were immunofluorescently labeled by the Fas or FasL antibody in one tissue section per patient. The reference structure was the number of DAPI-positive nuclei along the alveolar surface, which was kept constant for each tissue section (n = 25). The number of Fas- or FasL-immunopositive cells was divided by the number of DAPI-positive nuclei.
Double-Immunofluorescence Co-Localization of Fas and FasL
Twenty adjacent tissue sections to those used for single immunofluorescence localization of Fas or FasL were used for double-immunofluorescence localization of Fas and FasL. Co-localization was performed to determine whether Fas and FasL were expressed by the same cells. These tissue sections were incubated with both primary antibodies first, followed by the appropriate secondary antibodies, as described above. For Fas immunofluorescence localization, we used a 1:100 dilution of tyramide-fluorescein isothiocyanate conjugate (TSA kit, NEL701) for 10 minutes at room temperature. For FasL immunolocalization, we used a 1:100 dilution of tyramide-rhodamine conjugate (TSA kit, NEL702) for 10 minutes at room temperature. We counted the number of alveolar lining cells that were immunofluorescently colored orange, indicating co-localization of Fas and FasL in one tissue section per patient. The reference structure was the sum of the number of green cells (Fas-positive), red cells (FasL-positive), and orange cells (double labeled). That sum was kept constant for each tissue section (n = 25). The number of orange cells was divided by the sum of the green, red, and orange cells.
Double-Immunofluorescence Co-Localization of Fas or FasL with Epithelial Cell Markers
The results for the single immunofluorescence methods described above suggested that Fas and FasL were expressed by cells in the alveolar walls. Because Fas or FasL protein expression has been shown in airway epithelial cells of human lungs in situ 3 and epithelial cells isolated from the distal regions of the human lung in vitro, 25 we used two antibodies directed against epithelial cell antigens to identify the epithelial cells that express Fas or FasL in situ in the human lung. The two antibodies were cytokeratin, a marker of epithelial cells in general, and KL-6, a specific marker for alveolar type II epithelial cells. 34 The KL-6 antibody recognizes a glycoprotein of a mucin (MUC-1). KL-6 antigen was purified from human lung adenocarcinoma cells. 35
One set of 20 tissue sections each was incubated with anti-Fas antibody or anti-FasL antibody, as described above. Fas or FasL immunolocalization was amplified by a 1:100 dilution of tyramide-fluorescein isothiocyanate for 10 minutes at room temperature. All of the tissue sections were subsequently incubated with a 1:250 dilution of anti-cytokeratin mouse monoclonal antibody (C-2562; Sigma Chemical Co., St. Louis, MO) at room temperature for 2 hours, followed by incubation with anti-mouse IgG rhodamine conjugate (605-170; Roche Molecular Biochemicals, Indianapolis, IN) for 60 minutes at room temperature.
Another set of 20 tissue sections each was used to co-localize the specific marker of alveolar type II epithelial cells, KL-6, and Fas or FasL. Fas or FasL immunolocalization was amplified by a 1:100 dilution of tyramide-fluorescein isothiocyanate at room temperature for 10 minutes. All of the tissue sections were subsequently incubated with a 1:1000 dilution of anti-KL-6 mouse monoclonal antibody for 2 hours at room temperature, followed by incubation with anti-mouse IgG rhodamine conjugate for 60 minutes at room temperature, as described above.
We counted the number of alveolar lining cells that were immunofluorescently colored orange, indicating co-localization of Fas or FasL with either cytokeratin or KL-6 antibodies in one tissue section per patient. The reference structure was the number of red-stained cells, indicating localization of either cytokeratin or KL-6 antibody, along the alveolar surface, which we kept constant for each tissue section (n = 25). We divided the number of orange cells by the number of red cells.
Immunofluorescence Controls
Immunostaining controls included substitution of each primary with normal horse serum, substitution of each primary antibody with an irrelevant, species-matched, isotype-matched antibody (anti-insulin), and substitution of the secondary antibody with PBS.
Immunofluorescence Photography
We used a Zeiss AxioPhot photomicroscope equipped for epifluorescence microscopy. Photographs were recorded on color slide film (Kodak ASA 400; Eastman-Kodak, Rochester, NY), from which Cibachrome glossy color prints were prepared.
Localization of Markers of Apoptosis in the Lung
Markers of apoptosis were detected in adjacent tissue sections from the same set of 20 patients (one tissue section/patient). DNA strand breaks were identified in situ by end-labeling the breaks, using terminal dUTP nick-end labeling (TUNEL). Other molecular markers of apoptosis included caspase-3, Bax, BclII, and p53. Of these markers, all are proapoptotic, except for BclII, which is anti-apoptotic. TUNEL labeling was accomplished by using a commercial kit (220 582, Roche Diagnostics Corp., Indianapolis, IN), according to the manufacturer’s instructions. Caspase-3 (1:150 optimal dilution, NCL-CPP32p; Novocastra Laboratories Ltd., Newcastle on Tyne, UK), Bax (1:500 optimal dilution, B3428; Sigma), BclII (1:50 optimal dilution, NCL-bcl-2; Novocastra), and p53 (1:50 optimal dilution, NCL-p53-DO14, Novocastra) were revealed by antigen retrieval, as described above. TSA signal amplification, also described above, was used for caspase-3, BclII, and p53. Bax signal was detected by a standard immunoperoxidase method (ABC Elite Standard kit, PK-6100; Vector Laboratories Inc.). Secondary antibodies were goat anti-rabbit biotin conjugated for Bax and caspase-3 (BA-1000, Vector Laboratories, Inc.) and horse anti-mouse biotin conjugated for BclII and p53 (BA-2000, Vector Laboratories, Inc.). TUNEL, Bax, BclII, and p53 were visualized using horse-radish peroxidase/diaminobenzidine, whereas caspase-3 was visualized using alkaline phosphatase-red. Gill’s no. 3 hematoxylin was used as the counterstain for TUNEL, caspase-3, and Bax. No counterstain was used for BclII or p53 because the chromogen signal was subtle. Tissue detail was detected using differential interference contrast optics. Immunostaining controls included substitution of each primary antibody with blocking buffer and substitution of each secondary antibody with PBS. The immunostained tissue sections for markers of apoptosis were photographed using a Zeiss AxioPhot microscope equipped with a bright-field, high-resolution color digital camera (ProgRes 3012; Jenoptik, Jena, Germany). Digital images were prepared for illustration, without altering color or image detail.
We counted the number of alveolar lining cells that were immunohistochemically colored, indicating TUNEL, caspase-3, Bax, BclII, or p53 labeling, in one tissue section per patient. The reference structure was the number of nuclei along the alveolar surface, which we kept constant for each tissue section (n = 25). We divided the number of colored cells by the number of nuclei.
Statistical Analysis
Statistical analysis was done using SPSS 6.1.6 for Macintosh (SPSS, Chicago, IL) or StatView 5 for Windows (SAS Institute, Cary, NC). Nonparametric data were analyzed using the Mann-Whitney U-test or the Kruskal-Wallis test with Dunn’s test for multiple comparisons. 36 Parametric data were analyzed using paired t-test or analysis of variance with Student-Newman-Keuls test for multiple comparisons. 36 Categorical variables were compared using chi-square analysis. 36 Statistical significance was defined as P ≤ 0.05.
Results
Soluble Fas and FasL Concentrations in Pulmonary Edema Fluid and Plasma
Patient Characteristics
Fifty-one patients with ALI or ARDS and 40 control patients with hydrostatic pulmonary edema are included in this part of the study. Demographic and clinical characteristics of the patients are summarized in Table 1 ▶ . Patients with ALI or ARDS were significantly younger and had more organ failures, shorter duration of unassisted ventilation, and higher hospital mortality than patients with hydrostatic pulmonary edema. The most common cause of ALI or ARDS was sepsis (pulmonary or nonpulmonary) (47%), followed by drug overdose (14%), pneumonia without sepsis (10%), multiple transfusions (10%), aspiration (10%), and other causes (10%). The most common cause of hydrostatic pulmonary edema was congestive heart failure (36%), followed by acute myocardial infarction (31%), volume overload (18%), and other causes (15%).
Table 1.
Clinical Characteristics of 51 Patients with ALI or ARDS and 40 Patients with Hydrostatic Pulmonary Edema
| Clinical characteristics | ALI or ARDS (n = 51) | Hydrostatic pulmonary edema (n = 40) |
|---|---|---|
| Age (years) | 44 ± 16* | 60 ± 20 |
| Male gender | 59% | 43% |
| Caucasian | 60% | 67% |
| Current smoker | 24% | 27% |
| SAPSII score | 51 ± 19 | 46 ± 14 |
| Number of organ failures | 1.8 ± 1.3* | 1.2 ± 1.0 |
| Lung Injury Score | 2.9 ± 0.7 | 2.8 ± 0.7 |
| Edema/plasma protein ratio | 1.0 ± 0.2* | 0.5 ± 0.1 |
| Hospital mortality | 57%* | 24% |
| Days of unassisted ventilation | 8 ± 11* | 16 ± 11 |
Data are mean ± SD or percent of total patients.
*P < 0.05 compared to hydrostatic pulmonary edema group.
Soluble Fas and FasL Concentrations in Pulmonary Edema Fluid and Plasma
For patients with ALI or ARDS, the concentrations of both soluble Fas (Figure 1) ▶ and soluble FasL (Figure 2) ▶ were significantly higher in undiluted pulmonary edema fluid than in matched plasma, indicating local production in the alveolar compartment. By comparison, for patients with hydrostatic pulmonary edema, the concentrations of both soluble Fas (Figure 1) ▶ and soluble Fas ligand (Figure 2) ▶ were similar in undiluted pulmonary edema fluid and matched plasma. In addition, when patients with ALI or ARDS were compared to those with hydrostatic pulmonary edema, soluble Fas and FasL concentrations were higher in pulmonary edema fluid from patients with ALI or ARDS. Plasma concentrations of soluble Fas and FasL were not different between patients with ALI or ARDS and hydrostatic pulmonary edema.
Figure 1.

Box plot summary of undiluted pulmonary edema fluid and simultaneous plasma concentrations of soluble Fas protein in 51 patients with ALI or ARDS compared to 40 control patients with hydrostatic pulmonary edema. Horizontal line represents the median, box encompasses the 25th to 75th percentiles, and error bars encompass the 10th to 90th percentiles. *, P < 0.05 versus all other groups. **, P < 0.05 versus hydrostatic edema.
Figure 2.

Box plot summary of undiluted pulmonary edema fluid and simultaneous plasma concentrations of soluble Fas ligand protein in 51 patients with ALI or ARDS compared to 40 control patients with hydrostatic pulmonary edema. Horizontal line represents the median, box encompasses the 25th to 75th percentiles, and error bars encompass the 10th to 90th percentiles. *, P < 0.05 versus all other groups.
In contrast to our original hypothesis, soluble Fas was in excess of soluble FasL by >200-fold in undiluted pulmonary edema fluid from the patients with ALI or ARDS. The median concentration of soluble Fas protein was 27 ng/ml compared to 125 pg/ml (0.125 ng/ml) for soluble FasL protein.
Association between Higher Soluble Fas Protein Concentrations and Clinical Variables
The concentration of soluble Fas protein in both undiluted pulmonary edema fluid and plasma was associated with the overall severity of illness in patients with ALI or ARDS. Soluble Fas concentrations were higher in patients with hypotension, shock, sepsis, and higher SAPS II scores (Table 2) ▶ . Soluble Fas concentrations were also higher in patients with renal failure (Table 2) ▶ or multiorgan system failure when two or more organ systems were involved (Figure 3) ▶ . Soluble Fas concentrations were not associated with hospital mortality or duration of mechanical ventilation. By contrast, the concentration of soluble FasL in either undiluted pulmonary edema fluid or plasma was not significantly associated with any clinical variables or outcome measures.
Table 2.
Edema Fluid and Plasma-Soluble Fas Concentrations Are Higher in Patients with ALI or ARDS and Higher Severity of Illness, Sepsis, Shock, or Renal Failure
| Clinical characteristics | Edema fluid-soluble Fas (ng/ml) | P value | Plasma-soluble Fas (ng/ml) | P value |
|---|---|---|---|---|
| SAPS II | ||||
| <45 | 17 (3–117) | 0.027 | 11 (4–72) | 0.0035 |
| ≥45 | 53 (2–201) | 24 (5–99) | ||
| Sepsis | ||||
| No | 22 (5–201) | 0.15 | 12 (5–72) | 0.028 |
| Yes | 48 (2–195) | 21 (4–99) | ||
| Systolic blood pressure | ||||
| ≥70 | 25 (3–201) | 0.023 | 13 (4–72) | 0.0086 |
| <70 | 53 (2–195) | 26 (9–99) | ||
| Renal failure | ||||
| No | 17 (2–67) | 0.0024 | 12 (6–34) | 0.0008 |
| Yes | 49 (3–201) | 23 (4–99) |
Data are median (range).
Figure 3.

Box plot summary of undiluted pulmonary edema fluid concentration of soluble Fas protein versus the number of organ system failures in 51 patients with ALI or ARDS. Patients were divided into two groups: those that had less than two organ system failures at the time of enrollment and those that had two or more organ system failures at study enrollment. Horizontal line represents the median, box encompasses the 25th to 75th percentiles, error bars encompass the 10th to 90th percentiles, and closed circles represent outliers.
Immunofluorescence Localization of Fas and FasL Protein Expression
Patient Characteristics
Ten patients who died with ALI or ARDS and 10 control patients who died without pulmonary disease are included in this portion of the study. These patients were a different group from those who were used to measure the concentration of soluble Fas and FasL in pulmonary edema fluid and plasma. Demographic and clinical characteristics of the patients are summarized in Table 3 ▶ . Patients who died with ALI or ARDS were younger, had more organ failures, and longer duration of assisted ventilation than patients who died without pulmonary disease. The most common causes of ALI or ARDS were sepsis (pulmonary or nonpulmonary) (4 of 10) and pneumonia without sepsis (3 of 10). The serious medical problems of the control patients who died of nonpulmonary causes were cardiovascular (ischemic cardiomyopathy, ruptured thoracic aortic aneurysm, or heart failure), alcoholic cirrhosis/pancreatitis, Parkinson’s disease, or gunshot wound. The serious medical problems of two of the control patients were unavailable because they were participants in the organ donor/transplant program at the University of Utah.
Table 3.
Clinical Characteristics of 10 Patients Who Died with ALI or ARDS and 10 Patients Who Died without Pulmonary Disease
| Clinical characteristics | ALI or ARDS (n = 10) | Nonpulmonary (n = 10) |
|---|---|---|
| Age (years) | 43 ± 18 | 52 ± 24 |
| Male gender | 75%* | 16%‡ |
| Caucasian | 100%† | 100%‡ |
| Current smoker | 18%† | 7%‡ |
| SAPSII score | 68 ± 14† | 51 ± 14‡ |
| Number of organ failures | 2.6 ± 0.9 | 1.8 ± 1.1‡ |
| Lung Injury Score | 3.5 ± 0.4† | 1.4 ± 1.1‡ |
| Hospital mortality | 100% | 100% |
| Days intubated, with assisted ventilation | 11 ± 15† | 2 ± 3‡ |
Data are mean ± SD or percent of total patients.
*P < 0.05 compared to nonpulmonary group.
†Information was not available for 1 patient who was an organ donor.
‡Information was not available for 2 patients, both of whom were in the organ transplant program.
Lung Histopathology
The lungs of the patients who died of ALI or ARDS had characteristic pathological features, such as accumulation of leukocytes, especially neutrophils, in the alveolar air spaces (see Figures 5 to 7 ▶ ▶ ▶ ) and alveolar edema fluid. These features were not evident in the lung tissue sections from the patients who died of nonpulmonary causes (Figures 4f and 5h) ▶ ▶ .
Figure 5.
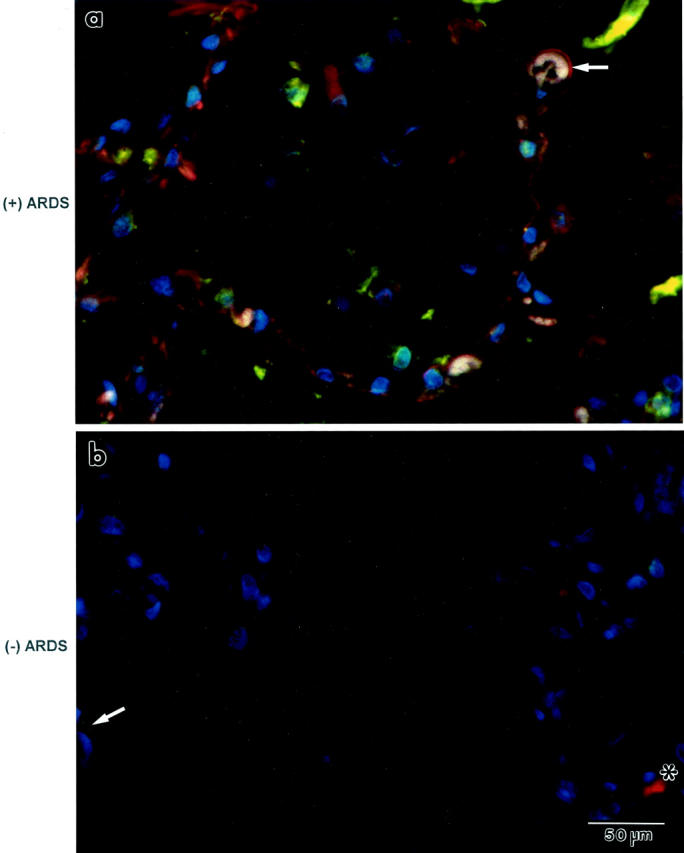
Immunofluorescence co-localization of Fas and FasL protein expression in lung tissue sections from a patient who died from ALI or ARDS [identified as “(+) ARDS”; a] or from another patient who died without pulmonary disease [identified as “(−) ARDS”; b]. a: Cells that line the alveolar walls (arrow) and in the alveolar walls are orange, indicating co-localization of Fas (green) and FasL (red) proteins. b: Only a few cells are immunostained either green (Fas, arrow) or red (FasL, *) in the lung tissue section from a patient who died without pulmonary disease. Nuclei are blue because DAPI was used as the counterstain. Both panels are the same magnification.
Figure 6.
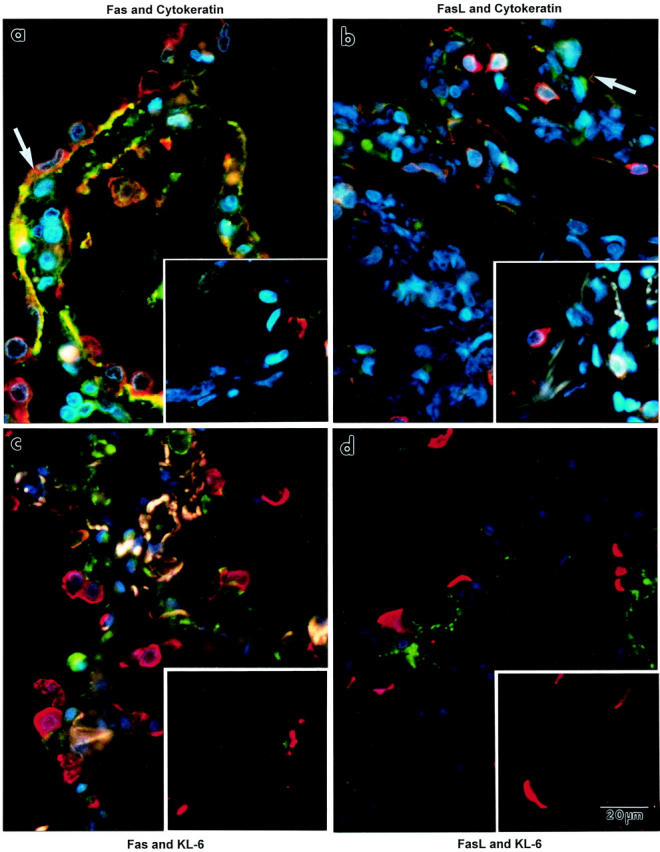
Immunofluorescence co-localization of Fas or FasL protein (both are green) with epithelial cell markers (red) in lung tissue sections from patients. The left column shows lung tissue sections that were immunostained for Fas. The right column shows lung tissue sections that were immunostained for FasL. The large picture of each pair shows a lung tissue section from a patient who died with ALI or ARDS. The inset of each pair shows a lung tissue section from a patient who died without pulmonary disease. a and b: Co-localization of cytokeratin (red immunofluorescence). In a, many of the epithelial cells that line the alveolar walls express orange immunofluorescence (arrow), indicating co-localization of cytokeratin and Fas proteins. Similar immunofluorescence attributes are displayed for cytokeratin and FasL proteins (b). c and d: Co-localization of KL-6, a specific marker for alveolar type II epithelial cells (red immunofluorescent crescents). KL-6 protein is exclusively expressed by cuboidal cells that are scattered along the alveolar wall perimeter. Neither Fas nor FasL (green) co-localized with KL-6. Little immunofluorescence staining for either Fas or FasL (green) was detected in the lung tissue sections from the patients who died without pulmonary disease (insets). The whitish fluorescence in the alveolar walls in c is because of autofluorescence of elastic fibers. Nuclei are blue, owing to the DAPI counterstain. All of the panels are the same magnification.
Figure 7.
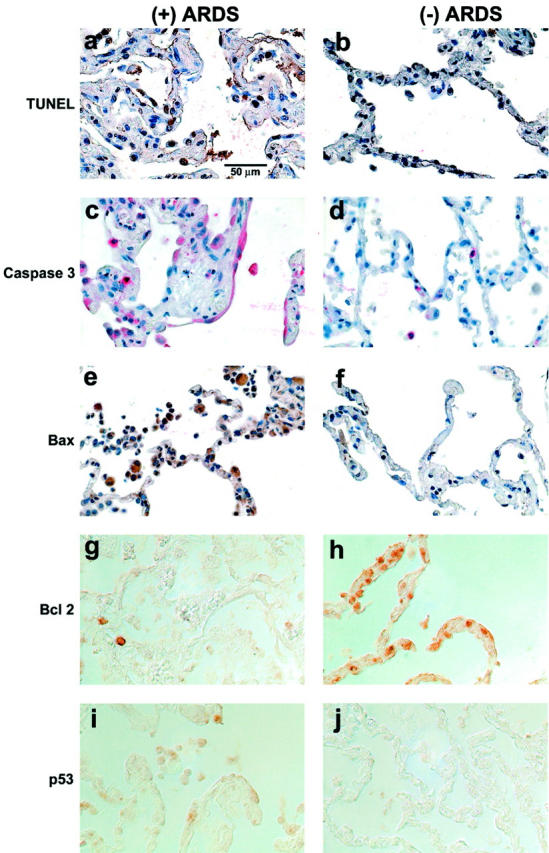
Localization of markers of apoptosis in lung tissue sections from patients. The left column shows lung tissue sections from a patient who died with ALI or ARDS [identified as “(+) ARDS”]. The right column shows lung tissue sections from a patient who died without pulmonary disease [identified as “(−) ARDS”]. a to f: Tissue sections that are counterstained with hematoxylin. g to j: Tissue sections that were imaged using differential interference contrast optics, without counterstain, because the epithelial cell immunostain reaction product was subtle. The rows of pictures are matched for one marker of apoptosis: TUNEL (a and b), caspase-3 (c and d), Bax (e and f), BclII (g and h), and p53 (i and j). Cells lining, and in, the alveolar walls demonstrate more TUNEL-labeled nuclei (arrow), caspase-3-labeled cytoplasm (arrow), Bax-labeled cytoplasm (arrow), and p53-labeled cytoplasm (arrow) in the tissue sections from the patients who died with ALI or ARDS compared to the patient who died without pulmonary disease. On the other hand, BclII-labeled cells lining, and in, the alveolar walls are more prominent in the tissue sections from the patient who died without pulmonary disease compared to the patient who died with ALI or ARDS, as expected. All of the panels are the same magnification.
Figure 4.

Immunofluorescence localization of Fas or FasL protein expression in lung tissue sections from patients who died from ALI or ARDS [identified as “(+) ARDS”]. a: Fas immunofluorescence is green. Blue fluorescence is DAPI, a counterstain for nuclei. Cells that line and are located in the alveolar walls are green. Many of the green-stained cells have attenuated cytoplasm that outlines the alveolar wall (arrows). c: FasL immunofluorescence is green. Cells in the alveolar walls, and along their surface (arrows), are green. b and d: Negative immunostaining controls. When either the primary Fas antibody (b) or the secondary antibody (d) was replaced with buffer, green immunofluorescence was absent. The four panels are the same magnification.
Fas and FasL Protein Immunolocalization in the Lung
The immunofluorescence results shown in Figures 4 and 6 ▶ ▶ were selected because they are typical for the 10 patients each who died with ARDS or because of nonpulmonary causes. For the lung tissue sections from the patients who died with ARDS, Fas or FasL immunofluorescence staining was displayed by cells that lined and were located in the alveolar walls (Figure 4, a and c) ▶ , by cells in the alveolar air spaces (Figure 4, a and c) ▶ , and by cells lining the distal airways (not shown). The immunopositive cells that outlined the alveolar walls had attenuated cytoplasmic extensions (arrows in Figure 4, a and c ▶ ). These immunostaining patterns were absent when either the primary antibody was substituted by buffer (Figure 4b) ▶ , when the secondary antibody was substituted by buffer (Figure 4d) ▶ , or when either primary antibody was substituted by an irrelevant, species-matched, isotype-matched antibody (anti-insulin; not shown). The frequency of Fas-immunopositive cells that lined the alveolar walls was significantly greater in the lung tissue sections from the patients who died with ALI or ARDS (16 ± 6%, mean ± SD) compared to those who died without pulmonary disease (7 ± 4%, P < 0.05 by Mann-Whitney U-test). Likewise, the frequency of FasL-immunopositive cells for the same two groups were significantly different (14 ± 5% versus 8 ± 5%, respectively, P < 0.05). We did not assess labeling frequency for epithelial cells that lined distal airways because their number was too variable among the tissue sections.
The results shown in Figure 4, a and c ▶ , suggested that the same cell types expressed Fas and FasL protein in the patients who died with ALI or ARDS. Confirmation of this suggestion required performing double immunofluorescence (co-localization) of Fas and FasL. Fas (green) and FasL (red) co-localized (orange) to cells lining the alveolar walls (arrows in Figure 5a ▶ ) and in the alveolar walls. Cells in the air spaces, on the other hand, expressed either Fas or FasL. Cells that lined the distal airways also were orange (not shown). These immunostaining patterns were evident infrequently in the lung tissue sections from the patients who died of nonpulmonary causes (Figure 5b) ▶ . The frequency of Fas- and FasL-immunopositive (orange) cells that lined the alveolar walls was significantly greater in the lung tissue sections from the patients who died with ALI or ARDS (12 ± 5%) compared to those who died without pulmonary disease (3 ± 3%, P < 0.05).
The results shown in Figures 4 and 5 ▶ ▶ suggested that the Fas- and FasL-expressing cells included alveolar epithelial cells. To confirm this possibility, we used an antibody against cytokeratin in combination with an anti-Fas or anti-FasL antibody (Figure 6 ▶ ; a to d). Cytokeratin (red) immunostain highlighted the epithelial cells that lined the alveolar walls (and distal airways, not shown), regardless of whether the tissue sections were from the patients who died with ALI or ARDS [Figure 6, a ▶ (Fas) or c (FasL)] or without pulmonary disease (Figure 6, b ▶ (Fas) or d (FasL)]. Alveolar epithelial cells in the lung tissue sections from the patients who died with ALI or ARDS had orange immunofluorescence staining, indicating co-localization of cytokeratin and Fas or FasL. Such co-localization was only occasionally evident in the lung tissue sections from the patients who died without pulmonary disease (Figure 6, b and d) ▶ . The frequency of Fas- and cytokeratin-immunopositive cells that lined the alveolar walls was significantly greater in the lung tissue sections from the patients who died with ALI or ARDS (16 ± 7%, P < 0.05) compared to those who died without pulmonary disease (5 ± 4%). Likewise, the frequency of FasL- and cytokeratin-immunopositive cells for the same two groups was significantly different (14 ± 6% versus 4 ± 2%, respectively; P < 0.05).
The double-immunofluorescence results shown in Figure 6, a and c ▶ , demonstrated that alveolar epithelial cells expressed Fas or FasL in the lungs of patients who died with ALI or ARDS. To determine whether cuboidal alveolar epithelial cells expressed Fas and FasL protein, we used an anti-KL-6 antibody. The co-localization results are shown in Figure 6 ▶ ; e to h. In the lung tissue sections from the patients who died with ALI or ARDS, Fas (green in Figure 6e ▶ ) or FasL (green in Figure 6g ▶ ) immunostain was evident in squamous epithelial cells that lined the alveolar walls. However, only the apical cytoplasm of cuboidal cells (and distal airway epithelial cells, not shown) were immunostained red, indicating localization of KL-6 protein. The red immunostain was very intense, raising the possibility that this stain masked surface labeling of Fas or FasL. Thus, Fas or FasL did not appear to co-localize with KL-6 in the epithelial lining of the alveolar walls. A similar absence of co-localization was observed in the lung tissue sections from the patients who died without pulmonary disease.
The Fas and FasL immunohistochemical results raised the possibility that apoptosis was greater in the lung tissue sections of the patients who died with ALI or ARDS compared to those who died without pulmonary disease. Therefore, we used immunohistochemistry to qualitatively assess activation of the apoptotic cascade in situ (Figure 7) ▶ . Epithelial cells that lined the alveolar walls and cells that were located in the alveolar walls had more TUNEL-labeled nuclei, caspase-3-labeled cytoplasm, Bax-labeled cytoplasm, and p53-labeled cytoplasm in the tissue sections from the patients who died with ALI or ARDS compared to those who died without pulmonary disease. On the other hand, BclII-labeled cells that lined the alveolar walls, or were located in the alveolar walls, were more prominent in the tissue sections from the patients who died without pulmonary disease compared to those who died with ALI or ARDS. The latter observation is consistent with the anti-apoptotic function of BclII. When we compared labeling frequency in the lung tissue sections from the patients who died with ALI or ARDS compared to those who died without pulmonary disease (Mann-Whitney U-test), we found significantly more labeling for TUNEL (10 ± 4% versus 3 ± 3%, P < 0.05), caspase-3 (11 ± 5% versus 3 ± 4%, P < 0.05), Bax (9 ± 6% versus 3 ± 4%, P < 0.05), and p53 (9 ± 4% versus 3 ± 3%, P < 0.05). Conversely, the labeling frequency for BclII was significantly less (3 ± 3% versus 18 ± 6%, P < 0.05) in the lung tissue sections from the patients who died with ALI or ARDS compared to those who died without pulmonary disease.
Discussion
Apoptosis mediated by Fas/FasL interaction participates in embryonic development, normal cell turnover, immune regulation, and tumor cell death. 37,38 The Fas/FasL system has also been implicated in human disease processes, including pulmonary disorders. However, the role of this system in ALI or ARDS is poorly defined. Accordingly, the present study investigated both the soluble and cellular expression of the Fas/FasL system in patients with ALI or ARDS. The major findings are summarized as follows. First, higher concentrations of both soluble Fas and soluble FasL were measured in the pulmonary edema fluid of patients with ALI or ARDS compared to control patients with hydrostatic pulmonary edema. In addition, the concentrations of both soluble Fas and soluble FasL were higher in the pulmonary edema fluid of patients with ALI or ARDS compared to simultaneous plasma samples, indicating local release in the lung. Soluble Fas concentrations were associated with worse clinical outcomes. Contrary to our initial hypothesis, soluble Fas was in excess of soluble FasL. Second, both Fas and FasL were immunolocalized in the lung to a greater extent in patients who died with ALI or ARDS than in patients who died without pulmonary disease. Both proteins were expressed by epithelial cells that lined the alveolar walls, as well as by inflammatory cells in the air spaces and cells in the alveolar walls that had the location and morphology of endothelial cells. Immunohistochemical markers of apoptosis also occurred more frequently among those same cell types in the lung tissue sections from the patients who died with ALI or ARDS. Combined, these findings indicate that local up-regulation of the Fas/FasL system occurs in the alveolar epithelium and is associated with greater apoptosis and worse clinical outcome. Thus, local up-regulation of the Fas/FasL system and activation of the apoptotic cascade seem to be mechanisms by which the alveolar epithelium is injured in humans with ALI or ARDS.
The role of the Fas/FasL system in ALI has not been well defined. Theoretically, the induction of apoptosis could play a role in both the injury and the repair processes. Excessive apoptosis might contribute to injury to both the alveolar epithelial and lung endothelial barriers. In a mouse model of ALI, intratracheal lipopolysaccharide administration led to alveolar epithelial and endothelial apoptosis by a Fas/FasL-dependent mechanism. 39 In another mouse model, intranasal treatment with a Fas-activating monoclonal antibody was associated with neutrophil infiltrates, alveolar septal thickening, hemorrhage, and TUNEL labeling in the alveolar septa and air spaces. 40 Moreover, alveolar type II epithelial cell apoptosis was confirmed ultrastructurally. In patients with sepsis, apoptosis has been postulated to be a major mechanism of end-organ damage and has been demonstrated in the intestinal epithelium and the spleen of patients dying from sepsis. 41 Activation of the Fas/FasL system could also contribute to lung fibrosis after lung injury. Hagimoto and colleagues 42 reported that ligation of Fas antigen in the lung of mice by administration of an inhaled anti-Fas antibody led to pulmonary fibrosis. The Fas/FasL system has also been implicated in lung fibrosis caused by bleomycin administration in mice. 43 By contrast, inadequate apoptosis might impair the resolution of type II cell hyperplasia 44 or the removal of activated inflammatory cells, leading to inadequate repair or prolonged inflammation. Matute-Bello and colleagues 45 have shown that neutrophils in BAL fluid from patients with ARDS have an abnormally low rate of apoptosis because of the anti-apoptotic effects of the BAL fluid that was retrieved from patients with ARDS. Failure of neutrophil apoptosis could lead to prolonged inflammation or neutrophil necrosis with release of harmful cellular contents.
Only two studies have examined the role of Fas and FasL in clinical ALI and ARDS. In one study of BAL fluid from patients with septic ARDS, mRNA for a number of molecules associated with apoptosis, including Fas and FasL, was up-regulated in the cellular component of the BAL fluid. 24 In the other study, Matute-Bello and colleagues 4 demonstrated that BAL fluid from patients at risk for ARDS or with established ARDS contained soluble FasL. Soluble FasL concentrations in the BAL fluid were higher in patients that went on to die. Furthermore, BAL fluid from patients with ARDS was proapoptotic for human distal bronchial epithelial cells in vitro, an effect that was mediated by Fas/FasL interaction. 4 However, that study did not assess concentrations of soluble Fas, nor did it identify the cell types that express Fas and FasL in situ in the human lung.
An unexpected finding of our study was that soluble Fas was in excess of soluble Fas ligand in the pulmonary edema fluid of patients with ALI or ARDS. We initially hypothesized that soluble FasL would be in excess, based on the report that BAL fluid from ARDS patients was proapoptotic for distal bronchial epithelial cells. Our results indicate that assessment of the apoptotic environment in ALI and ARDS will not be straightforward. Soluble Fas was in excess of soluble FasL, a finding that one would expect to inhibit the induction of apoptosis by soluble FasL. 7 In addition, the contributions of membrane-bound FasL must be factored in the overall pro- or anti-apoptotic balance. When one considers that a variety of other mechanisms can induce or inhibit apoptosis, 46 the complexity increases and is reminiscent of other systems involved in ALI and ARDS. For example, although neutrophil elastases were initially thought to be important mediators of tissue destruction in ALI and ARDS, 47 subsequent studies revealed that elastase inhibitors were present in excess in the lungs of ARDS patients. 48,49 Similarly, although proinflammatory cytokines likely play an important role in the lung inflammation of ALI and ARDS, inhibitors of these cytokines and anti-inflammatory cytokines co-exist in the lungs of ARDS patients. 50
A novel observation of our study is identification of the alveolar epithelial cells that express Fas and FasL proteins in situ in the human lung. Both proteins were expressed by alveolar epithelial cells that were positive for pancytokeratin. We further tried to confirm that Fas or FasL protein are expressed by alveolar type II epithelial cells, 40 using an antigen (KL-6) that identifies alveolar type II epithelial cells. 51 These double-immunofluorescence results suggest that the Fas- and FasL-positive cells were alveolar type I epithelial cells rather than mature type II cells. Previous studies of human lung tissue have localized Fas and/or FasL mRNA and protein expression in situ in human bronchial epithelial cells, 3,21,52 human distal epithelial cells in general, 21 and alveolar type II epithelial cells. 44 Similar localization has been described in rodent lung 53 and rabbit lung. 40 In vitro studies, likewise, have identified expression of both Fas and FasL proteins by epithelial cells derived from airways 3 and alveolar walls. 54 Our results from double-immunofluorescence localization of Fas or FasL and KL-6 antibodies, however, suggested that neither Fas nor FasL were expressed by alveolar type II epithelial cells. We believe that co-localization was masked by the great intensity of the KL-6 immunofluorescence stain.
Another new aspect of our study is correlation of a number of markers of the apoptotic cascade in situ in lung tissue sections from the same lungs. 55,56 Markers that were increased in the patients who died with ALI or ARDS included TUNEL, caspase-3, Bax, and p53. Expression of BclII was decreased, which is expected because BclII is anti-apoptotic. 57 Therefore, our results reveal evidence that up-regulation of the Fas/FasL system and activation of the apoptotic cascade are mechanisms of acute and persistent lung damage in ALI or ARDS, and may contribute to leakage of fluid and proteins into the alveolar air space and other pathological consequences.
Our study has some limitations. First, the edema fluid and plasma samples were obtained from a different set of patients than the autopsy samples of lung tissue. Thus, we cannot be certain that the results for the edema fluid and plasma samples are related to the results for the immunolocalization analyses. This problem came about because the only way to accurately sample the undiluted soluble factors present in the lung is to aspirate undiluted pulmonary edema fluid. The optimal time for sampling is early, before alveolar edema had resolved. By contrast, optimal and adequate amounts of tissue for structural analysis could only be obtained at autopsy, which occurred later in the course of ALI or ARDS, and only in those patients who died. However, comparison of the clinical characteristics of the two sets of patients (compare Tables 1 and 3 ▶ ▶ ) indicates that the groups were similar. Furthermore, despite the difference in timing of the two sets of samples, the Fas/FasL system was clearly activated in both sets of patients. A second limitation is the tissue analysis required using autopsy tissue. One might expect that cell death would be accelerated in autopsy samples. However, antigens for proapoptotic markers (TUNEL, caspase-3, Bax, and p53) were not obvious, whereas an anti-apoptotic marker (BclII) was obvious, in the lung tissue sections from the patients who died of nonpulmonary causes. These two groups of markers were increased and decreased, respectively, in the lung tissue sections from the patients who died with ALI or ARDS. Thus, the Fas/FasL immunofluorescence results for the patients who died with ALI or ARDS are likely not influenced by the use of autopsy samples. A third limitation is that the relative contribution of soluble Fas and soluble FasL proteins by alveolar epithelial cells versus inflammatory cells in the alveolar air spaces cannot be determined from the tissue studies. Given that both Fas and FasL proteins are expressed by cells in the alveolar walls and in the adjacent air spaces, apoptosis of alveolar epithelial cells may be mediated via a paracrine pathway between activated leukocytes (neutrophils and alveolar macrophages) and alveolar epithelial cells, 54 in addition to autocrine or juxtacrine pathways by epithelial cells. Finally, although this study provides convincing evidence that Fas and FasL protein expression is up-regulated in ALI or ARDS, whether activation of the Fas/FasL system in the lungs of patients with ALI or ARDS is helpful or harmful in the evolution or resolution of clinical lung injury remains to be determined.
In conclusion, this study provides the first comprehensive assessment of the expression of both soluble and lung cellular forms of Fas and FasL proteins, as well as markers of activation of the apoptotic cascade, in patients with ALI or ARDS in both the pulmonary edema fluid and the injured lung tissue itself. Our results suggest that activation of the Fas/FasL system, associated with activation of the apoptotic cascade, among cells in the alveolar walls is persistent during ALI or ARDS. Persistent activation of both pathways may explain in part the persistent leakage of edema fluid into the alveolar air spaces of patients who have severe ALI or ARDS.
Acknowledgments
We thank Dr. Edward Klatt (Department of Pathology, University of Utah) and Dr. Elizabeth Hammond (Department of Pathology, LDS Hospital) for providing surplus tissue from autopsy procedures; and Dr. John Michael (Department of Medicine, University of Utah), Dr. James Orme (Department of Medicine, LDS Hospital), and Ms. Donna Pope (Department of Medicine, LDS Hospital) for identifying autopsies and for guiding the retrieval of demographic and clinical information from medical records.
Footnotes
Address reprint requests to Kurt H. Albertine, Ph.D., Department of Pediatrics, University of Utah, 30 North 1900 East, Salt Lake City, UT 84132-2202. E-mail: kurt.albertine@hsc.utah.edu.
Supported, in part, by National Institutes of Health grants HL50153 (SCOR in Acute Lung Injury to K. H. A., M. F. S., Z. W., G. A. Z.), T35 HL07744 (to K. H. A., M. F. S.), HL51856 (to M. A. M.), HL51854 (to M. A. M.), and HL70521 (to L. B. W.).
References
- 1.Matthay MA, Wiener-Kronish JP: Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis 1990, 142:1250-1257 [DOI] [PubMed] [Google Scholar]
- 2.Bachofen M, Weibel ER: Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis 1977, 116:589-615 [DOI] [PubMed] [Google Scholar]
- 3.Hamann KJ, Dorscheid DR, Ko FD, Conforti AE, Sperling AI, Rabe KF, White SR: Expression of Fas (CD95) and FasL (CD95L) in human airway epithelium. Am J Respir Cell Mol Biol 1998, 19:537-542 [DOI] [PubMed] [Google Scholar]
- 4.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Kiener S, Mongovin S, Chi EY, Jonas M, Martin TR: Soluble Fas-ligand induces epithelial cell apoptosis in humans with acute lung injury (ARDS). J Immunol 1999, 163:2217-2225 [PubMed] [Google Scholar]
- 5.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S: The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991, 66:233-243 [DOI] [PubMed] [Google Scholar]
- 6.Suda T, Takahashi T, Golstein P, Nagata S: Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 1993, 75:1169-1178 [DOI] [PubMed] [Google Scholar]
- 7.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD: Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science 1994, 263:1759-1762 [DOI] [PubMed] [Google Scholar]
- 8.Tanaka M, Itai T, Adachi M, Nagata S: Downregulation of Fas ligand by shedding. Nat Med 1998, 4:31-36 [DOI] [PubMed] [Google Scholar]
- 9.Tanaka M, Suda T, Takahashi T, Nagata S: Expression of the functional soluble form of human fas ligand in activated lymphocytes. EMBO J 1995, 14:1129-1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arase H, Arase N, Saito T: Fas-mediated cytotoxicity by freshly isolated natural killer cells. J Exp Med 1995, 181:1235-1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagafuji K, Shibuya T, Harada M, Mizuno SI, Takenaka K, Miyamoto T, Okamura T, Gondo H, Niho Y: Functional expression of Fas antigen (CD95) on hematopoietic progenitor cells. Blood 1995, 86:883-889 [PubMed] [Google Scholar]
- 12.Rouvier E, Luciani MF, Golstein P: Fas involvement in Ca(+2)-independent T cell-mediated cytotoxicity. J Exp Med 1993, 177:195-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyaizu N, Adachi Y, Hashimoto F, McCluskey TW, Hosaka N, Kayagaki N, Yagita H, Pahwa S: Monocytes express Fas ligand upon CD4 cross-linking and induce CD4+ T cell apoptosis: a possible mechanism of bystander cell death in HIV infection. J Immunol 1997, 158:2456-2463 [PubMed] [Google Scholar]
- 14.Savill J, Haslett C: Granulocyte clearance by apoptosis in the resolution of inflammation. Semin Cell Biol 1995, 6:385-393 [DOI] [PubMed] [Google Scholar]
- 15.Liles WC, Kiener PA, Ledbetter JA, Aruffo A, Klebanoff SJ: Differential expression for Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med 1996, 184:429-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmad R, Menezes J, Knafo L, Ahmad A: Activated human platelets express Fas-L and induce apoptosis of Fas-positive tumor dells. J Leukoc Biol 2001, 69:123-128 [PubMed] [Google Scholar]
- 17.Lynch DH, Ramsdell F, Alderson MR: Fas and FasL in the homeostatic regulation of immune responses. Immunol Today 1995, 16:569-574 [DOI] [PubMed] [Google Scholar]
- 18.Sato K, Kimura F, Nakamura Y, Murakami H, Yoshida M, Tanaka M, Nagata S, Kanatani Y, Wakimoto N, Nagata N, Motoyoshi K: An aggressive nasal lymphoma accompanied by high levels of soluble Fas ligand. Br J Haematol 1996, 94:379-382 [DOI] [PubMed] [Google Scholar]
- 19.Tanaka M, Suda T, Haze K, Nakamura N, Sato K, Kimura F, Motoyoshi K, Mizuki M, Tagawa S, Ohga S, Hatake K, Drummond AH, Nagata S: Fas ligand in human serum. Nat Med 1996, 2:317-322 [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi S, Yamaoka M, Okuyama M, Nitoube J, Fukui A, Shirakabe M, Shirakawa K, Nakamura N, Tomoike H: Elevated circulating levels and cardiac secretion of soluble Fas ligand in patients with congestive heart failure. Am J Cardiol 1999, 83:1500-1503 [DOI] [PubMed] [Google Scholar]
- 21.Kuwano K, Miyazaki H, Hagimoto N, Kawasaki M, Fujita M, Kuitake R, Kaneko Y, Hara N: The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am J Respir Cell Mol Biol 1999, 20:53-60 [DOI] [PubMed] [Google Scholar]
- 22.Kuwano K, Hagimoto N, Kawasaki M, Nakamura N, Shirakawa K, Maeyama T, Hara N: Expression of FasL and Fas protein and their soluble form in patients with hypersensitivity pneumonitis. Int Arch Allergy Immunol 2000, 122:209-215 [DOI] [PubMed] [Google Scholar]
- 23.Kuwano K, Kawasaki M, Maeyama T, Hagimoto N, Nakamura N, Shirakawa K, Hara N: Soluble form of fas and fas ligand in BAL fluid from patients with pulmonary fibrosis and bronchiolitis obliterans organizing pneumonia. Chest 2000, 118:451-458 [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H: Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med 2000, 161:237-243 [DOI] [PubMed] [Google Scholar]
- 25.Wen LP, Madani K, Fahrni JA, Duncan SR, Rosen GD: Dexamethasone inhibits lung epithelial cell apoptosis induced by IFN-gamma and Fas. Am J Physiol 1997, 273:L921-L929 [DOI] [PubMed] [Google Scholar]
- 26.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Le Gall JR, Morris A, Spragg R: The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994, 149:818-824 [DOI] [PubMed] [Google Scholar]
- 27.Fein A, Grossman RF, Jones JG, Overland E, Pitts L, Murray JF, Staub NC: The value of edema protein measurements in patients with pulmonary edema. Am J Med 1979, 67:32-38 [DOI] [PubMed] [Google Scholar]
- 28.Verghese GM, Ware LB, Matthay BA, Matthay MA: Alveolar epithelial fluid transport and the resolution of clinically severe hydrostatic pulmonary edema. J Appl Physiol 1999, 87:1301-1312 [DOI] [PubMed] [Google Scholar]
- 29.Ware LB, Matthay MA: Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 2001, 163:1376-1383 [DOI] [PubMed] [Google Scholar]
- 30.Le Gall JR, Lemeshow S, Saulnier F: A new Simplified Acute Physiology Score (SAPSII) based on a European/North American multicenter study. JAMA 1993, 270:2957-2963 [DOI] [PubMed] [Google Scholar]
- 31.Murray JF, Matthay MA, Luce JM, Flick MR: An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis 1988, 138:720-723 [DOI] [PubMed] [Google Scholar]
- 32.Doyle RL, Szaflarski N, Modin GW, Wiener-Kronish JP, Matthay MA: Identification of patients with acute lung injury. Predictors of mortality. Am J Respir Crit Care Med 1995, 152:1818-1824 [DOI] [PubMed] [Google Scholar]
- 33.Imaizumi TA, Albertine KH, Jicha DL, McIntyre TM, Prescott SM, Zimmerman GA: Human endothelial cells differentially express and secrete ENA-78 and IL-8 when stimulated with inflammatory cytokines. Am J Respir Cell Mol Biol 1997, 17:181-192 [DOI] [PubMed] [Google Scholar]
- 34.Kohno N, Kyoizumi S, Awaya Y, Fukuhara H, Yamakido M, Akiyama M: New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest 1989, 96:68-73 [DOI] [PubMed] [Google Scholar]
- 35.Kohno N, Akiyama M, Kyoizumi S, Hakoda M, Kobuke K, Yamakido M: Detection of soluble tumor-associated antigens in sera and effusions using novel monoclonal antibodies, KL-3 and KL-6, against lung adenocarcinoma. Jpn J Clin Oncol 1988, 18:203-216 [PubMed] [Google Scholar]
- 36.Zar JH: Biostatistical Analysis, ed 2 1984:pp 122-205 Prentice-Hall, Inc., Englewood Cliffs
- 37.Nagata S: Apoptosis by death factor. Cell 1997, 88:355-365 [DOI] [PubMed] [Google Scholar]
- 38.Rathmell JC, Thompson CB: The central effectors of cell death in the immune system. Annu Rev Immunol 1999, 17:781-828 [DOI] [PubMed] [Google Scholar]
- 39.Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kooguchi K, Fujiwara I, Nakajima H: Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide induced lung injury in mice. Am J Respir Crit Care Med 2001, 163:762-769 [DOI] [PubMed] [Google Scholar]
- 40.Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC: Fas (CD95) induces alveolar epithelial cell apoptosis in vivo. Implications for acute pulmonary inflammation. Am J Pathol 2001, 158:153-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE: Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 1999, 27:1230-1251 [DOI] [PubMed] [Google Scholar]
- 42.Hagimoto N, Kuwano K, Miyazaki H, Kunitake R, Fujita M, Kawasaki M, Kaneko Y, Hara N: Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of fas antigen. Am J Respir Cell Mol Biol 1997, 17:272-278 [DOI] [PubMed] [Google Scholar]
- 43.Kuwano K, Hagimoto N, Kawasaki M, Yatomi T, Nakamura N, Nagata S, Suda T, Kunitake R, Maeyama T, Miyazaki H, Hara N: Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J Clin Invest 1999, 104:13-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bardales RH, Xie SS, Schaefer RF, Hsu SM: Apoptosis is a major pathway responsible for the resolution of type II pneumocytes in acute lung injury. Am J Pathol 1996, 149:845-852 [PMC free article] [PubMed] [Google Scholar]
- 45.Matute-Bello G, Liles WC, Radella FI, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR: Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med 1997, 156:1969-1977 [DOI] [PubMed] [Google Scholar]
- 46.Stefanec T: Endothelial apoptosis: could it have a role in the pathogenesis and treatment of disease? Chest 2000, 117:841-854 [DOI] [PubMed] [Google Scholar]
- 47.McGuire WW, Spragg RG, Cohen AB, Cochrane CG: Studies on the pathogenesis of the adult respiratory distress syndrome. J Clin Invest 1982, 69:543-553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sallenave JM, Donnely SC, Grant IS, Robertson C, Gauldie J, Haslett C: Secretory leukocyte proteinase inhibitor is preferentially increased in patients with acute respiratory distress syndrome. Eur Respir J 1999, 13:1029-1036 [DOI] [PubMed] [Google Scholar]
- 49.Lee WL, Downey GP: Leukocyte elastase. Physiological functions and role in acute lung injury. Am J Respir Crit Care Med 2001, 164:896-904 [DOI] [PubMed] [Google Scholar]
- 50.Martin TR: Cytokines and the acute respiratory distress syndrome (ARDS): a question of balance. Nat Med 1997, 3:272-273 [DOI] [PubMed] [Google Scholar]
- 51.Koh T, Aynsley-Green A, Tarbit M, Eyre JA: Neural dysfunction during hypoglycaemia. Arch Dis Child 1988, 63:1353-1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Druilhe A, Wallaert B, Tsicopoulos A, Lapae Silva JR, Tillie-Leblond I, Tonnel AB, Pretolani M: Apoptosis, proliferation, and expression of Bcl-2, Fas, and Fas ligand in bronchial biopsies from asthmatics. Am J Respir Cell Mol Biol 1998, 19:747-757 [DOI] [PubMed] [Google Scholar]
- 53.Fine A, Anderson NL, Rothstein TL, Williams MC, Goochuico BR: Fas expression in pulmonary alveolar type II cells. Am J Physiol 1997, 273:L64-L71 [DOI] [PubMed] [Google Scholar]
- 54.Serrao KL, Fortenberry JD, Owens ML, Harris FL, Brown LA: Neutrophils induce apoptosis of lung epithelial cells via release of soluble Fas ligand. Am J Physiol 2001, 280:L298-L305 [DOI] [PubMed] [Google Scholar]
- 55.Auten RL, Whorton MH, Mason SN: Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed newborn rat lung. Am J Respir Cell Mol Biol 2002, 26:391-397 [DOI] [PubMed] [Google Scholar]
- 56.Albertine KH, Plopper CG: Perspective: DNA oxidation or apoptosis: will the real culprit of DNA damage in hyperoxic lung injury please stand up? Am J Respir Cell Mol Biol 2002, 26:381-383 [DOI] [PubMed] [Google Scholar]
- 57.Hockenbery D, Zuther M, Hickey W, Nahn M: BCL2 protein is topographically restricted in tissue characterized by apoptotic cell death. Proc Natl Acad Sci USA 1991, 88:6961-6965 [DOI] [PMC free article] [PubMed] [Google Scholar]