

Abstract
Studies of adenomatous polyposis coli (APC) mutations in familial adenomatous polyposis (FAP) have focused on large bowel disease. It has been found that: 1) germline APC mutations around codon 1300 are associated with severe colorectal polyposis; 2) somatic APC mutations in colorectal tumors tend to cluster approximately between codons 1250 and 1450; and 3) patients with germline mutations close to codon 1300 tend to acquire somatic mutations (second hits) in their colorectal polyps by allelic loss, whereas the tumors of other FAP patients have truncating second hits. Using new and published data, we have investigated how germline and somatic APC mutations influence the pathogenesis of upper gastrointestinal polyps in FAP. We have compared the results with those from colorectal disease. We found that somatic mutations in upper gastrointestinal polyps cluster approximately between codons 1400 and 1580. Patients with germline APC mutations after codon 1400 tend to show allelic loss in their upper gastrointestinal polyps; the tumors of other patients have truncating somatic mutations after codon 1400. Finally, patients with germline mutations after codon 1400 tend to have more severe duodenal polyposis (odds ratio, 5.72; 95% confidence interval, 1.13 to 28.89; P = 0.035). Thus, in both upper gastrointestinal and colorectal tumors, a specific region of the APC gene is associated with severe disease, clustering of somatic mutations, and loss of the wild-type allele. However, the region concerned is different in upper gastrointestinal and colorectal disease. The data suggest that loss of all APC SAMP repeats is probably necessary for duodenal and gastric tumorigenesis in FAP, as it is in colonic tumors. Compared with colonic tumors, however, retention of a greater number of β-catenin binding/degradation repeats is optimal for tumorigenesis in upper gastrointestinal FAP.
Most Mendelian cancer syndromes predispose to tumors of more than one anatomical site. Familial adenomatous polyposis (FAP) is no exception. Although the predominant features of FAP are profuse colonic polyposis and colorectal carcinoma, most patients also develop adenomas of the duodenum 1 and many develop neoplastic gastric lesions (fundic polyps or adenomas). 2 With the development of more effective prophylactic surgery for colonic disease, upper gastrointestinal polyposis, especially duodenal disease, has become a major cause of mortality for FAP patients. The severity of duodenal disease varies from no polyps at all to multiple lesions and carcinoma, and is often measured by the semiquantitative Spigelman score. 3 Duodenal polyposis varies in its severity both within and among families.
FAP results from germline adenomatous polyposis coli (APC) mutations (Figure 1) ▶ . Clinical data show that FAP patients with germline mutations at codon 1309 have exceptionally severe colonic polyposis. 4 It is also well established that somatic APC mutations in colonic FAP polyps occur nonrandomly, usually being found in the colorectal mutation cluster region (MCR) (approximately codons 1250 to 1450). 5 It is also notable that in FAP patients, the site of the germline mutation determines the type of somatic mutation (second hit) in colorectal polyps: 6 germline mutations close to codon 1300 are associated with allelic loss and germline mutations elsewhere are associated with truncating somatic mutations in the MCR. Together with corroborative evidence from studies on sporadic colorectal tumors, these data show that simple inactivation of APC function is not optimally selected. Rather, mutations that truncate the protein close to codon 1300 provide the strongest selective advantage to the cell in which they occur. 6 Such mutations result in stable, truncated APC proteins 7 in which all of the SAMP repeats—involved in axin/conductin binding and β-catenin degradation—are lost and only 1 of the 20-amino acid β-catenin-binding/degradation repeats is retained. 8
Figure 1.

The APC protein, showing major identified domains and their position by amino acid.
It is not known whether or not upper gastrointestinal disease in FAP shows a dependence on APC mutation which is similar to that found for colorectal disease. The existence of an association between germline APC genotype and the severity of upper gastrointestinal polyposis is controversial, 9,10 and no large study has been performed. Recently, Bjork and colleagues 11 studied 40 patients with known germline APC mutations and found that patients with Spigelman stage IV duodenal disease or carcinoma tended to have germline mutations after codon 1051. A small number of previous studies has analyzed the spectrum of intragenic somatic mutations in upper gastrointestinal polyps from FAP patients. A single study of 64 duodenal and 11 gastric FAP polyps found high somatic mutation frequencies at codons 1450, 1462 to 1465, and 1554 to 1556, which the authors concluded to be similar to colorectal tumors. 12 In a more recent study, however, codon 1554 to 1556 was suggested as a mutational hot-spot for gastric polyps. 2 No study has examined whether or not the site or type of the somatic APC mutation in upper gastrointestinal polyps depends on the patient’s germline mutation.
We have screened 49 duodenal polyps from 26 FAP patients for somatic mutations and allelic loss. We have determined whether or not the type of somatic mutation in upper gastrointestinal polyps depends on the site of the germline APC mutation. We have combined our data with those from previous studies to test whether or not there exists a somatic MCR for upper gastrointestinal tumors in FAP. Finally, we have extended our study to a much larger sample set to test the hypothesis that germline mutations within the putative upper gastrointestinal MCR are associated with more severe disease.
Materials and Methods
Forty-nine fresh-frozen duodenal polyps from 26 patients with classical colonic FAP were collected from St. Marks Hospital (median diameter, 3 mm; range, 1 to 6 mm). In tandem with a similar study on early colonic adenomas, 6 hematoxylin and eosin-stained frozen sections of each polyp were reviewed to confirm basic histological features of adenomas—almost all were mildly dysplastic—and a minimum of ∼50% neoplastic cells in each lesion, as far as morphological assessment allowed. All polyps that did not meet these criteria were excluded from the analysis. Paired constitutional DNA was derived from blood or normal colonic tissue. Extraction of DNA from the tumor and normal tissue was performed using the Qiagen (Hilden, Germany) Tissue Extraction Kit and from blood using standard methods.
For assessing allelic loss at APC, polymorphic microsatellite markers close to the APC gene (D5S346 and D5S656) were chosen from public databases (for example, http://genome.ucsc.edu). The polymerase chain reaction (PCR) typically contained 20 to 100 ng DNA, 50 mmol/L KCl, 0.5 to 2.5 mmol/L MgCl2, 10 mmol/L Tris-HCl, 0.1% Triton, 2.5 μg of bovine serum albumin, 0.2 mmol/L of each dNTPs, 10 pmol of each oligonucleotide, and 1.25 U Taq DNA polymerase (Promega). The PCR reaction consisted of an initial step of 94°C for 4 minutes, then 40 cycles of 1 minute at 94°C, 1 minute at the appropriate annealing temperature, and 1 minute at 72°C in a PTC-225 Peltier thermal cycler (MJ Research). Microsatellites were analyzed for allelic loss using the Genotyper program (ABI). Allelic loss at each marker locus was considered to be present if the area under one allelic peak in the tumor was less than 0.5× or greater than 2× that of the other allele, after correcting for the relative allelic areas using the constitutional DNA.
Owing to relatively small amounts of DNA available from each polyp, APC mutation screening was restricted to regions F, G, H, I, and J of exon 15 (approximately codons 1147 to 1693). Each of these regions was amplified in the PCR using previously published primers 13 (or slight modification thereof, details available from authors). Samples were screened for mutations by capillary based fluorescence-single stranded conformational polymorphism analysis (F-SSCP) using the ABI 3100 system. Briefly, fluorescently labeled oligonucleotides were used to generate labeled PCR products of which 1 to 2 μl were combined with 1 μl of size standard, 10.5 μl of deionized formamide, and 0.5 μl of 0.3 N sodium hydroxide. Samples were denatured and analyzed on 2% Genescan polymer with 10% glycerol at 18°C, 22°C, 26°C, 30°C, and 35°C. Data were analyzed using ABI Genescan and Genotyper software. Direct sequencing in forward and reverse orientations was performed on a new PCR product from those samples that showed a mobility shift or extra banding on F-SSCP, alongside a normal control.
For assessment of germline genotype-phenotype associations in duodenal FAP, data were collected from a series of patients with classical colonic disease, from age 25, who were undergoing routine upper gastrointestinal surveillance at St. Mark’s Hospital. Patients were examined with a side-viewing endoscope between April 1989 and March 2000. Duodenal severity was retrospectively assessed from the most recent endoscopy and histology reports and scored by Spigelman stage. We examined the relationship between carefully assessed esphago-gastro-duodenoscopy findings, patient demographic characteristics (age at endoscopy, sex), and site of the germline APC mutation. A group of 245 patients with classical colonic FAP, of whom 129 had known germline APC mutations, was analyzed; patients without identified mutations were included in the analysis because they provided useful information, for example as regards the age dependence of disease severity. Statistical computations were performed using STATA (Version 7.0; Stata Corporation, College Station, TX). We performed logistic regression analysis to examine the relationship between duodenal FAP severity (the response variable) and the explanatory variables age, sex, and APC mutation position. Age was computed as an integer, days of life. APC mutations were grouped by functional protein domain (pre-armadillo region, codons 168 to 453, n = 32; armadillo repeat region, codons 454 to 1019, n = 15; β-catenin-binding region, codons 1020 to 1168, n = 27; post-β-catenin binding, codons 1169 to 1250, n = 13; colorectal MCR, codons 1250 to 1400, n = 32; postcolorectal MCR, codons 1400 to 1580, n = 10) and then coded as binary dummy variables for computational analysis.
Results
Somatic mutations, including allelic loss, were detected in 9 of the 49 adenomatous duodenal polyps studied (Table 1) ▶ . Notably, all six truncating mutations were distal to codon 1390 and all but one were distal to codon 1400 (Figure 2) ▶ . Additionally, three polyps from one patient showed allelic loss at APC; in each case, the germline wild-type allele was lost (Figure 2) ▶ . Subject to the limitations of this sample size, no significant associations were found between the site or type of the germline or somatic mutation and either polyp size or morphology (details not shown). Our data (Table 1) ▶ did, however, suggest that patients with germline mutations later in the APC gene tended to show allelic loss in their duodenal polyps, whereas other patients’ tumors harbored truncating mutations. This association was formally significant (Fisher’s exact test, P < 0.012), using a cutoff point for the germline mutation of codon 1400 (corresponding to the end of the second β-catenin binding/degradation repeat in APC) and assuming that allelic losses in different tumors from the same patient were independent events.
Table 1.
Upper Gastrointestinal Tumours from FAP Patients with Identified Germline Mutation and Somatic Mutation [Truncating Change or Allelic Loss (LOH)]
| Tumor | Patient | Germline mutation | Somatic mutation | |
|---|---|---|---|---|
| This study | 1 | FSE | 1392 | Q1447X |
| 2 | FON | 1309 | 1554 (4698–4699insA) | |
| 3 | OET | 1154 | S1392X | |
| 4 | FTS | 1061 | R1450X | |
| 5 | VCR | 1061 | 1554 (4698–4699insA) | |
| 6 | SET | 1194 | R1450X | |
| 7 | JWI | 1464 | LOH | |
| 8 | JWI | 1464 | LOH | |
| 9 | JWI | 1464 | LOH | |
| Andersen et al 17 | 1 | 1339 | 1513 | |
| 2 | 739 | 1556 | ||
| 3 | 1465 | LOH | ||
| 4 | 1465 | 1350 | ||
| 5 | 1157 | 1358 | ||
| Gallinger et al 16 | 1 | 1309 | 1465 | |
| 2 | 1309 | 1464 | ||
| Bapat et al 15 | 1 | 1309 | 1465 | |
| 2 | 1309 | 1464 | ||
| Abraham et al 2 | 1 | 1055 | 1554 | |
| 2 | 1309 | 1554 | ||
| 3 | 1309 | 1554 | ||
| 4 | 1309 | 1554 | ||
| 5 | 1055 | 1554 | ||
| 6 | 1055 | 1554 | ||
| 7 | 1546 | LOH | ||
| 8 | 1546 | LOH | ||
| 9 | 1546 | LOH | ||
| 10 | 1059 | 1554 | ||
| 11 | 1030 | LOH |
| Germline <1400 | Germline >1400 | Total | |
|---|---|---|---|
| LOH | 1 | 7 | 8 |
| No LOH | 20 | 1 | 21 |
| Total | 21 | 8 | 29 |
Top:Summary of data from this study and previously published data. Abraham et al 2 analyzed gastric fundus polyps. All other studies analyzed duodenal polyps. Only polyps with an identified second hit are used for this analysis so that potential methodological problems, for example from contaminating nonneoplastic tissue, are minimized. Germline mutation position and the site of somatic mutation are shown. For this study only, the precise nature of the somatic mutation is shown; specific types of other mutations can be found in the references cited, although all are truncating.
Bottom: The association between allelic loss and germline mutation after codon 1400 is highly significant (P < 5 × 10−5). The association remains significant if the somatic LOH events or truncating mutations are assumed not to be independent and the analysis is performed by patient rather than by tumour (Fisher’s exact test, P < 0.016).
Figure 2.
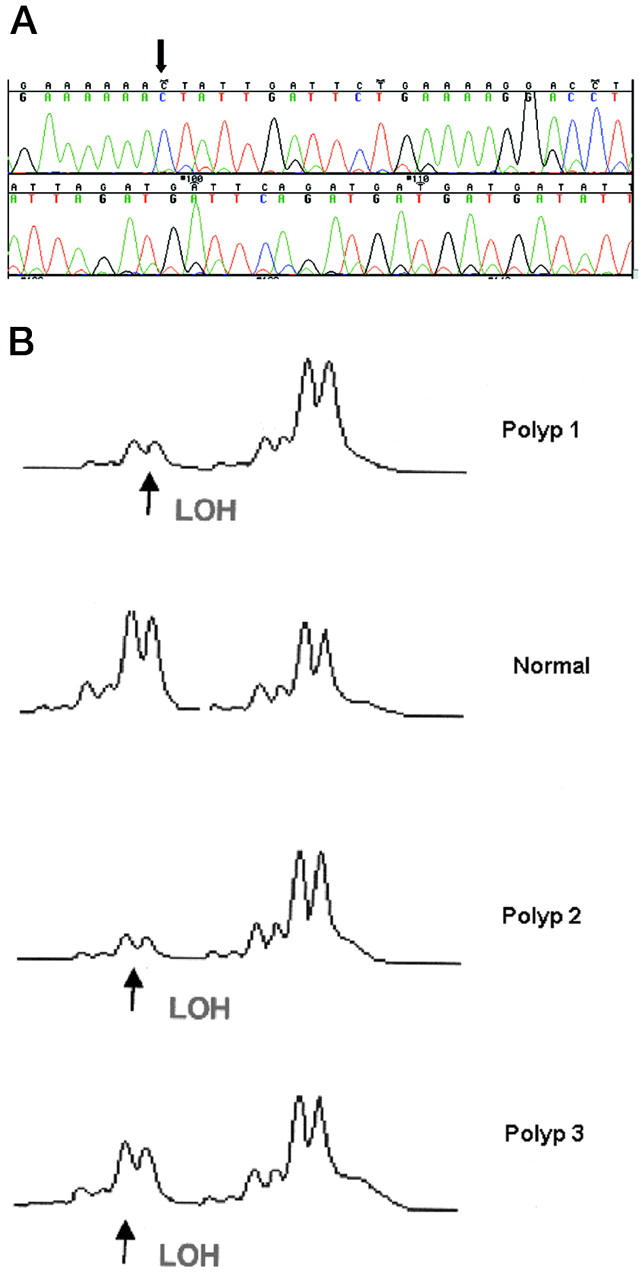
APC mutations. A: 4698-4699insA at codon 1554 (arrows) from duodenal polyp from patient VCR. Note that the frame-shifted sequence is reduced in intensity compared with the wild-type sequence because this polyp contained ∼50% neoplastic tissue and hence mutant allele dosage is only ∼25% of the total. B: Loss of wild-type allele at D5S656 in three duodenal polyps from patient JWI with a germline mutation at codon 1464.
We then tested our finding of a first hit-second hit association in upper gastrointestinal FAP by analyzing previously published data on APC mutations. 14 Although nearly 80 truncating somatic mutations have been detected in upper gastrointestinal polyps in FAP, the germline APC mutation has only been reported in a few of these cases. Moreover, many studies have not analyzed tumors for both truncating mutations and allelic loss. The studies that have reported both germline mutations and second hits in upper gastrointestinal FAP polyps (total of 29 tumors) are summarized in Table 1 ▶ ; 2,15-17 for the purposes of this analysis, we combined data from duodenal (including peri-ampullary) adenomas, gastric adenomas, and gastric fundic polyps. Confirming our own data, there was a highly significant tendency for tumors from patients with germline mutations after codon 1400 to show allelic loss (Fisher’s exact test, P < 0.00004). The tumors of other patients tended to harbor truncating mutations after codon 1400.
Using the analogy of colorectal polyposis, we then tested the hypothesis that somatic APC mutations in upper gastrointestinal FAP tended to cluster after codon 1400. Inspection of the location of somatic APC mutations reported in duodenal and gastric polyps 14 showed clear clustering between codons 1400 and 1580 (Figure 3 ▶ , Table 2 ▶ ); 74% of mutations occurred in this region, which we term the “upper gastrointestinal MCR.” There was a highly significant difference between the distributions of mutations in tumors from the upper and lower gastrointestinal tracts (Table 2 ▶ , Figure 3 ▶ ). This difference was also seen when gastric and duodenal tumors were compared separately with the colorectal tumors (Table 2) ▶ . There was no significant difference between the location of mutations in gastric and duodenal tumors (Fisher’s exact test, P > 0.07).
Figure 3.

Comparison between frequencies of truncating somatic mutations within different regions of the APC gene in colorectal (filled bars) and upper gastrointestinal tumors (open bars) from FAP patients and sporadic cases.
Table 2.
Difference Between Location of Truncating Somatic APC Mutations in Colorectal and Upper Gastrointestinal Polyps from FAP Patients and Sporadic Cases
| Colorectal | Gastric | Duodenal | |
|---|---|---|---|
| Codons 1-1200 | 89 | 11 | 2 |
| Codons 1201-1399 | 230 | 3 | 4 |
| Codons 1400-1580 | 232 | 33 | 23 |
Colorectal versus all upper gastrointestinal: χ22 = 33.2, P < 0.0001.
Colorectal versus gastric: χ22 = 23.0, P < 0.0001.
Colorectal versus duodenal: χ22 = 14.7, P < 0.001 (Yates’ correction).
Extending the analogy with colorectal tumors in FAP, 6 our results suggested that germline APC mutations distal to approximately codon 1400 might be associated with more severe upper gastrointestinal polyposis; such patients comprise fewer than 10% of all FAP cases (although they may be overrepresented in molecular studies of upper gastrointestinal tumors, possibly because they do have more severe disease). We therefore analyzed 245 FAP patients (119 male and 126 female, 129 with known germline mutations) who had been screened routinely for duodenal polyposis. Mean age at most recent endoscopy was 42.0 years (median, 40.2; inter-quartile range (IQR), 32.7 to 51.3 years). One hundred and ten patients had mild disease (Spigelman score 0, I, II), and 135 had severe disease (score III, IV, carcinoma). 18 A statistically significant association was observed between disease severity and age, with a mild tendency for disease to be more severe [odds ratio (OR), = 1.033; P = 0.007] in older patients (Table 3) ▶ . As predicted by our analysis of somatic mutations, mutations within the postcolorectal MCR (after codon 1400) were indeed associated with the greatest risk of severe disease (OR = 5.72, P = 0.035), after correction for age and sex. Additionally, however, raised risk was also observed for patients carrying mutations within the pre-armadillo region (OR = 2.55, P = 0.034) and β-catenin-binding region (OR = 3.27, P = 0.014), with reduced risk for germline mutations within the post-β-catenin-binding region (OR = 0.190, P = 0.039).
Table 3.
Summary Table of Multiple Logistic Regression Analysis of Explanatory Variables for Probability of Severe Duodenal FAP
| Variable | Odds ratio | Standard error | Z | P | 95% CI |
|---|---|---|---|---|---|
| Age (year) | 1.033 | 0.012 | 2.70 | 0.007 | 1.01–1.06 |
| Sex | 1.025 | 0.283 | 0.09 | 0.929 | 0.57–1.76 |
| Pre-armadillo | 2.552 | 1.129 | 2.12 | 0.034 | 1.07–6.08 |
| Armadillo | 1.275 | 0.719 | 0.43 | 0.666 | 0.42–3.85 |
| β-Catenin | 3.273 | 1.586 | 2.45 | 0.014 | 1.27–8.46 |
| Post β-catenin | 0.190 | 0.153 | −2.06 | 0.039 | 0.04–0.92 |
| Colorectal MCR | 1.616 | 0.670 | 1.16 | 0.248 | 0.72–3.64 |
| Post-colorectal MCR | 5.715 | 4.725 | 2.11 | 0.035 | 1.13–28.89 |
Parameters were estimated by the method of maximum likelihood (log likelihood = −153.8). The likelihood ratio test (full-model versus constant-only model) was χ2 = 29.40, P = 0.0003. Apart from age and sex, variable refers to the position of the germline mutation within the APC gene.
Discussion
We have shown that there is a somatic APC MCR for upper gastrointestinal tumors that is located approximately between codons 1400 and 1580 (Table 1) ▶ . The type of somatic mutation in upper gastrointestinal polyps depends on the site of the germline APC mutation, with germline mutations after codon 1400 associated with allelic loss in tumors. As a counterpoint to these findings, germline mutations after codon 1400 are associated with more severe duodenal polyposis. Mutations in other regions of the APC gene may also be associated with different severities of upper gastrointestinal disease. Knowledge of the position of the germline APC mutation may benefit the clinical management of duodenal and gastric FAP. For example, the interval of screening could be influenced by the mutation position, with more frequent screening being offered to certain patients, particularly those with germline APC mutations distal to codon 1400.
Analysis and interpretation of these data are inevitably subject to certain caveats. First, we have found an apparently low frequency of somatic APC mutations. Our data are, however, consistent with previous studies. 16,17 The low mutation detection rate probably results in part from studying small lesions, which may have contained less neoplastic material than estimated by morphology (for example, sections taken may have been unrepresentative of the neoplastic content of the polyp as a whole, or normal crypts may have been entrapped within adenomas 19 ) and from screening only part of the APC gene for mutations. However, F-SSCP is generally recognized to be highly sensitive and we detected all previously identified germline mutations and known common polymorphisms in the regions we analyzed. We cannot, therefore, entirely exclude the possibility that early FAP tumors may start to grow without a second hit. 20
Second, many studies have been unable to analyze the whole of the APC gene for genetic changes and/or have focused on specific regions of the gene, with many studies of colorectal tumors failing to analyze mutations 3′ of codon 1500. Moreover, different methods of mutation detection have been used for different regions of the gene (for example, protein truncation test for exon 15 and SSCP or denaturing gradient gel electrophoresis for other exons). Perhaps the best standardized comparison of colorectal and upper gastrointestinal tumors is provided by the data of Toyooka and colleagues 12 and Miyaki and colleagues 21 who screened for mutations in exons 5 to 9 and 13 to 15I in upper gastrointestinal tumors and, as far as can be ascertained, analyzed the same region by the same methods in colorectal tumors. These authors’ data show 73 somatic mutations in colorectal tumors before codon 1400, compared with 107 mutations after this codon; for upper gastrointestinal tumors, the numbers are zero and 47 mutations, respectively, a highly significant difference from the colorectal lesions (Fisher’s exact test, P < 7 × 10−10).
Third, we have combined data from duodenal and gastric tumors when analyzing first hit-second hit associations using previously published data. The very similar distributions of somatic mutations in gastric and duodenal tumors (Table 2) ▶ support the use of this approach. Furthermore, we have combined data from gastric fundic polyps and gastric adenomas in our analyses, based on evidence that both these lesions harbor second hits at APC. 2 Fourth, our use of codon 1400 as a proximal boundary for the MCR and for association with allelic loss is heuristic and inevitably an approximation, although it does correspond to a functional domain of APC. We have also made the plausible assumption that the distal boundary for somatic mutations APC is at approximately codon 1580, before the APC SAMP repeats, although some families with germline mutations distal to this site have developed severe duodenal disease. 9 Fifth, the site of the germline mutation evidently only explains part of the variation in the severity of duodenal FAP; modifying genes, environment, and chance may also have effects.
Despite these reasons for caution, our data from upper gastrointestinal FAP tumors consistently show that the same region of the APC gene (approximately codons 1400 to 1580) forms the somatic MCR, harbors mutations associated with allelic loss, and contains germline mutations associated with severe disease. Although this region seems to be similar in upper gastrointestinal FAP tumors and desmoids, 6 the corresponding region in colorectal tumors lies in the 5′ direction, around codon 1300. 4-6 We suspect that the reason for the different regions in colorectal and upper gastrointestinal tumors may lie in the effects of the resulting truncated proteins on β-catenin levels within the cell. 22,23 Tumorigenesis may benefit most from a level of β-catenin that is raised above normal, yet is not excessive. 22 It is likely that a truncated APC species with an extra β-catenin-binding/degradation repeat, as would typify mutant APC in upper gastrointestinal polyps and desmoids relative to colorectal tumors, would result in a lower level of β-catenin in the cell. Thus, the progenitor cells of in upper gastrointestinal polyps and desmoids would require a lower, specific level of β-catenin for them to produce a tumor. Other explanations for the differing MCRs include effects on stability of the truncated protein, 7 although explanations in terms of hypermutability of certain regions of APC 24 seem less likely.
Associations between first and second hits seem, therefore, to be a general feature of the APC tumor suppressor gene. It is likely that this phenomenon results because different selective advantages are associated with different APC genotypes. Tumors with the optimal genotypes grow more rapidly and are more likely to come to clinical attention than tumors with suboptimal genotypes. As we have hypothesized for colorectal disease, one of the reasons for the greater severity of upper gastrointestinal disease in patients with mutations after codon 1400 may be that susceptible cells from these individuals can readily acquire a strongly selected genotype by allelic loss, which occurs, by chance, more frequently than specific truncating mutations after codon 1400. 22 A particularly interesting aspect of investigating these phenomena and their causes will be to determine why tumors arising from different tissues show different patterns of APC mutation. The answer to this question may help to explain what is perhaps the great conundrum of tumorigenesis, namely why genes with widespread expression are only associated with tumors of specific sites.
Acknowledgments
We thank Nicholas A. Wright for invaluable help with histological review of polyps; the Equipment Park, Imperial Cancer Research Fund, for microsatellite genotyping gel running; and the APC Mutation Database (http://perso.curie.fr/thierry. soussi/apc.html), which was essential for this study.
Footnotes
Address reprint requests to Ian Tomlinson, Cancer Research UK, Molecular and Population Genetics Laboratory, 44, Lincoln’s Inn Fields, London WC2A 3PX, UK. E-mail: i.tomlinson@cancer.org.uk.
C. G., H. L., and M. C. contributed equally to this work.
References
- 1.Spigelman A, Phillips R: Surveillance of the duodenum in patients with familial adenomatous polyposis. Gut 1998, 42:144-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham SC, Nobukawa B, Giardiello FM, Hamilton SR, Wu TT: Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic adenomatous polyposis coli gene alterations. Am J Pathol 2000, 157:747-754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spigelman AD, Talbot IC, Penna C, Nugent KP, Phillips RK, Costello C, DeCosse JJ: Evidence for adenoma-carcinoma sequence in the duodenum of patients with familial adenomatous polyposis. The Leeds Castle Polyposis Group (Upper Gastrointestinal Committee). J Clin Pathol 1994, 47:709-710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nugent K, Phillips R, Hodgson S, Cottrell S, Smith-Ravin J, Pack K, Bodmer W: Phenotypic expression in familial adenomatous polyposis: partial prediction by mutation analysis. Gut 1994, 35:1622-1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y: Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992, 1:229-233 [DOI] [PubMed] [Google Scholar]
- 6.Lamlum H, Ilyas M, Rowan A, Clark S, Johnson V, Bell J, Frayling I, Efstathiou J, Pack K, Payne S, Roylance R, Gorman P, Sheer D, Neale K, Phillips R, Talbot I, Bodmer W, Tomlinson I: The type of somatic mutation at APC in FAP is determined by the site of the germline mutation: a new facet to Knudson’s ‘two-hit’ hypothesis. Nat Med 1999, 5:1071-1075 [DOI] [PubMed] [Google Scholar]
- 7.Dihlmann S, Gebert J, Siermann A, Herfarth C, von Knebel Doeberitz M: Dominant negative effect of the APC1309 mutation: a possible explanation for genotype-phenotype correlations in familial adenomatous polyposis. Cancer Res 1999, 59:1857-1860 [PubMed] [Google Scholar]
- 8.Polakis P: The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta 1997, 1332:F127-F147 [DOI] [PubMed] [Google Scholar]
- 9.Leggett BA, Young JP, Biden K, Buttenshaw RL, Knight N, Cowen AE: Severe upper gastrointestinal polyposis associated with sparse colonic polyposis in a familial adenomatous polyposis family with an APC mutation at codon 1520. Gut 1997, 41:518-521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedl W, Caspari R, Sengteller M, Uhlhaas S, Lamberti C, Jungck M, Kadmon M, Wolf M, Fahnenstich J, Gebert J, Moslein G, Mangold E, Propping P: Can APC mutation analysis contribute to therapeutic decisions in familial adenomatous polyposis? Experience from 680 FAP families. Gut 2001, 48:515-521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjork J, Akerbrant H, Iselius L, Bergman A, Engwall Y, Wahlstrom J, Martinsson T, Nordling M, Hultcrantz R: Periampullary adenomas and adenocarcinoma in familial adenomatous polyposis: cumulative risks and APC gene mutations. Gastroenterology 2001, 121:1127-1135 [DOI] [PubMed] [Google Scholar]
- 12.Toyooka M, Konishi M, Kikuchi-Yanoshita R, Iwama T, Miyaki M: Somatic mutations of the adenomatous polyposis coli gene in gastroduodenal tumors from patients with familial adenomatous polyposis. Cancer Res 1995, 55:3165-3170 [PubMed] [Google Scholar]
- 13.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M: Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66:589-600 [DOI] [PubMed] [Google Scholar]
- 14.Beroud C, Soussi T: APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res 1996, 24:121-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bapat B, Odze R, Mitri A, Berk T, Ward M, Gallinger S: Identification of somatic APC gene mutations in periampullary adenomas in a patient with familial adenomatous polyposis (FAP). Hum Mol Genet 1993, 2:1957-1959 [DOI] [PubMed] [Google Scholar]
- 16.Gallinger S, Vivona AA, Odze RD, Mitri A, O’Beirne CP, Berk TC, Bapat BV: Somatic APC and K-ras codon 12 mutations in periampullary adenomas and carcinomas from familial adenomatous polyposis patients. Oncogene 1995, 10:1875-1878 [PubMed] [Google Scholar]
- 17.Norheim Andersen S, Lovig T, Fausa O, Rognum TO: Germline and somatic mutations in exon 15 of the APC gene and K-ras mutations in duodenal adenomas in patients with familial adenomatous polyposis. Scand J Gastroenterol 1999, 34:611-617 [DOI] [PubMed] [Google Scholar]
- 18.Tomlinson IPM, Neale K, Talbot IC, Spigelman AD, Williams CB, Phillips RKS, Bodmer WF: A modifying locus for familial adenomatous polyposis may be present on chromosome 1p35–p36. J Med Genet 1996, 33:268-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Novelli MR, Williamson JA, Tomlinson IPM, Elia G, Hodgson SV, Talbot IC, Bodmer WF, Wright NA: Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 1996, 272:1187-1190 [DOI] [PubMed] [Google Scholar]
- 20.Bodmer W: Familial adenomatous polyposis (FAP) and its gene, APC. Cytogenet Cell Genet 1999, 86:99-104 [DOI] [PubMed] [Google Scholar]
- 21.Miyaki M, Konishsi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, Muraoka M, Takahashi H, Amada Y, Fukayama M, Maeda Y, Iwama T, Mishima Y, Mori T, Koike M: Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumours. Cancer Res 1994, 54:3011-3020 [PubMed] [Google Scholar]
- 22.Lamlum H, Papadopoulou A, Ilyas M, Rowan A, Gillet C, Hanby A, Talbot I, Bodmer W, Tomlinson I: APC mutations are sufficient for the growth of early colorectal adenomas. Proc Natl Acad Sci USA 2000, 97:2225-2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HC, Wheeler JM, Kim JC, Ilyas M, Beck NE, Kim BS, Park KC, Bodmer WF: The E-cadherin gene (CDH1) variants T340A and L59 9V in gastric and colorectal cancer patients in Korea. Gut 2000, 47:262-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spirio L, Samowitz W, Robertson J, Robertson M, Burt R, Leppert M, White R: Alleles of APC modulate the frequency and classes of mutations that lead to colon polyps. Nat Genet 1998, 20:385-388 [DOI] [PubMed] [Google Scholar]