

Abstract
To investigate the role of integrin α7 in muscle pathology, we used a “candidate gene” approach in a large cohort of muscular dystrophy/myopathy patients. Antibodies against the intracellular domain of the integrin α7A and α7B were used to stain muscle biopsies from 210 patients with muscular dystrophy/myopathy of unknown etiology. Levels of α7A and α7B integrin were found to be decreased in 35 of 210 patients (∼17%). In six of these patients no integrin α7B was detected. Screening for α7B mutation in 30 of 35 patients detected only one integrin α7 missense mutation (the mutation on the second allele was not found) in a patient presenting with a congenital muscular dystrophy-like phenotype. No integrin α7 gene mutations were identified in all of the other patients showing integrin α7 deficiency. In the process of mutation analysis, we identified a novel integrin α7 isoform presenting 72-bp deletion. This isoform results from a partial deletion of exon 21 due to the use of a cryptic splice site generated by a G to A missense mutation at nucleotide position 2644 in integrin α7 cDNA. This spliced isoform is present in about 12% of the chromosomes studied. We conclude that secondary integrin α7 deficiency is rather common in muscular dystrophy/myopathy of unknown etiology, emphasizing the multiple mechanisms that may modulate integrin function and stability.
Integrins are transmembrane heterodimers of two different subunits, α and β, associated by non-covalent interactions. In humans, at least 18 different α and 8 different β subunits are known, resulting in 22 distinct heterodimers. 1,2 The α7 subunit is mainly expressed in skeletal and cardiac muscle, while the β1 chain is expressed throughout the body and associated with other different α subunits. 3-6 In adults, the alternative β1D isoform is restricted to skeletal and cardiac muscle. 7-9
Integrin α7β1 in skeletal and cardiac muscle binds via its extracellular domain to laminin α2 and α4 10-12 and via its cytoplasmic domain possibly to α-actinin and talin ensuring continuity of structure and signaling between the cytoskeleton and the basal lamina. 13,14 Integrins have a prominent role in myogenesis, 15,16 differentiation, cell migration, and cell-cell interactions. 2,17-19
Integrin α7 in skeletal muscle localizes at the sarcolemma, at the neuromuscular junctions, and, most prominently, at the myotendinous junctions where it provides an anchorage for laminin α2, conferring mechanical stability and traction resistance to the skeletal muscle fibers. 5,9,20,21 The expression of several integrin α7 isoforms modulates the integrin α7β1 ligand affinity and signaling specificity during myogenesis. 20 All integrin isoforms are encoded by a single α7 gene (ITGA7) located on chromosome 12q13 22 and result from alternatively spliced variants. 20,23
In humans, different extracellular and cytoplasmic domain isoforms have been described. 23,24 The α7X1 and α7X2 extracellular isoforms result from the alternative splicing of exons 5 or 6, in a variable region nearby the ligand binding site. 5,24,25 The X2 isoform is the main variant expressed in adult muscle fibers. 24,26 The cytoplasmic isoform α7A, 20,24,27 corresponding to the normal exon 26 splicing, is temporarily up-regulated during myogenesis, while the α7B variant, the longest isoform resulting from the splicing out of exon 26, is the main isoform expressed in skeletal and cardiac muscle, smooth muscle cells, spleen, liver, and brain. 20,28
To study the involvement of α7 integrin during myogenesis and its role in muscle integrity and function, a null allele of the gene for the α7 integrin subunit has been generated. 29 Mice homozygous for the mutation showed features of a progressive muscular dystrophy in their muscle, suggesting that integrin α7 represents an indispensable linkage between the muscle fiber and the extracellular matrix, 29 particularly at the myotendinous junction. To further support the role of integrin in connecting muscle cells with the surrounding extracellular matrix, it has been shown that α7β1 is up-regulated in Duchenne muscular dystrophy (DMD) patients and in its animal model, mdx mice, to compensate for the reduced dystrophin-mediated linkage of fibers with the basal lamina. 11,30 In merosin-negative congenital muscular dystrophy (CMD) and in its animal model, dy/dy mice, α7 integrin appears to be significantly reduced suggesting that lack of laminin α2 (α2β1γ1) may down-regulate the expression or change the location of integrin α7 gene. 11,12,30
The cloning of the full-length human ITGA7 cDNA 5,26,31 allowed the identification of the first three patients with primary α7 integrin deficiency. 31 The three patients reported were affected with congenital myopathy with variable clinical phenotype, and all showed a complete absence of integrin α7 in their muscle biopsies due to primary integrin α7 nonsense/splicing mutations or to a down-regulation of integrin α7 mRNA. 31 Here we report a study of a large series of muscle biopsies from patients affected with unclassified muscular dystrophy/myopathy tested for integrin α7 deficiency by combined integrin α7 protein and gene studies.
Materials and Methods
Patients
Our muscle biopsy tissue bank at the Neuromuscular Center of the University of Padova was screened to search for patients meeting the following criteria: muscle weakness and/or hypotonia; muscle histopathology consistent with a myopathic or dystrophic process; and normal dystrophin, α-sarcoglycan, calpain, dysferlin, and laminin α2 in their muscle biopsies. Inflammatory myopathies, neurogenic atrophies, and metabolic or mitochondrial myopathies were a priori excluded. Two hundred ten patients met the clinical and histopathological selection criteria and were chosen for integrin α7 screening. One hundred thirteen were muscular dystrophies, 24 CMD, and 73 carried a histopathological diagnosis of undetermined myopathy.
Case Report
Patient E.S. was delivered by Caesarian section at the 39th gestational week due to the threat of miscarriage. The child was oxygen-dependent and mechanically ventilated since birth. He presented with hypotonia and with hip, wrist, and ankle contractures. Karyotype study was normal. A brain computed tomography (CT) scan showed cortical atrophy and white matter signal abnormalities. Creatine kinase (CK) was 507 U/L (normal <250) and electromyography (EMG) myopathic. At one month of age a muscle biopsy was consistent with a congenital muscular dystrophy. Dystrophin, α-sarcoglycan, and laminin α2 studies in his muscle biopsy were normal. The patient died at 13 months of age from respiratory failure. Integrin α7 was markedly reduced in the patient’s muscle biopsy.
Integrin α7 Immunohistochemistry
Serial cryosections, 4-μm thick, were fixed for 5 minutes in 100% ice-cold-acetone, air dried, and preincubated with phosphate-buffered saline containing 2% bovine serum albumin and 5% goat serum as blocking agent. 31 Polyclonal anti-integrin α7B antibody (1:500) directed against the intracellular domain of the protein, 12 polyclonal anti-integrin α7A antibody (1:500) directed against the COOH terminal peptide of the protein, polyclonal anti-β1D (1:500), 12 and monoclonal antibody directed against the carboxyl terminus region of the laminin α2 (1:1000) 32 (Chemicon, Temecula, CA) were used to incubate the sections for 2 hours at room temperature. Appropriate Cy-3-conjugated (anti-rabbit or anti-mouse) secondary antibody (1:100) (Caltag Lab, Burlingame, CA) were used. The visualization of mounted sections was done on Zeiss Axioskop photomicroscope. Hematoxylin and eosin staining was done on parallel cryosections to check tissue integrity and histopathology.
Muscle biopsies were scored as integrin α7B negative, integrin α7B markedly reduced, or integrin α7B slightly reduced (based on the amount of integrin α7 immunostaining detected in each muscle biopsy). The same criteria were used to score α7A immunostaining.
Integrin α7β Mutation Studies
RNA extraction
Approximately 50 mg of frozen muscle biopsy tissue was homogenized using a Kinematica Polytron PT 2100 homogenizer. RNA was extracted using Trizol (Invitrogen, Carlsbad, CA) accordingly to the manufacturer’s instructions and stored in RNase-free water at −80°C.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
For cDNA synthesis, ∼1–2 micrograms of RNA was reverse transcribed using oligo-dT primers as previously described. 33 cDNA was boiled to denature and inactivate reverse transcriptase. Eighteen sets of overlapping primers were designed to cover the entire 3414 bp of the integrin α7 coding sequence and the 5[prime]-UTR region (Table 1) ▶ . PCR reactions contained about 50 ng of cDNA, 50 ng of each primer, 100 nmol/L of each dNTP, and 0.2 μCi α-[32P] dATP in 12.5 μl total volume. Reaction conditions were 10 minutes at 94°C to denature; 30 seconds at 94°C, 30 seconds at 65°C, 30 seconds at 72°C for 35 cycles, with an extension of 10 minutes at 72°C for all primer sets except primer sets 1212F-1451R and 2068F-2245R where the annealing temperature was 50°C.
Table 1.
Integrin α7 Primers Sequence
| Primer set | |
|---|---|
| -56F, 5′-TTGGGGCGTGCGAGATTTC-3′ | 249R, 5′-CTGCCCAGGAAGAGCCAGGGC-3′ |
| 32F, 5′-GGGCCTCCGGGATTTGCTAC-3′ | 279R, 5′-AGCGAAGAGGCCTCCAGTGC-3′ |
| 201F, 5′-GAGCTGGCTGCTGGTGGGTGC-3′ | 467R, 5′-GTCTCCAGGATCTGGTCCAC-3′ |
| 410F, 5′-TTGTTACCTGTGCACACCGAT-3′ | 676R, 5′-GCAACCCCTTCCAATTATAGG-3′ |
| 608F, 5′-CCGCCTTCTCCCCTGATAG-3′ | 841R, 5′-AGCTCAGCTCTTCTGCACG-3′ |
| 807F, 5′-GGGGAAAGGTCTGGTGCG-3′ | 1022R, 5′-GCACCCACTATCAGGTCTGGCC-3′ |
| 980F, 5′-TGACCTCAACAGTGATGGCTG-3′ | 1282R, 5′-CCTGTGAAGGTTTGGCGAC-3′ |
| 1212F, 5′-CTTTGATGGTGATGGGAAAGTC-3′ | 1451R, 5′-GCAATAGAGACCTCATGGGAG-3′ |
| 1391F, 5′-ACACCGCAGTGCTCTTCAGGGC-3′ | 1648R, 5′-GGTTACGGCTCAGGAACGTC-3′ |
| 1581F, 5′-GTTAGATGCGGACACAGACCG-3′ | 1894R, 5′-AGTGGATCTCTGCCCGCTGG-3′ |
| 1850F, 5′-TCCTCAATGCCCACCAGC-3′ | 2083R, 5′-GCAGGTTGGTGACCATCA-3′ |
| 2068F, 5′-GTGGATGGAACAACAGCCC-3′ | 2245R, 5′-CACACTCAACATGGGAGGC-3′ |
| 2207F, 5′-TCTGCCTGTCCAATGAGAATG-3′ | 2464R, 5′-CAGAGAAGAAGAGTTGCTGGG-3′ |
| 2407F, 5′-ATTGAGCTGCCACTGTCCATTG-3′ | 2644R, 5′-CAACCTGCATTGGGTACAGC-3′ |
| 2607F, 5′-TGCCAATGGGAAGTGGTTG-3′ | 2887R, 5′-GGCAGATGAACACCACACAG-3′ |
| 2806F, 5′-CCAGTGTCCTCTGCTGAGAAG-3′ | 3075R, 5′-GTATACCATCACTGGGATCACTG-3′ |
| 3017F, 5′-CCTCCATAAAGAACTTGATGCTCC-3′ | 3251R, 5′-GGAATCTTCACCGCATGG-3′ |
| 3223F, 5′-GTGCCCCAGTACCATGCGGTG-3′ | 3391R, 5′-GCCCATCGGGGCCCAGCTCG-3′ |
Single Strand Conformational Polymorphism Analysis
Three different single strand conformational polymorphism (SSCP) conditions 33 were used for screening for integrin α7 mutation. Conformers were re-amplified using the original amplification primer and PCR conditions as previously described 33 and directly sequenced on automatic sequencer (CRIBI Biotechnology Center, Department of Biology, University of Padova).
The integrin α7 mutations identified were confirmed with the appropriate restriction endonuclease digestion if a gain or loss of restriction site was detected or with appropriate PCR primers designed to cover the mutated region and PCR product size-fractioned with denaturing polyacrylamide gel electrophoresis. Primers used to amplify exon 15 and exon 21 were as following: ex 15F 5′-TGCCCCTTATCTATGTCTCC-3′; ex 15R 5′-GAGAGGGCTTTCTCTCAATCC-3′; ex 21F 5′-TCCCCTCATACTCTCTTTTTCC-3′, and ex 21R 5′-AACTCCCCATAACATCTGTAACC-3′.
Integrin α7 Gene Expression Studies
RNA extraction and cDNA synthesis were done as described above. Biopsies studied were from the 30 patients screened for integrin α7 mutation and from 10 integrin α7-positive myopathic control muscle biopsies.
Primer sets for co-amplification of integrin α7 RNA and α-sarcoglycan RNA were designed, and for each set the forward primer was synthesized with fluorescein-linked 5′ ends (Invitrogen). Primer sequences were as follows: α-sarcoglycan ADH3F: 5′fluorescein-CTGCTCAACGTCACCTCTG-3′; ADH3R: 5′-CCGGCACTGACTTATCCAC-3′; and integrin α7 ITGA7.9F: 5′fluorescein-TTGATGGTGATGGGAAAGTCTTCA-3′; and ITGA7.12R: 5′-CAGACCCTTAGGTCCACACAGACC-3′.
Multiplex RT-PCR and quantitation of fluorescent PCR products were done as previously described. 34 Briefly, cDNA corresponding to approximately 20 ng of total RNA was amplified with primers mixed for 22 cycles using standard conditions (denaturation at 95°C for 10 minutes, at 94°C for 1 minute, at 62°C for 1 minute, at 72°C for 1 minute, for 22 cycles; and extension at 72°C for 10 minutes). Fluorescent RT-PCR products were denatured and loaded on an ABI automated sequencer (Applied Biosystems, Foster City, CA) using a ROX/fluorescein matrix standard. ROX labeled markers were included as internal size standards. Peak areas corresponding to expected RT-PCR product sizes were determined, and peak area ratios of integrin α7 relative to internal control (α-sarcoglycan) calculated. Two separate RT-PCR measurements were done for each patient’s biopsy. Statistical significance was done by Student’s t test using all calculated ratios.
Results
Histological Features of 35 Integrin α7-Deficient Patients
The entire database, about 5000 muscle biopsies, was searched for patients meeting the histopathological diagnosis of muscular dystrophy of unknown etiology (normal dystrophin, α-sarcoglycan, calpain, dysferlin, and laminin α2). About 560 muscle biopsies, collected over 2 consecutive years, were searched for undetermined myopathy. Two hundred ten patients were selected for integrin α7 screening from the database search of muscle biopsies. One hundred thirty seven muscular dystrophy and 73 myopathies were tested with integrin α7 immunofluorescence. Muscle biopsies from patients diagnosed with muscular dystrophy showed marked fiber size variation, increased central nuclei, degeneration and regeneration, and a marked increase in the endomysial and perimysial connective tissue with fatty infiltration. Common features in the undetermined myopathy group of muscle biopsies included mild fiber size variation and scattered central nuclei, with little or absent muscle fiber degeneration or regeneration. Fibrofatty infiltration was absent or minimal.
Antibodies directed against the α7A, α7B, and the β1D subunit of integrin were used to characterize integrin expression in patients’ muscle biopsies. In parallel, laminin α2 immunostaining was done in each muscle biopsy to check the integrity of the muscle fibers. Three percent (6 of 210) of the muscle biopsies studied were completely deficient for integrin α7B (Table 2 ▶ ; Figure 1G ▶ ) and about 14% (29 of 210) showed a (variable) reduction of integrin α7B immunostaining (Table 2 ▶ ; Figure 1, M and Q ▶ ).
Table 2.
Integrin α7 Immunofluorescence Results in 210 Muscle Biopsies
| Undetermined muscular dystrophy (n = 137) | Undetermined myopathy (n = 73) | Total (n = 210) | |||||
|---|---|---|---|---|---|---|---|
| <2 yr | >2 yr | Total | <2 yr | >2 yr | Total | ||
| Integrin α7 negative | 2 (1%) | 4 (3%) | 6 (4%) | 0 | 0 | 0 | 6 (3%) |
| Integrin α7 markedly reduced | 3 (2%) | 5 (4%) | 8 (6%) | 4 (5%) | 7 (10%) | 11 (15%) | 19 (9%) |
| Integrin α7 slightly reduced | 1 (0.7%) | 6 (4%) | 7 (5%) | 1 (1%) | 2 (3%) | 3 (4%) | 10 (5%) |
| Integrin α7 normal | 13 (9%) | 103 (75%) | 116 (84%) | 4 (5%) | 55 (75%) | 59 (80%) | 175 (83%) |
Percentage values were rounded to nearest 5.
Figure 1.
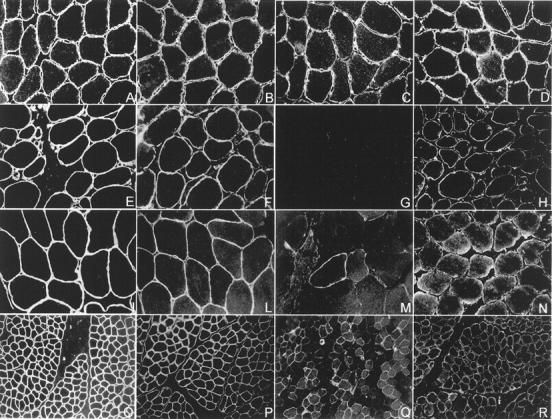
Integrin immunofluorescence studies. Transverse cryosections of muscle-biopsy specimen from a normal control and three patients with integrin deficiency were stained with antibodies directed against the intracellular domain of integrin α7B, integrin α7A, integrin β1D, and laminin α2. A–D show a normal muscle biopsy, E–H show a muscle biopsy showing complete integrin α7 deficiency, and I–R show muscle biopsies from two partial integrin α7-deficient patients. Integrin α7B immunostaining shows a uniform staining of the periphery of each myofiber in the control muscle (C), where a muscle biopsy showing a complete absence of immunoreactivity is shown in G. M: Integrin α7 is severely reduced. The majority of the muscle fibers show a barely detectable immunostaining, but a few, scattered fibers are strongly integrin α7-positive and show a slight cytoplasmic α7B-positive immunostaining. Q: A different pattern of integrin α7 partial deficiency is shown. Integrin α7-positive and -negative fibers are scattered in a mosaic-like pattern. The integrin-positive fibers show a α7B-positive cytoplasmic staining. Integrin α7A was reduced (H, N, R) and integrin β1D mildly reduced (F, L, P) in the complete integrin α7 deficiency muscle biopsy and in both the partial integrin α7 deficiency specimens in comparison with the control muscle (D and B). Laminin α2 was normal in all muscle biopsies (A, E, I, O).
Six muscle biopsies showing complete integrin α7B deficiency were all undetermined muscular dystrophy (Figure 1G) ▶ . Three were diagnosed as CMD and 3 as limb-girdle muscular dystrophy. Marked integrin α7B reduction (ie, variable and barely detectable integrin α7 immunostaining in the majority of the muscle fibers with or without some scattered myofibers showing patchy but positive immunostaining) (Figure 1M) ▶ was present in about 6% (8 of 137) of the muscular dystrophies and in 15% (11 of 73) of the myopathies studied (Table 2) ▶ . A slight reduction in integrin α7B (ie, variable and faint integrin α7 immunostaining and/or a mosaic-like pattern) (Figure 1Q) ▶ was detected in 5% (7 of 137) of muscular dystrophies and in about 4% (3 of 73) of unclassified myopathy (Table 2) ▶ . Interestingly, the integrin α7-positive muscle fibers, in the subset of α7B partial deficiency, often showed a integrin α7-positive cytoplasmic immunostaining. Integrin α7A was reduced (Figure 1, H, N, R) ▶ , and integrin β1D mildly reduced (Figure 1, F, L, P) ▶ in all muscle biopsies showing integrin α7B deficiency. In the six muscle biopsies showing complete integrin α7B deficiency, integrin α7A was more severely decreased than in the partial deficiency. Laminin α2 was normal in all biopsies studied (Figure 1, E, I, O) ▶ .
Integrin α7B deficiency was present in about 39% (11 of 28) (Table 2) ▶ of muscle biopsies from patients younger than 2 years, and only in 13% (24 of 182) (Table 2) ▶ from patients older than 2 years. To determine whether integrin α7B deficiency in younger patients was a manifestation of integrin α7B being expressed at a lower level at a younger age, we selected a patient with a marked reduction of integrin α7B in muscle at 1 year of age who had two subsequent muscle biopsies obtained at 2 and 6.3 years of age. Integrin α7B immunostaining was performed in parallel with laminin α2 and integrin β1D in the three-muscle biopsy. As shown in Figure 2 ▶ , the patient’s muscle biopsy performed at 2 years of age (Figure 2C) ▶ contained scattered myofibers with a faint and variable integrin α7B immunostaining. Rare fibers showed a patchy and barely detectable integrin α7B immunostaining in the muscle biopsy performed at 6.3 years of age (Figure 2F) ▶ . Integrin β1D was mildly reduced (Figure 2, B and E) ▶ , and laminin α2 was normal in these biopsies (Figure 2, A and D) ▶ .
Figure 2.

Integrin α7 deficiency in a two-year-old patient is not rescued over time. Cryosections from muscle biopsies performed at 2 years (A–C), and 6.3 years of age (D–F) in the same patient were stained with an antibody directed against the intracellular domain of the integrin α7B (C and F), integrin β1D (B and E), and laminin α2 (A and D). In C (a muscle biopsy performed at 2 years of age), integrin α7B is faintly expressed in a minority of the patient’s muscle fibers. In the muscle biopsy performed at 6.3 years of age the number of integrin α7-positive fibers is dramatically reduced and strongly integrin α7-positive fibers are no longer detectable (F). Integrin β1D was mildly reduced in both of the patient’s muscle biopsies (B and E) and laminin α2 was normal (A and D).
Integrin α7 Mutation Studies
Adequate muscle tissue for RNA extraction was available for 30 of 35 muscle biopsies showing integrin α7B deficiency. Twenty were muscular dystrophies (including the six samples showing complete α7B deficiency) and 10 were myopathies. These 30 muscle biopsies were selected for integrin α7 mutation screening. RNA was extracted and RT-PCR/SSCP was performed using 18 sets of overlapping primers covering the entire integrin α7 coding sequence.
Integrin α7 Polymorphisms
Several conformers were identified and directly sequenced. Seven integrin α7 polymorphisms were identified. All of the identified polymorphisms were in the extracellular domain of the protein. Of the seven polymorphisms identified, six were silent nucleotide changes: G285T (Pro95Pro), G351A (Glu117Glu), A366G (Gln122Gln), G810A (Gly270Gly), T2307C (Ser769Ser), and C3018G (Ser1006Ser). An A1952G nucleotide change resulted in a histidine to an arginine amino acid change (His651Arg). The A1952G nucleotide change was present in 32 of 70 (46%) of the chromosomes studied.
Integrin α7 Alternatively Spliced Isoform
A unique conformer was detected with primer set 2607F-2868R in a single patient. Sequencing showed an in-frame deletion mutation starting at nucleotide position 2641 to nucleotide position 2712 of the integrin α7 coding sequence (Figure 3) ▶ . The heterozygous in-frame deletion encompassed 72 bp in exon 21. This in-frame deletion detected at the cDNA level was absent at the genomic DNA level suggesting that the recognition of a cryptic splice site within exon 21 may be responsible for the skipping of about 40% of exon 21 directly to exon 22. To further characterize this in-frame deletion we performed SSCP on exon 21 in genomic DNA from the patient and from 60 controls. Identical conformers were detected in the patient and in 14 of 120 chromosomes studied (∼12%). Sequence analysis of the conformers showed a G to A substitution at position 2644 in cDNA (Figure 3) ▶ . The G2644A base change resulted in a glutamic acid to lysine amino acid change (E822K) and it caused a gain of MseI restriction digestion site. Appropriate restriction digestion and RFLP analysis confirmed the mutation in the genomic DNA of the patient.
Figure 3.

Identification of a novel alternatively spliced integrin α7. Top: SSCP analysis of RT-PCR product for primer set 2607F-2868R showed a unique conformer in a patient (asterisk). Direct sequencing of this conformer showed a 72-bp in-frame deletion mutation starting at nucleotide position 2641 to nucleotide 2712 (Δ2641–2712) of the integrin α7 coding sequence corresponding to about 40% of exon 21. The Δ2641–2712 deletion was absent in the genomic DNA of the patient. SSCP analysis of exon 21 (bottom) showed a G to A substitution at position 2644 of the integrin α7 cDNA. The G2644A nucleotide change caused the creation of a cryptic donor splice site within exon 21.
Interestingly, when the G2644A mutated exon 21 sequence was searched for a splice site using a Splice Site Finder program (supported by Genet.sickkids.on.ca) a cryptic donor splice site sequence was identified (AG/GTTA2644AG) (Figure 4) ▶ . When the score of the cryptic donor sequence was calculated using an algorithm based on the matrix compiled by Shapiro and Senapathy, 35 the cryptic donor splice site AG/GTTA2644AG) scored 71.0 where the wild-type donor sequence (TG2712/GTGAGG) obtained 84.3 (accordingly to a Splice Site Score Calculator program supported by Genet. sickkids.on.ca).
Figure 4.

Identification of a novel cryptic donor splice site in integrin α7 exon 21. Full length and alternatively spliced integrin α7 are shown. Full length integrin α7 is the major transcript in adult skeletal muscle and results from the expected splicing from exon 21 to exon 22 (wild-type integrin α7 in the figure). The alternatively spliced integrin α7 derives from the use of a cryptic splice site starting at position 2639 (AG/GTTA2644AG) of the integrin cDNA resulting in the skipping of about 40% of the exon 21. PCR/SSCP of exon 21, in the original patient where the alternatively spliced isoform was identified, showed a G to A nucleotide change at position 2644 (bold). Nucleotide sequence 5[prime] of the G2644A nucleotide substitution were consistent with a consensus donor splice site sequence: 2639A−262G−177/G+1100T+2100T+3xx. The G to A substitution at position + 4 of the intron 21 created a weak donor sequence according to an algorithm to identify splice site based on a Shapiro and Senapathy matrix. In the alternatively spliced integrin α7 the donor splice site sequence is shown. Numbers below each base give the frequency of occurrence. 49
Integrin α7 Mutation
One of the 30 patients studied showed a heterozygous integrin α7 mutation. Patient E.S. showed a unique conformer with primer set 1850F-2083R (Figure 4) ▶ . Direct sequencing of the aberrant conformers showed an heterozygous C to G nucleotide change at position 1969 in this patient (Figure 5) ▶ . The C1969G nucleotide change resulted in a arginine to glycine amino acid change at position 657 of the integrin α7 protein. The Arg657Gly is located in the region containing the major ADP-ribosylation site. The C1969G nucleotide change resulted in a loss of a SmaI restriction-enzyme site. To verify the frequency of this mutation in the control population, integrin α7 exon 15 was PCR-amplified, and the PCR products were digested with SmaI. Digestion fragments were present in all of 200 control chromosomes but only in one allele of the patient’s DNA (heterozygous mutation) (Figure 5 ▶ , bottom).
Figure 5.
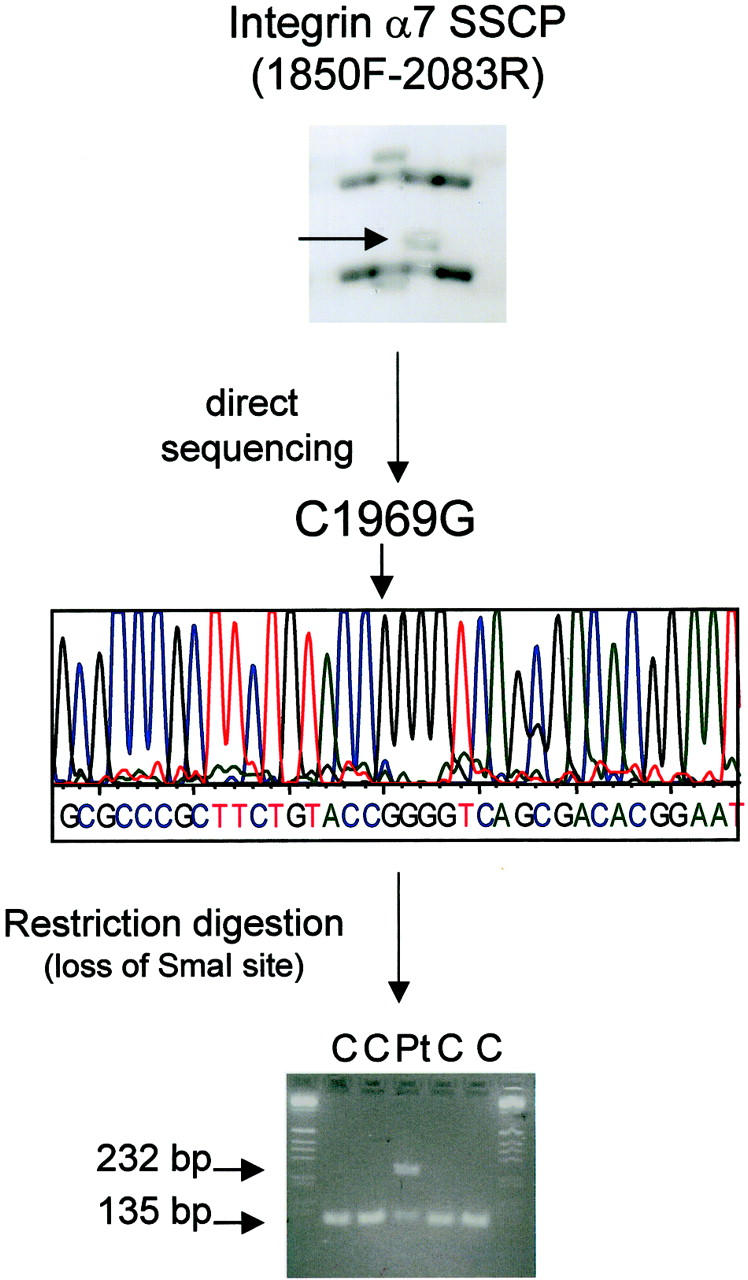
Screening of the integrin α7 coding sequence for potential mutations by SSCP showed a C1969G heterozygous nucleotide change resulting in an Arg657Gly amino acid change in patient E.S. Top: SSCP analysis of RT-PCR product for primer set 1850F-2083R showed a unique conformer in patient E.S. (The second conformer detected resulted in a A1952G nucleotide change (His651Arg). This polymorphism was present in 46% of the chromosomes studied). Direct sequencing of the aberrant conformer showed a C to G nucleotide change at position 1969 of the integrin α7 coding sequence (middle) resulting in an arginine to glycine amino acid change at position 657 of the integrin α7 protein. Nucleotide change C1969G resulted in a loss of SmaI restriction-enzyme site. Primers were designed to amplify exon 15 of the integrin α7 gene and the PCR product was digested with SmaI. Digestion fragments were present in 200 control chromosomes and only in one of the patient’s chromosomes (heterozygous mutation) (bottom).
Integrin α7B Gene Expression Studies
The same 30 patients used for integrin α7 mutation studies were selected for RNA studies. Ten muscle biopsies with myopathic histopathology but with normal integrin α7B immunofluorescence were chosen as controls.
RNA was isolated from patients’ muscle biopsies and 100 ng of total RNA was reverse transcribed into cDNA using oligo dT 10 primers. Approximately 20 ng of total cDNA was amplified using both integrin α7 and α-sarcoglycan primers. Quantitative multiplex fluorescent RT-PCR (QMF-RT/PCR) products (22 cycles) were done in duplicate for each sample and electrophoresed on an ABI automatic sequencer (373A) to quantitate signals using peak area. Integrin α7 versus α-sarcoglycan RNA ratios were determined. The patients’ muscle biopsies showed a normal amount of total integrin α7 RNA relative to controls in their muscles (Figure 6) ▶ . Mean integrin α7 versus α-sarcoglycan ratios were 24.2 ± 10 in the controls and 24.3 ± 11 in the patients’ group. Student’s t-test did not show any significant difference between patients’ and controls’ integrin α7 RNA levels (P = 0.8).
Figure 6.

Integrin α7-deficient patients show normal amount of integrin α7 mRNA. Shown are examples of automated sequencer traces of quantitative multiplex fluorescent RT-PCR of the integrin α7 relative to α-sarcoglycan. The levels of the integrin α7 RNA was similar in the patients relative to controls.
Discussion
Despite the recent advances in our understanding of the molecular basis of neuromuscular disorders, the underlying molecular defect can be identified only in a subset of the cases. A large cohort of patients with muscular dystrophy/myopathy cannot be assigned a specific molecular diagnosis. The goal of this study was to pursue an integrin α7 candidate protein and gene analysis in our large cohort of muscular dystrophy/myopathy patients of unknown etiology. This approach was chosen since candidate protein and gene studies have shown impressive progress in dissecting the heterogeneous limb-girdle dystrophy group. To date, dystrophies caused by α-, β-, and δ-sarcoglycan have all been defined via candidate gene and protein approaches. 36-40
The cloning of the integrin α7 gene, 31 the preferential expression of integrin α7 in skeletal muscle, 41-43 and the identification of causative integrin α7 mutations in a subset of Japanese patients presenting with congenital myopathy 31 made integrin α7 an ideal candidate gene for screening a large bank of patient muscle biopsies. Candidate protein and gene analyses were conducted by using integrin α7 immunofluorescence in 210 muscle biopsies of patients affected with muscular dystrophy or myopathy of unknown etiology and followed by direct mutation screening of muscle biopsy RNA in 30 patients in which integrin α7 deficiency was identified. No integrin α7 mutations were identified in 29 of 30 patients showing various degrees of integrin α7 deficiency ranging from complete to slight deficiency. Only in one patient presenting at birth with CMD and showing, by immunofluorescence, a marked reduction of integrin α7 in his muscle biopsy, an heterozygous C1969G missense mutation resulting in an arginine to a glycine amino acid change (Arg657Gly) was identified. Despite our efforts, we were unable to identify in this patient a second mutation, consistent with the presumed recessive inheritance observed in CMD. We feel that the C1969G nucleotide change may be a causative mutation based on several lines of evidence. First, the C1969G results in an arginine to a glycine amino acid change (Arg657Gly). Arginine is a large, charged amino acid with a molecular weight of 174, whereas glycine is a small, (molecular weight 75) uncharged, polar amino acid. It is unlikely that such a dramatic amino acid change would have no functional consequences in an heterodimeric protein. Second, the C1969G mutation was not observed in 200 control chromosomes and it is located in a region of documented functional importance. The Arg657Gly mutation lies in the region containing the major ADP-ribosylation site 44 that it is likely involved in the mechanisms modulating α7 integrin-mediated signaling pathways.
Several integrin α7 polymorphisms were also identified. In particular, we report a novel alternatively spliced integrin α7 isoform, generated from the use of a cryptic splice site in exon 21 (AG/GTTA2644AG), that was derived from a G2644A nucleotide change. The use of this cryptic splice site resulted in the skipping of about 40% of exon 21 directly to exon 22. This splice variant is present in about 12% of the chromosomes studied. A Senpathy score of at least 70 is required for donor splice site recognition. 45 The AG/GTTA2644AG cryptic donor site sequence scored only 71, thus the splicing theory would predict that the AG/GTTA2644AG sequence would be a weak donor site. Since both normal and cryptic mRNA are observed, both splice sites are used. However, the ratio between the normal and cryptic RNA favors the normal transcript (Figure 3) ▶ , suggesting that the utilization of the cryptic AG/GTTA2644AG splice is incomplete. It is difficult to predict the functional importance of this alternatively spliced integrin α7 isoform that removes 27 amino acids at a region where a major ADP-ribosylation site and a potential integrin leucyl-aspartyl-valine (LDV) binding site have been located. 26 While it is possible that the spliced variant may act as a modifying factor in a predisposing genetic background, it is also possible that it is without any functional and/or structural importance. Among the several integrin α7 isoforms previously reported, 5,20,23-28 the α7D, resulting from a partial deletion of exon 15 and the entire exon 16, is particularly interesting encompassing the major ADP-ribosylation site. 23,26 Unfortunately, the exact role of ADP-ribosylation on α7 integrins remains to be determined, 26 thus the potential functions of these isoforms are still largely unknown.
It has been reported that the expression of the cytoplasmic splice variant α7B is developmentally regulated, and that it is detectable in skeletal muscle only after the age of 2 years. 30 Since we used an antibody against the integrin α7B variant, we checked if the integrin α7 deficiency was related to the age of the patients at the time of the biopsy. Indeed, 39% (11 of 28) of the muscle biopsies of patients younger than 2 years compared to 13% (24 of 182) of patients older than 2 years showed integrin α7 deficiency. Even if this difference is significant, we feel that factors other than developmental stage may affect the sarcolemmal expression of the α7B integrin subunit in diseased skeletal muscle, as suggested by Cohn et al. 30 In the only patient where consecutive muscle biopsies were available, the integrin α7 deficiency detected in the muscle biopsy performed at 1 year of age was not rescued in the subsequent muscle biopsies done at 2 and 6.3 years of age. Moreover, 60% (17 of 28) of patients younger than 2 years showed normal integrin α7 immunostaining. The positive integrin α7 immunostaining detected in patients younger than 2 years of age may also be interpreted as some yet undefined compensatory mechanisms acting in dystrophic/myopathic muscle as suggested by the expression of integrin α7B in DMD and in the hypertrophic muscle fibers in SMA (spinal muscular atrophy) patients younger than 2 years. 30 To better investigate the discrepancy between the integrin α7 protein deficiency detected through immunofluorescence analysis of patients’ muscle biopsies and the lack of identification of integrin α7 gene mutations, we conducted a series of experiments.
Our inability to detect integrin α7 gene mutation in integrin α7-deficient muscle samples is not likely due to technical reasons. We cannot rule out that mutations in the promotor region or in some intronic sequences that we did not analyze, underlie the observed integrin α7 deficiency. However, we feel that our mutation detection system is quite sensitive (we calculated about 93% sensitivity in a previous study). 46 Since laminin α2 deficiency, both partial or total, is a well-known cause of secondary integrin α7 deficiency, 11,12,30 all of the 210 muscle biopsies included in this study were checked by laminin α2 immunofluorescence, and all were normal.
To determine whether the decreased accumulation of integrin α7 protein might be the result of protein instability or due to the lack of integrin α7 gene expression or RNA processing, we quantitated total RNA from muscle biopsies of integrin α7-deficient patients. QMF-RT/PCR has been shown to have a relatively low experimental error, and has the additional advantage of using internal control RNA for each test. 47 Our results showed normal integrin α7 mRNA levels in the patients studied suggesting that the loss of integrin α7 in their muscle biopsies occurs at the protein level. It is possible that the observed secondary integrin α7 deficiency may be the result of an incorrect processing and/or location of a normally synthesized integrin α7 protein in the patients’ muscles. The cytoplasmic integrin α7-positive immunostaining observed in some of our patients’ muscle biopsies further support the hypothesis of a integrin α7 moiety that is properly produced but is mislocated or unstable at the membrane level in absence, for example, of some yet unknown ligand. Quantitation of integrin α7, such as by immunoblotting analysis, may be informative, but unfortunately the small size of our diagnostic biopsies is a limiting factor. The biopsies are not sufficient to obtain a membrane-enriched pellet, and this limitation did not allow us to perform such an analysis in this study.
Taken together, our results suggest that secondary integrin α7 deficiency is rather common in muscular dystrophy/myopathy of unknown etiology. This is not surprising considering the central role played by α7β1D integrin in anchoring the intracellular cytoskeleton via actin, to the extracellular matrix, via laminin, and the potential for signal transduction possibly mediated by extracellular matrix, soluble growth factors, and/or associated transmembrane molecules. “Inside-out” and “inside-in” signals can mutually modulate the activation of cell proliferation and migration, and if this signaling is malfunctioning, the lack of appropriate cell matrix interaction can lead to cell death via apoptosis. 48 Analyses of mutant phenotypes provide further evidence of the key role of integrins in adhesion-mediated events in vertebrates. Germ-line mutations in genes coding for other laminin binding integrin result in disease. For example, mutations in either the α6 or the β4 subunit of the epithelial hemidesmosomes integrin result in a variant of the usually lethal skin blistering disorder, epidermolysis bullosa. α3 integrin in epithelia plays a direct role in the assembly of the basement membrane as shown by the α3-null mice that show basement membrane abnormalities in the kidney, lung, and skin and die soon after birth. 48 Further work is necessary to obtain more insight into the role of integrin-mediated adhesion and signaling and their modulating factors.
Footnotes
Address reprint requests to Elena Pegoraro, M.D. Ph.D., Neuromuscular Center, Department of Neurological and Psychiatric Sciences, University of Padova, 35128 Padova, Italy. E-mail: elena.pegoraro@unipd.it.
Supported by Italian Telethon grant 1114.
References
- 1.Horwitz AF: Integrins and health. Sci Am 1997, 276:68-75 [DOI] [PubMed] [Google Scholar]
- 2.Giancotti FG, Ruoslahti E: Integrin signaling. Science 1999, 285:1028-1032 [DOI] [PubMed] [Google Scholar]
- 3.Kramer RH, Vu MP, Cheng YF, Ramos DM, Timpl R, Waleh N: Laminin-binding integrin α7β1: functional characterization and expression in normal and malignant melanocytes. Cell Regul 1991, 2:805-817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von der Mark H, Durr J, Sonnenberg A, von der Mark K, Deutzmann R, Goodman SL: Skeletal myoblasts utilize a novel β1-series integrin and not α6β1 for binding to the E8 and T8 fragments of laminin. J Biol Chem 1991, 266:23593-23601 [PubMed] [Google Scholar]
- 5.Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ: H36-α7 is a novel integrin α chain that is developmentally regulated during skeletal myogenesis. J Cell Biol 1992, 117:643-657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burkin DJ, Kaufman SJ: The α7β1 integrin in muscle development and disease. Cell Tissue Res 1999, 296:183-190 [DOI] [PubMed] [Google Scholar]
- 7.van der Flier A, Kuikman I, Baudoin C, van der Neut R, Sonnenberg A: A novel β1 integrin isoform produced by alternative splicing: unique expression in cardiac and skeletal muscle. FEBS Lett 1995, 369:340-344 [DOI] [PubMed] [Google Scholar]
- 8.Belkin AM, Zhidkova NI, Balzac F, Altreda F, Tomatis D, Maier A, Tarone G, Koteliansky VE, Burridge K: β1D integrin displaces the β1A isoform in striated muscles: localization at junctional structures and signaling potential in non-muscle cells. J Cell Biol 1996, 132:211-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin PT, Kaufman SJ, Kramer RH, Sanes JR: Synaptic integrins in developing, adult, and mutant muscle: selective association of α1, α7A, and α7B integrins with the neuromuscular junction. Dev Biol 1996, 174:125-139 [DOI] [PubMed] [Google Scholar]
- 10.Yao CC, Ziober BL, Squillace RM, Kramer RH: α7 integrin mediates cell adhesion and migration on specific laminin isoforms. J Biol Chem 1996, 271:25598-25603 [DOI] [PubMed] [Google Scholar]
- 11.Hodges BL, Hayashi YK, Nonaka I, Wang W, Arahata K, Kaufman SJ: Altered expression of the α7β1 integrin in human and murine muscular dystrophies. J Cell Sci 1997, 110:2873-2881 [DOI] [PubMed] [Google Scholar]
- 12.Vachon PH, Xu H, Liu L, Hayashi Y, Arahata K, Reed JC, Wewer UM, Engvall E: Integrins (α7β1) in muscle function and survival: disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest 1997, 100:1870-1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otey CA, Pavalko FM, Burridge K: An interaction between α-actinin and the β1 integrin subunit in vitro. J Cell Biol 1990, 111:721-729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark EA, Brugge JS: Integrins and signal transduction pathways: the road taken. Science 1995, 268:233-239 [DOI] [PubMed] [Google Scholar]
- 15.Ocalan M, Goodman SL, Kuhl U, Hauschka SD, von der Mark K: Laminin alters cell shape and stimulates motility and proliferation of murine skeletal myoblasts. Dev Biol 1988, 125:158-167 [DOI] [PubMed] [Google Scholar]
- 16.Goodman SL, Risse G, von der Mark K: The E8 subfragment of laminin promotes locomotion of myoblasts over extracellular matrix. J Cell Biol 1989, 109:799-809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouslahti R, Pierschbacher MD: New perspectives in cell adhesion: rGD and integrins. Science 1987, 238:491-497 [DOI] [PubMed] [Google Scholar]
- 18.Hynes RO: Integrins: versatility, modulation, and signaling in cell adhesion. Cell 1992, 69:11-25 [DOI] [PubMed] [Google Scholar]
- 19.Schoenwaelder SM, Burridge K: Evidence for calpeptin-sensitive protein-tyrosine phosphatase upstream of the small GTPase Rho: a novel role for calpain inhibitor calpeptin in the inhibition of protein-tyrosine phosphatase. J Biol Chem 1999, 274:14359-14367 [DOI] [PubMed] [Google Scholar]
- 20.Song WK, Wang W, Stato H, Bielser DA, Kaufman SJ: Expression of α7 integrin cytoplasmic domains during skeletal muscle development: alternate forms, conformational change, and homologies with serine/threonine kinases and tyrosine phosphatases. J Cell Sci 1993, 106:1139-1152 [DOI] [PubMed] [Google Scholar]
- 21.Bao ZZ, Lakonishok M, Kaufman S, Horwitz AF: α7β1 integrin is a component of the myotendineous junction on skeletal muscle. J Cell Sci 1993, 106:579-590 [DOI] [PubMed] [Google Scholar]
- 22.Wang W, Wu W, Desai T, Ward DC, Kaufman SJ: Localization of the α7 integrin gene (ITGA7) on human chromosome 12q13: clustering of integrin and Hox genes implies parallel evolution of these gene families. Genomics 1995, 26:568-570 [DOI] [PubMed] [Google Scholar]
- 23.Vignier N, Moghadaszadeh B, Gary F, Beckman J, Mayer U, Guicheney P: Structure, genetic localization, and identification of the cardiac and skeletal transcripts of the human integrin α7 gene (ITGA7). Biochem Biophys Res Commun 1999, 260:357-364 [DOI] [PubMed] [Google Scholar]
- 24.Ziober BL, Vu MP, Waleh N, Crawford J, Lin CS, Kramer RH: Alternative extracellular and cytoplasmic domains of the integrin α7 subunit are differentially expressed during development. J Biol Chem 1993, 268:26773-26783 [PubMed] [Google Scholar]
- 25.Ziober BL, Chen YQ, Kramer RH: The laminin-binding activity of the α7 integrin receptor is defined by developmentally regulated splicing in the extracellular domain. Mol Biol Cell 1997, 8:1723-1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leung E, Lim SP, Berg R, Yang Y, Ni J, Wang S, Krissansen GW: A novel extracellular domain variant of the human integrin α7 subunit generated by alternative intron splicing. Biochem Biophys Res Commun 1998, 243:317-325 [DOI] [PubMed] [Google Scholar]
- 27.Collo G, Starr L, Quaranta V: A new isoform of the laminin receptor integrin α7β1 is developmentally regulated in skeletal muscle. J Biol Chem 1993, 268:19019-19024 [PubMed] [Google Scholar]
- 28.George-Weinstein M, Foster RF, Gerhart JV, Kaufman SJ: In vitro and in vivo expression of α7 integrin and desmin define the primary and secondary myogenic lineages. Dev Biol 1993, 156:202-229 [DOI] [PubMed] [Google Scholar]
- 29.Mayer U, Saher G, Fässler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Pöschl E, von der Mark K: Absence of integrin α7 causes a novel form of muscular dystrophy. Nat Genet 1997, 17:318-323 [DOI] [PubMed] [Google Scholar]
- 30.Cohn RD, Mayer U, Saher G, Herrmann R, van der Flier A, Sonnenberg A, Sorokin L, Voit T: Secondary reduction of α7B integrin in laminin α2-deficient congenital muscular dystrophy supports an additional transmembrane link in skeletal muscle. J Neurol Sci 1999, 163:140-152 [DOI] [PubMed] [Google Scholar]
- 31.Hayashi YK, Chou FL, Engvall E, Ogawa M, Matsuda C, Hirabayashi S, Yokochi K, Ziober BL, Kramer RH, Kaufman SJ, Ozawa E, Goto Y, Nonaka I, Tsukahara T, Wang J, Hoffman EP, Arahata K: Mutations in the integrin α7 gene cause congenital myopathy. Nat Genet 1998, 19:94-97 [DOI] [PubMed] [Google Scholar]
- 32.Leivo I, Engvall E: Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci USA 1988, 85:1544-1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pegoraro E, Marks H, Garcias CA, Crawford T, Mancias P, Connolly AM, Fanin M, Martinello F, Trevisan CP, Angelini C, Stella A, Scavina M, Munk RL, Servidei S, Bönnemann CC, Bertorini T, Acsadi G, Thompson CE, Gagnon D, Hoganson G, Carver V, Hoffman EP: Laminin α2 muscular dystrophy: genotype/phenotype study in 22 patients. Neurology 1998, 51:101-110 [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Pegoraro E, Menegazzo E, Gennarelli M, Hoop RC, Angelini C, Hoffman EP: Myotonic dystrophy: evidence for a possible dominant-negative RNA mutation. Hum Mol Genet 1995, 4:599-606 [DOI] [PubMed] [Google Scholar]
- 35.Shapiro MB, Senapathy P: RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 1987, 15:7155-7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tomè FMS, Romero NB, Fardeau M, Beckmann JS, Kaplan JC, Campbell KP: Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell 1994, 78:625-633 [DOI] [PubMed] [Google Scholar]
- 37.Bonnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally EM, Duggan DJ, Angelini C, Hoffman EP, Ozawa E, Kunkel LM: β-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet 1995, 11:266-273 [DOI] [PubMed] [Google Scholar]
- 38.Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, Tomè FMS, Fardeau M, Jackson CE, Beckmann JS, Campbell KP: β-Sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet 1995, 11:257-265 [DOI] [PubMed] [Google Scholar]
- 39.Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bonnemann CG, Gussoni E, Denton PH, Kyriakides T, Middleton L, Hentati F, Ben Hamida M, Nonaka I, Vance JM, Kunkel LM, Ozawa E: Mutations in the dystrophin-associated protein τ-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995, 270:819-822 [DOI] [PubMed] [Google Scholar]
- 40.Nigro V, de Sa Moreira E, Piluso G, Vainzof M, Belsito A, Politano L, Puca AA, Passos-Bueno MR, Zatz M: Autosomal recessive limb-girdle muscular dystrophy, LGMD2E, is caused by mutations in the δ-sarcoglycan gene. Nat Genet 1996, 14:195-198 [DOI] [PubMed] [Google Scholar]
- 41.Schuler F, Sorokin LM: Expression of laminin isoforms in mouse myogenic cells in vitro and in vivo. J Cell Sci 1995, 108:3795-3805 [DOI] [PubMed] [Google Scholar]
- 42.Echtermeyer F, Schober S, Poschl E, von der Mark H, von der Mark K: Specific induction of cell motility on laminin by α7 integrin. J Biol Chem 1996, 271:2071-2075 [DOI] [PubMed] [Google Scholar]
- 43.Yao CC, Breuss J, Pytela R, Kramer RH: Functional expression of the α7 integrin receptor in differentiated smooth muscle cells. J Cell Sci 1997, 110:1477-1487 [DOI] [PubMed] [Google Scholar]
- 44.Zolkiewska A, Moss J: Processing of ADP-ribosylated integrin α7 in skeletal muscle myotubes. J Biol Chem 1995, 270:9227-9233 [DOI] [PubMed] [Google Scholar]
- 45.O’Neil JP, Rogan PK, Cariello N, Nicklas JA: Mutations that alters RNA splicing of the human HPRT gene: a review of the spectrum. Mutat Res 1998, 411:179-214 [DOI] [PubMed] [Google Scholar]
- 46.Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Angelini C, Pegoraro E, McNally E, Bonnemann CG, Kunkel LM, Noguchi S, Ozawa E, Pendlenbury W, Oechler H, Waclawik A, Duenas DA, Hutchison T, Hausmanowa-Petrusewicz I, Fidzianska A, Bean SC, Haller JS, Bodensteiner J, Greco C, Pestronk A, Berardinelli A, Gelinas DF, Abram H, Kuncl RW: Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med 1997, 336:618-624 [DOI] [PubMed] [Google Scholar]
- 47.Zhou J, Hoffman EP: Pathophysiology of sodium channelopaties: studies of sodium channel expression by quantitative multiplex fluorescence polymerase chain reaction. J Biol Chem 1994, 269:18563-18571 [PubMed] [Google Scholar]
- 48.Belkin AM, Stepp MA: Integrins as receptors for laminins. Microsc Res Tech 2000, 51:280-301 [DOI] [PubMed] [Google Scholar]
- 49.Lewin B: Genes IV. 1990. Oxford University Press, Oxford, England