

The current dogma is that active stromal and vascular invasion by cancer cells themselves is essential to the metastatic process. 1 Yet, in this issue of The American Journal of Pathology, Sugino and co-workers 2 present evidence that, for some tumor models, tumor cells can gain access into blood vessels by intravasation of tumor cell nests surrounded by vascular endothelial cells, followed by intravascular tumor growth in the lung, without penetration of the vascular wall during either process. These investigators report a murine mammary tumor (MCH66) model of metastasis that does not require invasion into the vascular wall of the target organ. Observations from comparing a non-metastatic MCH66 clone (MCH66C8) and another highly invasive metastatic cell line (MCH416) suggested that high angiogenic activity and sinusoidal remodeling of tumor blood vessels were prerequisites for MCH66 metastasis. Moreover, by using differential cDNA analysis the authors identified several genes that were overexpressed by MCH66, including genes for pleiotrophin as well as extracellular matrix (ECM)-associated molecules, which may modulate the microenvironment toward neovascularization. Sugino and co-workers 2 conclude that, in some tumors, tumor angiogenesis plays a central role in the induction of invasion-independent metastasis. Clearly, these findings provide additional momentum for using anti-angiogenic models in screening and development of new therapeutic agents for cancer metastasis.
For a tumor to grow, the tumor cells must not only proliferate, but benign host tissue, especially new blood vessels, must also form around the tumor cells. In 1971, Folkman proposed that tumor growth is dependent on angiogenesis. 3 Moreover, he has advocated that tumor cells and blood vessels compose a highly integrated ecosystem, that endothelial cells can be switched from a resting state to one of rapid growth by a diffusible signals from tumor cells, (or inflammatory cells or stroma-derived products), and that anti-angiogenesis might be an effective anticancer therapy. 4 Indeed, there is abundant evidence that tumor growth and spread is angiogenesis dependent, that tumor cells can produce diffusible angiogenic regulatory molecules, and that angiogenesis antagonists can slow or prevent tumor growth. Much of this evidence as well as probable tumor angiogenic mechanisms have been summarized in a various reviews. 4-6
To metastasize, a tumor cell must gain access to the vasculature from the primary tumor, survive the circulation, escape immune surveillance, localize in the vasculature of the target organ, escape from (or grow from within) the vasculature into the target organ, and induce tumor angiogenesis. 7,8 Tumor spread is amplified when the new metastasis sheds additional tumor cells to form even more metastases by following the same sequence of events. 8 Angiogenesis is needed; because, without it, tumor cells would not shed into the circulation. 9-12 Greater numbers of tumor vessels increase the opportunity for tumor cells to enter the circulation. Using a transplantable mouse fibrosarcoma model, Liotta et al 11,12 showed that tumor cells shed into the bloodstream increased from roughly 103 cells per 24 hours on day 5 after tumor implantation to roughly 105 cells per 24 hours on day 15, an increase correlating closely with increasing intratumor microvessel density, especially when the intratumor microvessels were more than 30 microns in diameter (ie, sinusoid-sized blood vessels). Also, these studies revealed that the establishment of lung metastases is directly related to the number of tumor cells (especially cell clusters) shed into the circulation. 11,12 These elegant experiments were among the first to show that intratumor microvessel density can correlate with aggressive tumor behavior. Subsequently, many studies have shown that intratumoral microvessel density correlates with tumor aggressiveness of many different tumor types. 4,5,6
Tumor angiogenesis promotes tumor growth, because the new vessels allow exchange of nutrients, oxygen, and waste products by a crowded cell population for which simple diffusion of these substances across its outer surfaces is no longer adequate. In addition to this perfusion effect, endothelial cells release important paracrine growth factors for tumor cells. 13-16 The invasive chemotactic behavior of endothelial cells at the tips of growing capillaries is mediated in part by their secretion of various collagenases, urokinases, and plasminogen activator. 17,18 These degradative enzymes likely facilitate spread of tumor cells into and through the adjacent fibrin gel matrix and connective tissue stroma. Fox et al 17 found a significant association of urokinase-type plasminogen activator and plasminogen activator inhibitor-1 with intratumor microvessel density; and they concluded that the poor prognosis in breast carcinomas with relatively elevated urokinase-type plasminogen activator and plasminogen activator inhibitor-1 may result from an interaction between endothelial and tumor cells using the urokinase-type plasminogen activator enzyme system. Moreover, vascular endothelial cell growth factor (VEGF) (a potent angiogenic factor) can cause endothelial cells to express plasminogen activator, plasminogen activator inhibitor, interstitial collagenase, and procoagulant activity. 19 VEGF promotes extravasation of plasma fibrinogen, leading to fibrin deposition within the tumor matrix, which also promotes the ingrowth of macrophages, fibroblasts, and endothelial cells. 20 Newly formed proliferating capillaries have fragmented basement membranes and are leaky, making them more accessible to tumor cells than mature vessels. 21 These degradative enzymes may facilitate the escape of tumor cells into the tumor neovasculature. Indeed, the invading capillaries may actively participate in the metastatic process by engulfing and thus facilitating the entry of tumor cells into vascular spaces, allowing systemic spread. Thus, the additive impact of the perfusion and paracrine tumor effects plus the endothelial cell-derived invasion-associated enzymes all likely contribute to rapid tumor growth and to a much higher metastatic potential by facilitating entry of tumor cells into the blood vascular and lymphatic system. 4-6
The current and previous work of Sugino et al 2,22,23 add support for a new paradigm for intravasation of tumor cells into vessels. Their convincing observations show that, in some tumor models, tumor cells can gain access into blood vessels by intravasation of tumor cell nests surrounded by vascular endothelial cells, followed by intravascular tumor growth without penetration of the vascular wall during either process. But, to many scientists, tumor invasion and metastases only means that tumor cells must traverse or invade through their own basement membrane, penetrate or migrate through the connective tissue stroma, find lymphatic and blood vessels, burrow through the vessel basement membrane and endothelial cell layers, survive the intravascular compartment, lodge within the target organ, and burrow out of the blood vessel before developing into a new metastatic focus. This process is well illustrated one of the more popular basic pathology texts 1 and, indeed, it is likely true for many tumor models. Although sometimes intravascular tumor emboli are enclosed by endothelial cells, usually, tumor emboli encountered in general surgical pathology practice are not surrounded by endothelial cells, but rather a platelet/fibrin coat with no discernable endothelial cell covering (Figure 1, A and B) ▶ . But, even the latter process does not eliminate a probable significant contributing role of tumor angiogenic/endothelial cell activity. Activated endothelial cells are derived from a normal population of intact fully functional host cells and their “invasiveness” could reach optimum function, given proper stimuli. Moreover, I would suggest that the fully activated endothelial cell “invasiveness” would be much superior to the invasiveness of tumor cells, which suffer from significant genetic damage. Another dogma I have long questioned is that tumor cells must invade through their own basal lamina to spread. It seems more reasonable that, as tumors dedifferentiate, tumor cells lose their capacity to form a competent and/or complete basal lamina. And, when a distinctive, well-oriented basal lamina fails to form, tumor cells begin to grow in an unrestricted, disorganized pattern into adjacent connective tissue stroma, where they directly encounter activated endothelial cells and inflammatory cells.
Figure 1.
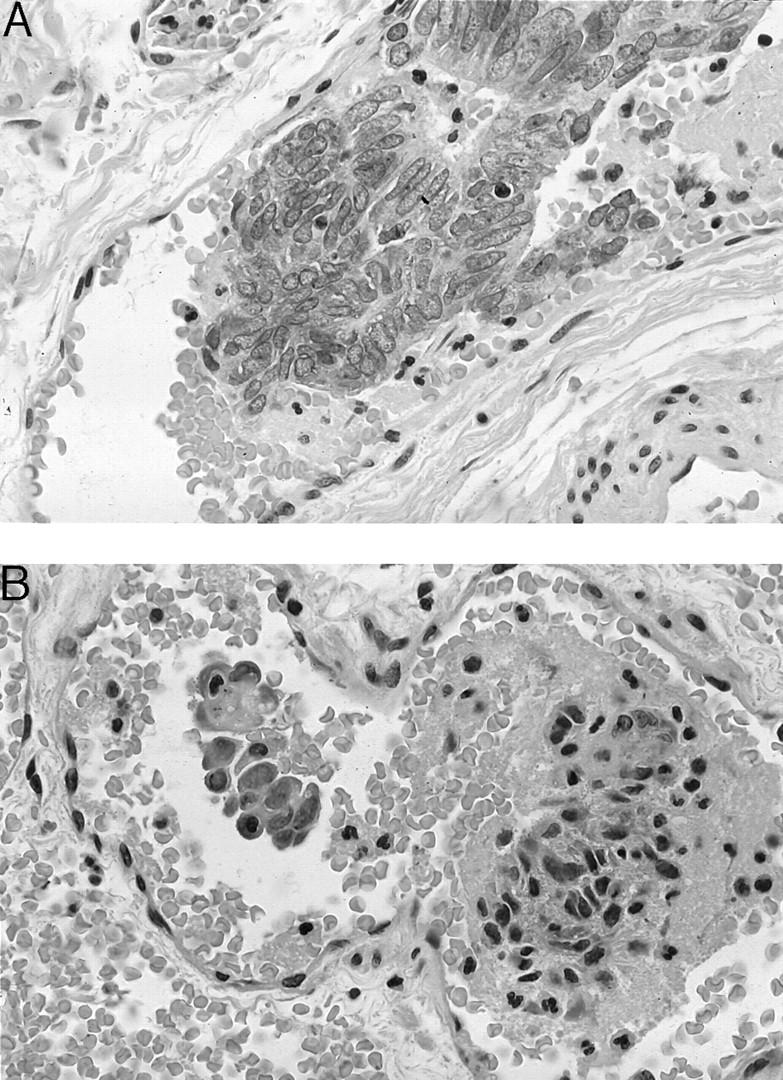
A: An example of an intravascular tumor embolus of endometrioid adenocarcinoma. Note the absence of a surrounding endothelial cell layer, but rather closely adherent fibrinoid material (presumably a platelet fibrin complex). B: A second example of an intravascular tumor embolus of a poorly differentiated endometrioid adenocarcinoma. As in A, note the absence of a surrounding endothelial cell layer, but rather closely adherent fibrinoid material (presumably a platelet fibrin complex).
Finally, I want to reemphasize some views that Dr. Thompson (Department of Pathology, University of Aberdeen Medical School, Aberdeen Royal Infirmary, Aberdeen, UK) has advocated in a recent editorial. 24 His views lend support to Sugino and co-workers findings 2,22,23 as well many of the above comments. He believes, as others do, that malignant tumor growth depends on angiogenesis, that there is a link between prognosis and increased tumor vascularity, that tumor invasion requires tissue destruction, that neovascularization must not only be protected but sustained (especially at the tumor edge), that for tumor survival the edge is the future and the center is history, and that angiogenesis is essential not only for tumor growth but also for tumor invasion. Likewise, I believe these are important considerations and that we need to expand the current paradigms regarding tumor growth and spread. Clearly, the tumor cell is not the only active cell in the growth and spread of tumors, but other host reactive cells, especially tumor angiogenic/endothelial cells, are critical in facilitating tumor growth and spread.
Footnotes
Address reprint requests to Noel Weidner, M.D., Professor and Director of Anatomic Pathology, University of California, San Diego, Medical Center, Hillcrest Hospital, Mail Code 8720, 200 West Arbor Drive, San Diego, CA 92103-8720. E-mail: noweidner@ucsd.edu.
References
- 1. ed 6 Cotran RS Kumar V Collins T eds. Neoplasia. Robbins Pathologic Basis of Disease 1999, :pp 260-327 WB Saunders, Philadelphia, PA [Google Scholar]
- 2.Sugino T, Kusakabe T, Hoshi N, Yamaguchi T, Kawaguchi T, Goodison S, Sekimata M, Homma Y, Suzuki T: An invasion-independent pathway of blood-borne metastasis: a new murine mammary tumor model. Am J Pathol 2002, 160:1970-1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkman J: Tumor angiogenesis: therapeutic implications. N Engl J Med 1971, 285:1182-1186 [DOI] [PubMed] [Google Scholar]
- 4.Weidner N: Commentary: intratumor microvessel density as a prognostic factor in cancer. Am J Pathol 1995, 147:9-19 [PMC free article] [PubMed] [Google Scholar]
- 5.Weidner N, Folkman J: Tumor vascularity as a prognostic factor in cancer. deVita VT Hellman S Rosenberg SA eds. Important Advances in Oncology 1996. 1996, Lippincott-Raven, Philadelphia, PA [PubMed]
- 6.Weidner N: Tumor vascularity: what does it tell us about the growth and spread of cancer? D’Amore PA Voest E eds. Tumor Angiogenesis and Microcirculation. 2001, Marcel Dekker, New York, NY
- 7.Fidler IJ, Gersten DM, Hart IR: The biology of cancer invasion and metastasis. Adv Cancer Res 1978, 28:149-250 [DOI] [PubMed] [Google Scholar]
- 8.Nicolson G: Cancer metastasis. Sci Am 1979, 240:66-76 [DOI] [PubMed] [Google Scholar]
- 9.Weiss L: Biophysical aspects of the metastatic cascade. Weiss L eds. Fundamental Aspects of Metastasis. 1976, :pp 51-70 Amsterdam, North Holland [Google Scholar]
- 10.Bernstein LR, Liotta LA: Molecular mediators of interactions with extracellular matrix components in metastasis and angiogenesis. Curr Opin Oncol 1994, 6:106-113 [DOI] [PubMed] [Google Scholar]
- 11.Liotta L, Kleinerman J, Saidel G: Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer Res 1974, 34:997-1004 [PubMed] [Google Scholar]
- 12.Liotta L, Saidel G, Kleinerman J: The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res 1976, 36:889-894 [PubMed] [Google Scholar]
- 13.Nicosia RF, Tchao R, Leighton J: Interactions between newly formed endothelial channels and carcinoma cells in plasma clot culture. Clin Exp Metastasis 1986, 4:91-104 [DOI] [PubMed] [Google Scholar]
- 14.Rak JW, Hegmann EJ, Lu C, Kerbel RS: Progressive loss of sensitivity to endothelium-derived growth inhibitors expressed by human melanoma cells during disease progression. J Cell Physiol 1994, 159:245-255 [DOI] [PubMed] [Google Scholar]
- 15.Hamada J, Cavanaugh PG, Lotan O: Separable growth and migration factors for large-cell lymphoma cells secreted by microvascular endothelial cells derived from target organs for metastasis. Br J Cancer 1992, 66:349-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folkman J: Angiogenesis and breast cancer. J Clin Oncol 1994, 12:441-443 [DOI] [PubMed] [Google Scholar]
- 17.Fox SB, Stuart N, Smith K, Brunner N, Harris AL: High levels of uPA and PA-1 are associated with highly angiogenic breast carcinomas. J Pathol 1993, 170(Suppl):388a [Google Scholar]
- 18.Moscatelli D, Gross J, Rifkin D: Angiogenic factors stimulate plasminogen activator and collagenase production by capillary endothelial cells. J Cell Biol 1981, 91:201a(Abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Dvorak HF, Senger DR: Increased expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in kidney and bladder carcinomas. Am J Pathol 1993, 143:1255-1262 [PMC free article] [PubMed] [Google Scholar]
- 20.Senger DR, Van De Water L, Brown LF, Nagy JA, Yeo T-K, Berse B, Jackman RW, Dvorak AM, Dvorak HF: Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev 1993, 12:303-324 [DOI] [PubMed] [Google Scholar]
- 21.Nagy JA, Brown LF, Senger DR, Lanir N, Van de Water L, Dvorak AM, Dvorak HF: Pathogenesis of tumor stroma generation: a critical role for leaky blood vessels and fibrin deposition. Biochim Biophys Acta 1989, 948:305-326 [DOI] [PubMed] [Google Scholar]
- 22.Sugino T, Kawaguchi T, Suzuki T: Sequential process of blood-borne lung metastases of spontaneous mammary carcinoma in C3H mice. Int J Cancer 1993, 55:141-147 [DOI] [PubMed] [Google Scholar]
- 23.Sugino T, Kawaguchi T, Suzuki T: Stromal invasion is not essential to blood-borne metastasis in mouse mammary carcinoma. Scientific Program Booklet of the 170th Meeting of The Pathological Society of Great Britain and Ireland, January, 1995 (Abstract 161)
- 24.Thompson WD: Editorial: tumor versus patient: vascular and tumour survival versus prognosis. J Pathol 2001, 193:425-426 [DOI] [PubMed] [Google Scholar]