

Abstract
Macrophage-derived foam cells in developing atherosclerotic lesions may potentially originate either from recruitment of circulating monocytes or from migration of resident tissue macrophages. In this study, we have determined the source of intimal macrophages in the apoE-knockout mouse flow-cessation/hypercholesterolemia model of atherosclerosis using a bone marrow transplantation approach. We also examined the time course and spatial distribution of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression to assess whether endothelial adhesion molecules were involved in recruitment of either circulating monocytes or resident macrophages. We used allelic variants of the mouse common leukocyte antigen (CD45) to distinguish host-derived and donor-derived white blood cells (WBCs) both in blood and in macrophage-rich carotid lesions. We found that the distribution of CD45 isoforms in lesions is similar to that of circulating WBCs, whereas the host-type CD45 isoform is more prevalent in resident adventitial macrophages. These data indicate that macrophage-derived foam cells in the lesion derive mainly from circulating precursors rather than from resident macrophages. The corresponding time course of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expression suggests that recruitment of circulating WBCs by endothelial adhesion molecules is likely to be more important during lesion initiation than during the later phase of rapid lesion growth.
Both in experimental models of atherosclerosis and in human disease, infiltration of macrophages into the arterial intima constitutes one of the earliest cellular events in the development of atherosclerotic lesions. 1-3 Macrophages in the lesion may be derived either from cells already resident in the arterial wall 4 or from circulating monocytes that have undergone diapedesis. Because the mechanisms of cellular recruitment and/or activation are likely to differ depending on the source of the inflammatory cells, determining the relative contribution to lesion growth of cells from these two sources may have important implications in devising successful strategies to slow the growth of lesions.
Although much convincing evidence demonstrates the importance of blood-borne monocytes in early lesion development, 1,5,6 the potential contribution of resident macrophages has been more difficult to assess. Experiments involving injection of labeled tracer monocytes can provide estimates for the rate of recruitment of circulating inflammatory cells but do not directly address the issue of mobilization of macrophages already present in arterial tissue. Moreover, alterations in adhesion molecule expression that can occur during monocyte isolation and labeling may modify the interaction between circulating monocytes and the vascular wall. The recent development of a polymerase chain reaction-based method of quantifying monocyte recruitment has shown promise of improved sensitivity and ease of quantitation compared to more traditional approaches such as labeling injected cells with radioisotopes or with fluorescent dyes, 6 but this method has not yet been adapted to the purpose of measuring resident macrophage recruitment. A recent study in which adventitial macrophages were fluorescently labeled in vivo by direct application of a dye solution provided evidence of a possible role for adventitial macrophage recruitment in the development of porcine coronary lesions. 4
In the present work, we have developed a novel approach to assess the relative contribution of resident versus circulating inflammatory cells to the development of macrophage-rich arterial lesions in the apoE knockout (KO) mouse using bone marrow transplantation (BMT). We have taken advantage of naturally occurring allelic variants of the mouse common leukocyte antigen (CD45 or Ptprc) to distinguish between host- and donor-derived white blood cells (WBCs) both in blood and in macrophage-rich carotid lesions. CD45, a transmembrane glycoprotein having intracellular tyrosine phosphatase activity, 7 is expressed ubiquitously by nonerythroid hematopoietic cells. Using specific antibodies to two CD45 isoforms, we have been able to differentiate tissue-resident (host-derived) inflammatory cells from blood-borne (bone marrow- or donor-derived) cells in developing carotid lesions.
Vascular cells produce a number of adhesion molecules, including intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule(VCAM)-1, and P-selectin, which are thought to play a role in the recruitment of inflammatory cells to developing atherosclerotic lesions. 8-11 To assess whether recruitment of resident and circulating WBCs may depend differently on presentation of adhesion molecules by vascular cells, we have also examined the time course and spatial distribution of ICAM-1 and VCAM-1 expression in carotid lesions in relation to inflammatory cell infiltration.
Materials and Methods
Materials
Monoclonal antibodies (mAbs) against mouse CD45.1 (clone A20), CD45.2 (clone 104), CD31 (PECAM-1; clone MEC 13.3), CD54 (ICAM-1; clone 3E2), CD106 (VCAM-1; clone 429), and Mac-3 (clone M3/84) were obtained from BD PharMingen (San Diego, CA). Nile Red and Alexa Fluor 488-streptavidin were purchased from Molecular Probes (Eugene, OR). Rhodamine Red X-conjugated secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). Monoclonal rat anti-bromodeoxyuridine (BrdU) was purchased from Abcam, Ltd. (Cambridge, UK). Polyclonal rabbit anti-human CD3ε that cross-reacts with mouse T lymphocytes and monoclonal mouse anti-smooth muscle α-actin (clone 1A4) were from Sigma Chemical Co. (St. Louis, MO). Injectable busulfan (Busulfex) was obtained from Orphan Medical (Minnetonka, MN). Custom primers for polymerase chain reaction were prepared by Invitrogen (Rockville, MD). An atherogenic diet containing 1.25 wt% cholesterol, 0.5 wt% cholate, and 35 kcal% fat was purchased from Research Diets (New Brunswick, NJ).
Animal Strains and Genotyping
ApoE KO mice in the C57BL/6 background (B6.129P2-Apoetm1Unc) and mice in the same background homozygous for the CD45.1 alloantigen (B6.SJL-PtprcaPep3b/BoyJ) were purchased from Jackson Laboratories (Bar Harbor, ME). ApoE KO mice homozygous for the CD45.1 alloantigen (ApoE PepBoys) were obtained by crossbreeding the two strains. These mice were confirmed as apoE KOs by polymerase chain reaction of genomic DNA. Mouse genomic DNA was isolated from 1-cm tail samples using the Wizard Genomic DNA kit (Promega, Madison, WI). ApoE KO genotyping was performed using the primer set recommended by the developers of this strain. 12 Phenotypic analysis of CD45 alloantigen expression was performed by flow cytometry of peripheral blood WBCs as described below.
Bone Marrow Transplantation
Groups of CD45.1 apoE KO recipient mice were transplanted with bone marrow obtained from CD45.2 apoE KO donor mice. Donor mice and recipient mice were sex- and age-matched. Before BMT, recipient mice were injected intraperitoneally with 20 μg of busulfan/g body weight for 4 consecutive days to ablate the host bone marrow. 13 Bone marrow was harvested by flushing the femurs and tibias of donor mice with RPMI 1640 supplemented with 1% fetal bovine serum, 1% l-glutamine, and penicillin/streptomycin (transplant medium). Harvested bone marrow cells were washed twice with the same medium, diluted to a final concentration of 2.5 × 107 cells/ml, and injected into recipient mice via the retro-orbital venous sinus 18 to 24 hours after the final busulfan injection. The extent of donor bone marrow engraftment was assessed at 2 and 6 weeks after transplant by flow cytometric analysis of peripheral blood WBCs.
Flow Cytometry
For flow cytometric analysis, peripheral blood WBCs were isolated by gravity sedimentation of whole blood on 3% dextran in Hanks’ balanced salt solution followed by red blood cell lysis by hypotonic shock. The WBCs were resuspended in fluorescence-activated cell sorting (FACS) staining medium (phenol red-free Hanks’ balanced salt solution supplemented with 6% fetal bovine serum and 0.01 mol/L Na2-ethylenediaminetetraacetic acid). Isolated peripheral WBCs were stained for 30 minutes at 4°C in the dark with fluorescein isothiocyanate-labeled anti-mouse CD45.2 and R-phycoerythrin-labeled anti-mouse CD45.1 (1:100) before FACS analysis, then washed once with FACS staining medium, and resuspended in the same medium. Samples were analyzed by flow cytometry on a Becton-Dickinson FACSCalibur (Becton-Dickinson, Mountain View, CA). Between 5 × 105 and 12 × 105 total events were collected for analysis of engraftment of each transplanted mouse. Flow cytometric data were gated to exclude residual red blood cells and cell aggregates. Fractional engraftment was calculated as (CD45.2-positive WBCs)/(CD45.2-positive WBCs + CD45.1-positive WBCs) × 100%.
Experimental Model of Atherosclerosis
Approximately 8 weeks after BMT, formation of arterial lesions was induced in recipient mice by ligation of the left common carotid artery according to our previously published method 14 and the mice were placed on an atherogenic diet. Groups of four to eight mice were sacrificed by means of CO2 asphyxiation after 3, 7, or 14 days on diet. The 0-day group consisted of mice that had received BMT but that had not been ligated. For analysis of cellular proliferation in carotid lesions, groups of three to four mice at each time point were injected intraperitoneally with the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) (250 μl of 10 mg/ml in normal saline) at 12 hours and 1 hour before sacrifice. Blood samples were obtained from the retro-orbital venous sinus for analysis of total plasma cholesterol using the Infinity Cholesterol Reagent (Sigma Diagnostics). After sacrifice, the vasculature was cleared of blood by brief pressure perfusion with normal saline via the left ventricle, with outflow through the severed vena cava. Tissue samples (spleen, liver, lung, carotids) were dissected out, embedded in OCT medium, and frozen in liquid nitrogen with 2-methylbutane as heat transfer fluid. Both carotids were removed together with the aortic arch and embedded in a vertical orientation. All animal protocols were approved by the Emory University Institutional Animal Care and Use Committee.
Tissue Preparation and Immunohistochemistry
Frozen sections were cut at 7 μm and collected on Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA). We sectioned the entire length of each carotid artery from its origin at the aortic arch to the vicinity of the ligation. Serial sections for immunohistochemistry were collected in groups of seven with intervals of 140 μm between the first section in each group. To determine which groups of serial sections encompassed the apex of the lesion, one section out of each group of 40 was examined by phase contrast microscopy.
Serial frozen sections of carotid lesions were fixed in ice-cold acetone and stained with biotinylated mAbs to CD45.1 or CD45.2 (1:20 dilution) followed by Alexa Fluor 488-labeled streptavidin to determine the distribution and relative abundance of host-type and donor-type WBCs, respectively. Nuclei were counterstained with Hoechst 33258. Sections from carotid lesions of apoE PepBoys that had not undergone BMT were stained identically for comparison. To assess the cellular composition of lesions in the transplanted mice, additional serial sections were immunostained with cell type-specific antibodies including anti-Mac3 for monocytes/macrophages, anti-smooth muscle α-actin for smooth muscle cells (SMCs), anti-CD3 for T lymphocytes, and anti-CD31 for endothelial cells. Some frozen sections were fixed in 4% paraformaldehyde and stained with Nile Red to visualize the distribution of lipids in the lesions.
Adhesion molecule expression was evaluated by staining acetone-fixed frozen sections with primary mAbs to ICAM-1 (1:50 dilution) or VCAM-1 (1:50 dilution) followed by Rhodamine Red X-labeled secondary antibodies for detection by immunofluorescence microscopy.
Cell proliferation in carotid lesions was analyzed by staining paraformaldehyde-fixed frozen sections using a rat mAb to BrdU (1:20) followed by detection with a Rhodamine Red X-labeled anti-rat IgG. Some sections were stained simultaneously with anti-BrdU and biotinylated anti-CD45.1 or anti-CD45.2 mAbs for analysis of host- and donor-specific WBC proliferation. Total intimal cell numbers were determined by semiautomated image analysis of Hoechst-stained nuclei. Intimal proliferation index was defined as (number of BrdU-stained intimal nuclei)/(total number of intimal nuclei) × 100%.
Image Analysis and Morphometry
For morphometric analysis, frozen sections obtained from the apex of the carotid lesion were stained with anti-CD31 to visualize the luminal boundary as defined by the endothelium. Luminal area was measured by tracing this boundary. The elastic laminae were visualized by means of their autofluorescence using a fluorescein isothiocyanate filter set. Internal elastic lamina (IEL) area was measured by tracing the contour of the IEL. Lesion (intimal) area was calculated as the difference between IEL area and luminal area. External elastic lamina (EEL) area was measured as the area encompassed by the outermost elastic lamina. Images were acquired using a Zeiss Axioskop fluorescence microscope equipped with a Photonic Science cooled CCD camera. All image analysis was performed using ImagePro Plus software (Media Cybernetics, Silver Spring, MD).
Statistical Procedures
Student’s t-test was used for comparison of means between groups at each time point. A value of P < 0.05 was considered significant.
Results
Mouse Breeding
We successfully established a strain of apoE KO mice homozygous for the rare CD45.1 (Ptprca) allele of the mouse common leukocyte antigen. The genotype (apoE KO) and the phenotype (CD45.1 homozygote) of these mice were confirmed by polymerase chain reaction (data not shown) and by FACS analysis of peripheral WBCs, respectively (Figure 1A) ▶ .
Figure 1.

Flow cytometric analysis of isolated peripheral WBCs demonstrates successful engraftment of CD45.2 donor bone marrow in CD45.1 recipient mice. A and B: Isolated peripheral WBCs stained with phycoerythrin-labeled anti-CD45.1 and fluorescein isothiocyanate-labeled anti-CD45.2 mAbs were analyzed by flow cytometry. A: In an apoE KO mouse homozygous for CD45.1, all peripheral WBCs stain positively for CD45.1. B: Six weeks after transplant with bone marrow from an apoE KO mouse homozygous for CD45.2, donor-type WBCs predominate in the circulation (cluster of points on x axis). Additional in vitro experiments with mixed populations of homozygous cells (data not shown) suggest that the double-labeled objects correspond to cell aggregates that account for 6% of gated events on average. These events have been excluded from the calculation of percentage engraftment.
Bone Marrow Transplantation
Engraftment of bone marrow transplanted from apoE KO CD45.2 homozygous donors to apoE KO CD45.1 homozygous recipients after busulfan conditioning followed a uniform time course as measured by flow cytometry of peripheral WBCs. By 2 weeks after transplant, FACS analysis indicated that 63 ± 14% (n = 33) of circulating WBCs were donor-derived (CD45.2-positive). Donor-derived hematopoietic engraftment increased steadily throughout time to a value of 84 ± 7% (n = 37) donor-type cells at 6 weeks after transplant (Figure 1B) ▶ , after which the values approached a plateau. Chimerism of peripheral WBCs at 8 weeks (81 ± 9%, n = 4) was not significantly different from that at 6 weeks. In this study, arterial lesion development was initiated by carotid ligation on average 8 weeks after BMT to ensure that donor-type cells predominated in the circulation.
We measured total plasma cholesterol levels in apoE KO mice that had received BMTs and compared them to levels in nontransplanted apoE KO mice to confirm that BMT in itself did not alter the response to an atherogenic diet. Plasma cholesterol levels were significantly elevated over baseline by 3 days on diet. After 14 days of atherogenic diet, the plasma cholesterol level was not significantly different in transplanted apoE KO mice than in nontransplanted mice (1700 ± 570 mg/dl, n = 9, versus 1830 ± 460 mg/dl, n = 5). Plasma cholesterol levels in transplanted apoE KO mice fed a standard chow diet also were not significantly different from those in nontransplanted mice on the same diet (data not shown).
Time Course of Macrophage-Rich Lesion Development
Carotid lesions developed rapidly in the ligated arteries of ApoE KO transplant mice fed an atherogenic diet (Figure 2) ▶ . No lesions were present in nonligated carotid arteries of the transplanted mice. By 3 days after ligation, inflammatory cell recruitment was evident in some but not all transplanted mice as a partial lining of adherent WBCs at the luminal surface. By 7 days after ligation, all transplanted mice had developed intimal lesions (Figure 2A) ▶ . Lesion area increased dramatically between 7 and 14 days after ligation (Figure 2B) ▶ , leading to nearly complete occlusion of the vessel lumen (Figure 2C) ▶ .
Figure 2.
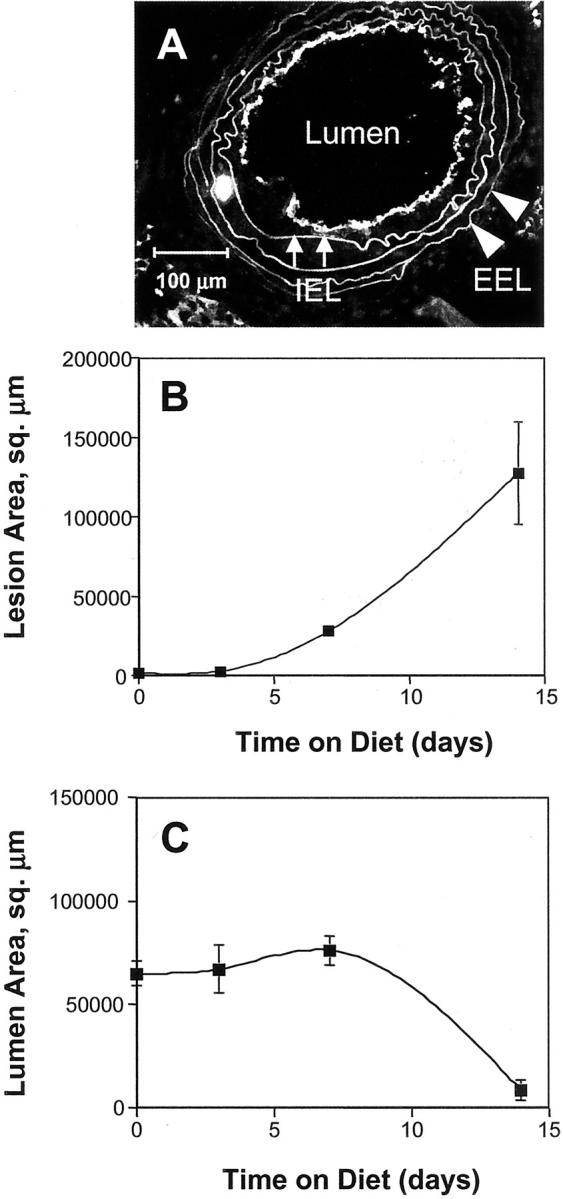
Development of macrophage-rich lesions in ligated carotid arteries of ApoE KO mice that have undergone BMT leads to lumenal occlusion after 14 days on hypercholesterolemic diet. A: Representative frozen section used for morphometric analysis. Scale bar, 100 μm. Morphometric parameters were measured by image analysis of frozen sections immunostained with anti-CD31 to visualize the endothelium as a boundary for the vessel lumen. Elastin autofluorescence was used to identify the IEL (small arrows) and the EEL (arrowheads). Average values of morphometric parameters were calculated for groups of four to six mice sacrificed at the indicated time points after carotid ligation. Morphometric analysis of lesion area (B) and lumen area (C). Error bars, 1 SEM.
Cellular Composition of Carotid Lesions
Immunohistochemical analysis of host and donor WBC distribution in carotid artery lesions of ApoE KO transplant mice showed that the majority of inflammatory cells in the lesions at 7 and 14 days after ligation were donor-derived (CD45.2-positive), as demonstrated by serial sections (Figure 3) ▶ . At 14 days, 85 ± 8% (n = 4) of intimal WBCs were donor-derived, as measured by digital image analysis of immunostained serial sections. Few host-derived WBCs were present in the developing lesions, although some were evident in the adventitia of the ligated arteries.
Figure 3.

Donor-derived inflammatory cells predominate in mouse carotid lesions initiated by flow cessation/hypercholesterolemia. Serial frozen sections were stained without primary antibody (negative controls) or with mAbs to CD45.2 (donor WBCs) or CD45.1 (host WBCs). Sections were counterstained with Hoechst dye (blue) to visualize cell nuclei. Left: Numerous donor-derived (CD45.2) WBCs were evident in the neointima at both 7 and 14 days after ligation. Cells expressing CD45.2 surface antigen have distinct green borders (inset, arrowheads). Middle: In contrast, few host-derived (CD45.1-positive) WBCs were visible in lesions at either time point. Right: In negative controls, only elastin autofluorescence appears green. Scale bar, 100 μm.
Cell type-specific immunohistochemical staining with antibody to Mac-3 demonstrated that the majority of inflammatory cells present in the carotid lesions of ligated, transplanted mice were monocyte/macrophages (Figure 4) ▶ . A few CD3-positive T lymphocytes were present as well (data not shown). At 7 and 14 days after ligation, macrophages in the lesion contained substantial amounts of lipid, indicative of their transition into foam cells. Double labeling with antibodies to Mac-3 and CD45.2 revealed that the distribution of host- and donor-derived macrophages in the vessel changed during the course of lesion development (Figure 4) ▶ . As expected, control animals that had not undergone BMT or carotid ligation showed no staining for donor-type macrophages. Host-type macrophages were identified only in the adventitial layer of the vessel. In transplanted mice before initiation of lesion development, adventitial macrophages comprised a mixture of host- and donor-derived cells. By 3 days after ligation, donor-type monocyte/macrophages had begun to adhere to the luminal endothelium. At 14 days, the majority of macrophages and macrophage-derived foam cells in the neointima of the left carotid were of donor origin, whereas examination of the nonligated, contralateral artery at the same time point revealed a mixture of host- and donor-type macrophages in the adventitia, similar to that observed in the left carotids before ligation. A quantitative analysis revealed that, before ligation, ∼55 ± 3% (n = 4) of adventitial WBCs originated from the donor bone marrow. The adventitial distribution of host- and donor-type WBCs was distinctly different from that in the circulation, where donor-derived cells constituted 78 ± 11% (n = 4) of circulating WBCs at 6 weeks after transplant in the same mice (P = 0.013, two-tailed paired t-test). Thus, before the initiation of atherosclerotic lesions, host-type cells accounted for a significantly greater fraction of tissue-resident, adventitial macrophages than of circulating monocytes.
Figure 4.

Distribution and source (host versus donor) of macrophages in mouse carotid lesions throughout a 14-day time course. Frozen sections of carotid lesions were double labeled with mAbs to Mac-3 and CD45.2 to identify macrophages and donor-type WBCs, respectively. The corresponding lipid distribution was visualized by Nile Red staining. Top left: In mice that had undergone BMT, both host-type (red) and donor-type (yellow) macrophages were evident in the carotid adventitia before ligation (0 day). Top right: By 3 days after ligation, donor-type monocyte/macrophages (yellow) had begun to adhere to the endothelium, initiating neointima formation. Scale bars, 20 μm. Row 2, left: Macrophages occupied ∼35% of the lesion area (red staining) at 14 days after ligation. Row 2, right: CD45.2 mAb recognizes an antigen on the cell surface, resulting in strong labeling along leukocyte borders (green areas). Row 3, left: Superimposing the panels in row 2 produces an image in which donor-type macrophages appear as orange to yellow cells with green or yellow borders. Double labeling demonstrates that the majority of macrophages in the lesion are donor-derived. Row 3, right: Lipid distribution at 14 days after ligation. Scale bars, 100 μm. Controls (bottom left) Only host-type macrophages (red) were present in the adventitia of nontransplanted apoE KO mice homozygous for CD45.1. Right: No lesion is evident in the nonligated, contralateral vessel of transplanted mice. Both host-type (red) and donor-type (yellow) macrophages appear in the adventitia. Scale bar, 100 μm.
Intimal proliferation index increased significantly between 7 and 14 days after ligation, from 3.7 ± 2.6% (n = 3) of intimal cells to 14.8 ± 2.8% (n = 4). Compared to baseline values, the total number of proliferating cells in the neointima was significantly elevated at 14 days after ligation (Table 1) ▶ . Many of these proliferating intimal cells stained positively for CD45.2, establishing their identity as donor-derived WBCs (Figure 5) ▶ . Proliferating donor-derived leukocytes were observed throughout the neointima, with no apparent spatial preference (Figure 5 ▶ ; additional data not shown).
Table 1.
Intimal Proliferation in ApoE Transplant Mice after Carotid Ligation
| Time on diet, days | BrdU-labeled intimal cells, mean ± SEM (n) |
|---|---|
| 0 | 0.2 ± 0.2 (4) |
| 3 | 7.0 ± 3.6 (4) |
| 7 | 7.3 ± 3.4 (3) |
| 14 | 87.2 ± 18.0* (4) |
*P < 0.05 relative to day 0.
Figure 5.

Donor-derived WBCs proliferate in the neointima of mouse carotid lesions. Frozen sections were immunostained with anti-BrdU mAb to identify proliferating cell nuclei (pink) and with anti-CD45.2 mAb to identify donor-derived WBCs (green outlines, arrowheads at right). Nuclei were counterstained with Hoechst 33258. Left: A cross-section of the entire vessel, with a boxed area indicating the region of the close-up highlighted at the right. Scale bar, 20 μm.
Adhesion Molecule Expression in Developing Carotid Lesions
To evaluate possible routes of inflammatory cell recruitment, we examined the time course of adhesion molecule expression in the ligated carotids by means of immunohistochemical staining for ICAM-1 and VCAM-1. The time course of expression and the spatial localization of ICAM-1 were distinctly different from those of VCAM-1 (Figure 6) ▶ . Before the initiation of lesions and at 3 days after ligation (Figure 6) ▶ , VCAM-1 was expressed at the luminal boundary of the vessel, where it co-localized with endothelial cells identified by CD31 staining. By 7 days after ligation, however, VCAM-1 expression had disappeared from the endothelium and had shifted to the media, where VCAM-1 staining co-localized with smooth muscle α-actin. In contrast, the endothelium expressed ICAM-1 throughout the course of carotid lesion development up to 14 days after ligation, although expression at later time points was diminished compared to baseline levels. Adventitial ICAM-1 expression was evident by 3 days after ligation and increased during the course of lesion progression. In addition, ICAM-1 staining was detected on cells in the deeper layers of the neointima during the later stages of lesion development.
Figure 6.
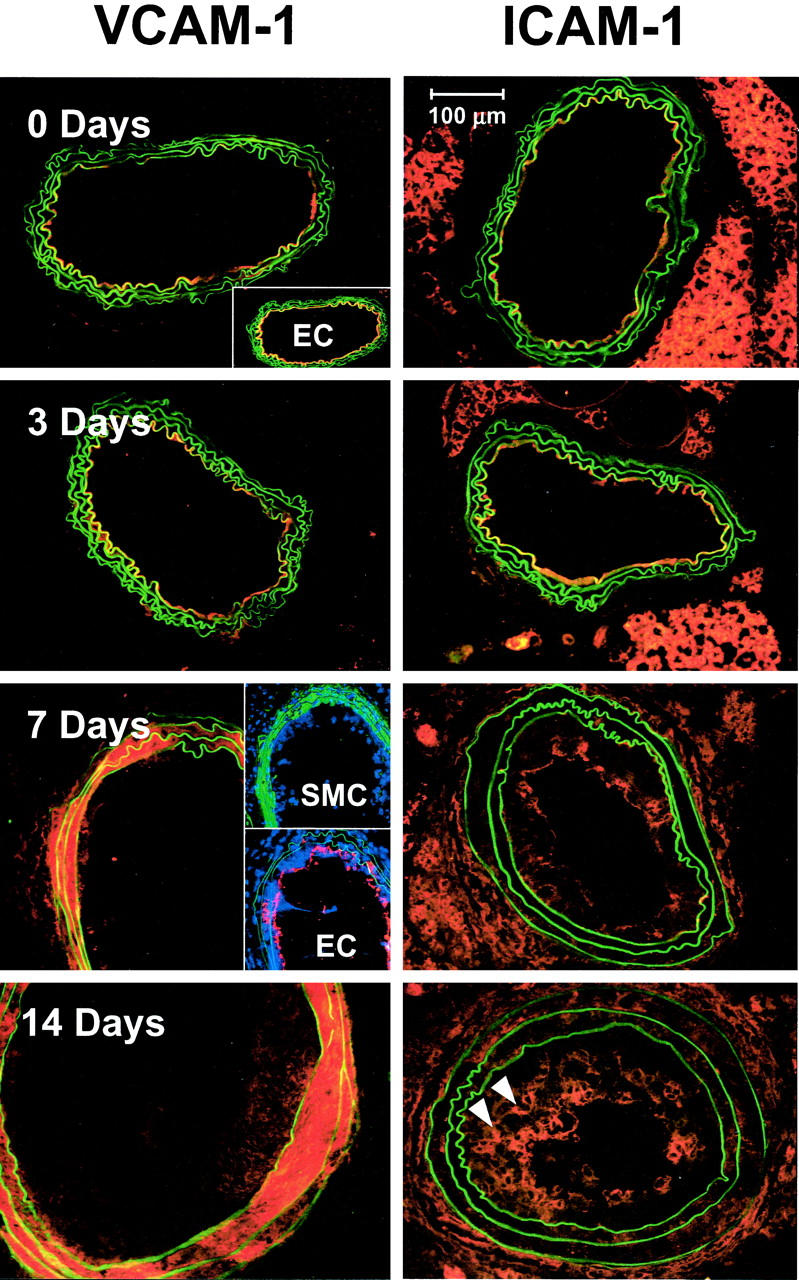
VCAM-1 and ICAM-1 expression in ligated mouse carotid arteries follow distinct time courses. Frozen sections were stained with mAb to either VCAM-1 (left) or ICAM-1 (right), followed by Rhodamine Red X-labeled secondary reagents. Before ligation and up to 3 days after ligation, VCAM-1 expression was confined to the luminal boundary of the vessel, where it co-localized with endothelial cells as demonstrated by CD31 staining (inset, top). By 7 days, VCAM-1 expression had disappeared from the endothelium (row 3, bottom inset) and had shifted to the media, where it co-localized with vascular SMCs, as demonstrated by staining for SM α-actin (green areas, top inset). This pattern continued up to 14 days after ligation (bottom). Before lesion initiation, ICAM-1 expression was confined to the luminal boundary of the artery (top right). By 3 days after ligation, some ICAM-1 staining could be detected in the adventitia as well. Adventitial staining became stronger at 7 and 14 days after ligation, coupled with a decrease in relative intensity of luminal staining. By 14 days after ligation, ICAM-1 was expressed by a subset of cells deeper within the neointima (arrowheads). Scale bar, 100 μm.
Discussion
Bone marrow transplantation in the apoE KO and LDL-receptor KO mouse models has been well established as a method for examining the influence of protein expression by macrophages on the development of atherosclerotic lesions. 15-17 Previous studies have shown that transplantation of syngeneic wild-type bone marrow into apoE KO recipients retards atherosclerotic lesion growth in these mice, partially rescuing the phenotype through production of apoE by donor-derived macrophages. 15 In the present study, however, we were interested in using the difference between donor- and host-derived cells primarily as a means to track inflammatory cell migration into developing lesions in vivo. We reasoned that this approach could be used to resolve definitively the longstanding issue of whether intimal macrophages in developing atherosclerotic lesions originate primarily from tissue-resident precursors or from circulating monocytes. We therefore aimed to develop a model in which BMT itself would have minimal effect on the course of atherosclerotic lesion development compared to that observed in nontransplanted apoE KO mice.
In the work that we report here, we have demonstrated the utility of a novel approach to track the recruitment of inflammatory cells into developing atherosclerotic lesions in the mouse carotid artery in vivo, using differences in CD45 (Ptprc) alleles to distinguish host- and donor-derived WBCs in a BMT model. This approach offers several advantages over protocols in which WBCs are labeled ex vivo and re-injected. First, the label (ie, antigenic difference between CD45.1 and CD45.2) is permanent and does not diminish in intensity in succeeding generations of cells descended from the original donors, unlike fluorescent dye labels that are diluted at each cell division. Secondly, our protocol avoids artifacts resulting from inadvertent activation of WBCs during isolation, labeling, and re-injection. The donor WBCs that function as tracers in this model have not been genetically manipulated in any manner that might alter their functionality. The difference between the Ptprca (CD45.1) and Ptprcb (CD45.2) alleles amounts to five amino acid changes in the extracellular region of the protein, 18 which confer altered antigenicity but have no other known effects on leukocyte function. The more common Ptprcb (CD45.2) allele occurs in both atherosclerosis-prone (C57BL/6) and atherosclerosis-resistant (eg, Balb/c) mouse strains, suggesting that CD45 allelic variation does not in itself confer an increased susceptibility to lesion development. Furthermore, we initiate lesion growth by carotid ligation after the donor bone marrow has already become well engrafted, ensuring that circulating inflammatory cells recruited into the developing lesion have been derived de novo by hematopoiesis. Although our approach does not provide a direct measurement of the instantaneous rate of WBC recruitment, this rate can be estimated by mathematical modeling using data obtained experimentally for counts of total WBCs, proliferating WBCs, and apoptotic/necrotic WBCs in the lesions as a function of time. Thus, we expect that data obtained using the methodology described in this article will prove to be complementary to results of studies using labeled and re-injected WBCs.
We chose to use busulfan preconditioning rather than total body irradiation to ablate the host bone marrow before BMT because the former approach offers several advantages over the latter. Busulfan-treated mice remain healthy both before and after BMT and do not suffer drastic weight loss as in the case of mice that have undergone total body irradiation. Because busulfan treatment is myeloablative but not immunosuppressive (circulating host T cells are not destroyed), 13 the transplanted mice resist adventitious infections much more readily than mice that have been irradiated, even without antibiotic treatment after transplant. These factors improve the survival rate of transplanted mice subjected to carotid ligation surgery.
The carotid ligation/hypercholesterolemia model has proven to be a useful tool for studying arterial lesion formation in the mouse in a number of contexts, owing to the rapid, reproducible, and spatially predictable course of lesion development. 14,19 Because of differences in the hemodynamic environment in which lesion growth occurs, this model cannot perfectly reproduce the process of spontaneous lesion development at vulnerable locations in the vasculature of the apoE KO mouse. Nevertheless, histological assessment confirms that, by 14 days after ligation, lesions initiated by this procedure in the apoE KO mouse display many of the features typical of advanced plaques in this model, 20 including involvement of multiple cell types (SMCs, macrophages, T lymphocytes), formation of cholesterol crystals, and differentiation of fibrous cap and lipid-rich core regions. Furthermore, the carotid ligation protocol is particularly well suited for mathematical analysis of lesion growth kinetics since the starting point (t = 0) of lesion development is well defined. Morphometric data obtained in the present study indicate that the time course of carotid lesion development and arterial remodeling closely follows that observed in our previous study in nontransplanted apoE KO mice. 21
In the mouse BMT model used in this study, replacement of host-type WBCs by donor-derived cells occurs much more slowly among macrophages in the carotid adventitia than among circulating monocytes. By 8 weeks after transplant, more than 80% of circulating WBCs originated from the donor bone marrow, whereas only ∼55% of adventitial WBCs were donor-derived. This difference in turnover rates allows us to infer the origin of WBCs in carotid lesions based on the relative distribution of host- and donor-type cells. If inflammatory cells in the lesion had originated primarily from resident adventitial macrophages, we would expect the ratio of host- to donor-type cells to reflect more closely that of the adventitia. We find, however, that the distribution of host- and donor-type cells in carotid lesions more nearly parallels the ratio in circulating WBCs, suggesting that most inflammatory cells in the lesions are in fact blood-derived.
Previous in vitro studies using blocking antibodies to endothelial adhesion molecules and leukocyte integrins have demonstrated that monocytes are able to use both VCAM-1 and ICAM-1 on the endothelium to migrate across an endothelial monolayer. 22,23 Recent studies by Cybulsky and colleagues 11 using transgenic mice that produce a mutant form of VCAM-1 lacking the fourth Ig domain (VCAM-1D4D) have provided evidence that VCAM-1 plays an important part in the recruitment of monocytes in vivo, particularly during the early stages of atherosclerotic lesion development. Our results suggest that, at early time points, both VCAM-1 and ICAM-1 play a role in monocyte recruitment to developing lesions in the carotid ligation/hypercholesterolemia model, whereas only ICAM-1 is likely to be involved in the later stages. At later time points (>7 days after ligation), VCAM-1 staining disappeared from the vessel lumen, whereas ICAM-1 continued to be expressed on the endothelium. Disappearance of VCAM-1 expression from the endothelium occurred concurrently with its appearance in the media, where VCAM-1 staining co-localized with SMCs. VCAM-1 expression by SMCs has been reported in animal models of atherosclerosis as well as in naturally occurring human lesions. 9,24,25 Such expression can be induced in vitro by treatment of SMCs with cytokines including interferon-γ, tumor necrosis factor-α, and interleukin-4, 24,26 suggesting that cytokine release by inflammatory cells migrating into the media may be responsible for induction of VCAM-1 on medial SMCs. Our observations indicate that the spatial pattern of adhesion molecule expression in developing lesions in the carotid ligation/hypercholesterolemia model is equivalent to that reported for spontaneously occurring lesions in apoE KO mice fed an atherogenic diet, 27 although the time course of lesion development is greatly accelerated in the ligated carotids.
Growth kinetics of atherosclerotic lesions can be described as the sum of terms corresponding to rates of cell migration, cell proliferation, and cell death (necrosis and/or apoptosis). Using Hoechst staining, we observed few pyknotic or fragmented nuclei in lesions induced in our model at time points up to 14 days (see Figures 3, 4, and 5 ▶ ▶ ▶ ; additional data not shown), implying that apoptosis was an infrequent event within the neointima during this time frame. 28 Furthermore, additional studies in which we injected propidium iodide in vivo (data not shown) revealed rare dead cells in the neointima (eg, one cell per 28 cross-sections examined). Therefore, as a first approximation, we have neglected the contribution of cell death to intimal lesion growth kinetics at time points up to 14 days. In the present study, lesion growth was greatest between 7 and 14 days on diet, when VCAM-1 was absent from the endothelium and ICAM-1 expression had diminished, indicating that inflammatory cell recruitment by either VCAM-1 or ICAM-1 does not drive the later, expansive phase of lesion growth. Because we have not examined the expression of other endothelial adhesion molecules, we cannot rule out the possibility that leukocyte recruitment by alternate routes contributes to lesion growth after 7 days. However, the dramatic increase in BrdU-labeled intimal cells by 14 days, coupled with the decrease in VCAM-1 and ICAM-1 expression, suggests that proliferation of inflammatory cells contributes significantly to lesion growth at later time points in this model. Macrophage proliferation within murine atherosclerotic lesions has previously been reported in the apoE*3-Leiden transgenic mouse, which expresses a human apoE allele associated with familial dysbetalipoproteinemia. 29 In summary, our data are consistent with a model in which initial recruitment of circulating inflammatory cells by endothelial adhesion molecule expression is followed by rapid lesion growth because of proliferation of these cells within the lesion environment. The BMT protocol described in this paper should prove useful for future studies investigating the recruitment of inflammatory cells in a variety of situations, including their contribution to lesions triggered by other interventions or to spontaneously developing lesions in atherosclerosis-prone mice. Such studies might be used to assess the potential effect of type of intervention, vascular location, or specific genetic mutations on the kinetics of inflammatory cell recruitment and lesion development.
Footnotes
Address reprint requests to Dr. Zorina S. Galis, Emory University School of Medicine, Division of Cardiology, Woodruff Memorial Bldg. Rm. 319, 1639 Pierce Rd., Atlanta, GA 30322. E-mail: zgalis@emory.edu.
Supported by the American Heart Association (grant EIA 0040087N to Z. S. G.), the National Institutes of Health (institutional training grant 5 T32 HL07745 07 to S. M. L.), the American Heart Association Southeast Regional Affiliate (postdoctoral fellowship 0120357B to S. M. L.), and the Emory University Summer Undergraduate Research Experience (SURE) Program (to H. L. P.).
References
- 1.Gerrity RG: The role of the monocyte in atherogenesis. I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Pathol 1981, 103:181-190 [PMC free article] [PubMed] [Google Scholar]
- 2.Joris I, Zand T, Nunnari JJ, Krolikowski FJ, Majno G: Studies on the pathogenesis of atherosclerosis. I. Adhesion and emigration of mononuclear cells in the aorta of hypercholesterolemic rats. Am J Pathol 1983, 113:341-358 [PMC free article] [PubMed] [Google Scholar]
- 3.Stary HC: The intimal macrophage in atherosclerosis. Artery 1980, 8:205-207 [PubMed] [Google Scholar]
- 4.Miyata K, Shimokawa H, Kandabashi T, Higo T, Morishige K, Eto Y, Egashira K, Kaibuchi K, Takeshita A: Rho-kinase is involved in macrophage-mediated formation of coronary vascular lesions in pigs in vivo. Arterioscler Thromb Vasc Biol 2000, 20:2351-2358 [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeld ME, Tsukada T, Gown AM, Ross R: Fatty streak initiation in Watanabe heritable hyperlipidemic and comparably hypercholesterolemic fat-fed rabbits. Arteriosclerosis 1987, 7:9-23 [DOI] [PubMed] [Google Scholar]
- 6.Kim C-J, Khoo JC, Gillotte-Taylor K, Li A, Palinski W, Glass CK, Steinberg D: Polymerase chain reaction-based method for quantifying recruitment of monocytes to mouse atherosclerotic lesions in vivo. Enhancement by tumor necrosis factor-α and interleukin-1β Arterioscler Thromb Vasc Biol 2000, 20:1976-1982 [DOI] [PubMed] [Google Scholar]
- 7.Charbonneau H, Tonks NK, Walsh KA, Fischer EH: The leukocyte common antigen (CD45): a putative receptor-linked protein tyrosine phosphatase. Proc Natl Acad Sci USA 1988, 85:7182-7186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel SS, Thiagarajan R, Willerson JT, Yeh ETH: Inhibition of α4 integrin and ICAM-1 markedly attenuate macrophage homing to atherosclerotic plaques in apoE-deficient mice. Circulation 1998, 97:75-81 [DOI] [PubMed] [Google Scholar]
- 9.Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD, Cybulsky MI: Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res 1999, 85:199-207 [DOI] [PubMed] [Google Scholar]
- 10.Ramos CL, Huo Y, Jung U, Ghosh S, Manka DR, Sarembock IJ, Ley K: Direct demonstration of P-selectin and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ Res 1999, 84:1237-1244 [DOI] [PubMed] [Google Scholar]
- 11.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos J-C, Connelly PW, Milstone DS: A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest 2001, 107:1255-1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N: Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA 1992, 89:4471-4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeager AM, Shinn C, Pardoll DM: Lymphoid reconstitution after transplantation of congenic hematopoietic cells in busulfan-treated mice. Blood 1991, 78:3312-3316 [PubMed] [Google Scholar]
- 14.Godin D, Ivan E, Johnson C, Magid R, Galis ZS: Remodeling of carotid artery is associated with increased expression of matrix metalloproteinases in mouse blood flow cessation model. Circulation 2000, 102:2861-2866 [DOI] [PubMed] [Google Scholar]
- 15.Boisvert WA, Spangenberg J, Curtiss LK: Treatment of severe hyper-cholesterolemia in apolipoprotein E-deficient mice by bone marrow transplantation. J Clin Invest 1995, 96:1118-1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, Atkinson JB, Linton MF: Increased atherosclerosis in mice reconstituted with apolipoprotein E null macrophages. Proc Natl Acad Sci USA 1997, 94:4647-4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr, MacRae L, Fazio S, Farese RV: Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1 J Clin Invest 2000, 105:711-719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zebedee SL, Barritt D, Epstein R, Raschke WC: Analysis of Ly5 chromosome 1 position using allelic differences and recombinant inbred mice. Eur J Immunogenet 1991, 18:155-163 [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Lindner V: Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol 1997, 17:2238-2244 [DOI] [PubMed] [Google Scholar]
- 20.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R: ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 1994, 14:133-140 [DOI] [PubMed] [Google Scholar]
- 21.Ivan E, Khatri J, Johnson C, Magid R, Godin D, Nandi S, Lessner SM, Galis ZS: Expansive arterial remodeling is associated with increased neointimal macrophage foam cell content: the murine model of macrophage-rich carotid artery lesions. Circulation 2002, in press [DOI] [PubMed]
- 22.Takahashi M, Ikeda U, Masuyama J-I, Kitagawa S-I, Kasahara T, Saito M, Kano S, Shimada K: Involvement of adhesion molecules in human monocyte adhesion to and transmigration through endothelial cells in vitro. Atherosclerosis 1994, 108:73-81 [DOI] [PubMed] [Google Scholar]
- 23.Meerschaert J, Furie MB: The adhesion molecules used by monocytes for migration across endothelium include CD11a/CD18, CD11b/CD18, and VLA-4 on monocytes and ICAM-1, VCAM-1, and other ligands on endothelium. J Immunol 1995, 154:4099-4112 [PubMed] [Google Scholar]
- 24.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P: Inducible expression of vascular cell adhesion molecule-1 by vascular smooth muscle cells in vitro and within rabbit atheroma. Am J Pathol 1993, 143:1551-1559 [PMC free article] [PubMed] [Google Scholar]
- 25.O’Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, Fishbein D, McCarty J, Ferguson M, Hudkins K, Benjamin CD, Lobb R, Alpers CE: Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest 1993, 92:945-951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barks JL, McQuillan JJ, Iademarco MF: TNF-α and IL-4 synergistically increase vascular adhesion molecule-1 expression in cultured vascular smooth muscle cells. J Immunol 1997, 159:4532-4538 [PubMed] [Google Scholar]
- 27.Zhou X, Hansson GK: Detection of B cells and proinflammatory cytokines in atherosclerotic plaques of hypercholesterolaemic apolipoprotein E knockout mice. Scand J Immunol 1999, 50:25-30 [DOI] [PubMed] [Google Scholar]
- 28.Pollman MJ, Yamada T, Horiuchi M, Gibbons GH: Vasoactive substances regulate vascular smooth muscle cell apoptosis. Countervailing influences of nitric oxide and angiotensin II. Circ Res 1996, 79:748-756 [DOI] [PubMed] [Google Scholar]
- 29.Lutgens E, Daemen M, Kockx M, Doevendans P, Hofker M, Havekes L, Wellens H, de Muinck ED: Atherosclerosis in APOE*3-Leiden transgenic mice. From proliferative to atheromatous stage. Circulation 1999, 99:276-283 [DOI] [PubMed] [Google Scholar]