

Abstract
To obtain a genomic portrait of heart failure derived from end-stage dilated cardiomyopathy (DCM), we explored expression analysis using the CardioChip, a nonredundant 10,848-element human cardiovascular-based expressed sequence tag glass slide cDNA microarray constructed in-house. RNA was extracted from the left ventricular free wall of seven patients undergoing transplantation, and five nonfailing heart samples. Cy3- and Cy5-labeled (and reverse dye-labeled) cDNA probes were synthesized from individual diseased or nonfailing adult heart RNA, and hybridized to the array. More than 100 transcripts were consistently differentially expressed in DCM >1.5-fold (versus pooled nonfailing heart, P < 0.05). Atrial natriuretic peptide was found to be up-regulated in DCM (19-fold compared to nonfailing, P < 0.05), as well as numerous sarcomeric and cytoskeletal proteins (eg, cardiac troponin, tropomyosin), stress response proteins (eg, HSP 40, HSP 70), and transcription/translation regulators (eg, CCAAT box binding factor, eIF-1AY). Down-regulation was most prominently observed with cell-signaling channels and mediators, particularly those involved in Ca2+ pathways (Ca2+/calmodulin-dependent kinase, inositol 1,4,5-trisphosphate receptor, SERCA). Most intriguing was the co-expression of several novel, cardiac-enriched expressed sequence tags. Quantitative real-time reverse transcriptase-polymerase chain reaction of a selection of these clones verified expression. Our study provides a preliminary molecular profile of DCM using the largest human heart-specific cDNA microarray to date.
Dilated cardiomyopathy (DCM) is characterized clinically by left ventricular dilatation, wall thinning, and homogeneous dysfunction of the myocardium leading to congestive heart failure. Genetically, DCM seems to evolve through primary mutations in the genes of the sarcomeric proteins. 1 However, recent evidence suggests that, despite distinct pathways leading to divergent endpoint phenotypes of each disease, there may exist some overlapping genetic modifiers leading to a conversion of one to the other. 2 How this occurs is under question; to understand this, a better knowledge of the molecular pathways and intermediary regulators is required.
Global analysis of gene expression has proven to be a fruitful means of examining the overall molecular portrait of a particular event as well as seeking out novel candidate transcripts that may play a role in formulating the phenotype or genotype of interest. By using this strategy, multiple genes and pathways in complex disorders can be visualized simultaneously, allowing for a feasible platform from which to investigate new and interesting genes. Using expressed sequence tag technology, our laboratory has generated a compendium of genes expressed in the human cardiovascular system, with the ultimate goal of assembling the intricacies of development and of disease, particularly the pathways leading to heart failure. 3 Through a computer-based in silico strategy, we have been able to identify—in a large scale—both known and previously unsuspected genetic modulators contributing to the growth of the myocardium from fetal through adult, and from normal to a perturbed hypertrophic phenotype. In contrast a gene-by-gene approach in elucidating the genes and mechanisms involved is time-consuming and cumbersome.
Recently, microarray technology has been used as a means of large-scale screening of vast numbers of genes—if not whole genomes—that possess differential expression in two distinct conditions. Although new and exciting developments have arisen in such fields as cancer 4 and yeast, 5 advances in understanding the complexity of cardiovascular disease, 6 specifically DCM, have been limited. One recent study examined gene expression in two failing hearts using oligo-based arrays. 7 Although the GeneChip® (Affymetrix, Santa Clara, CA) offers a carefully controlled systematic method of analysis, its current lack of user flexibility in its design hinders novel gene discovery currently available in tissue-specific arrays. Our laboratory has taken advantage of our vast previously acquired resources and has constructed what we believe to be the first ever custom-made cardiovascular-based cDNA microarray, which we term the “CardioChip.” 8 Its practicality and flexibility has allowed us to conceptualize the molecular events surrounding end-stage heart failure.
Materials and Methods
PCR Product Generation and Construction of the 10,848-Element cDNA Array
In the process of our large-scale sequencing project, PCR products were generated from human fetal and adult heart, familial hypertrophic cardiomyopathy heart and vascular cDNA libraries, as described previously. 9-13 After sequence similarity searching using BLAST 14 in the “nr” and “dbest” databases, a nonredundant set of clones was compiled, assigned to individual wells of 96-well microplates (Corning) and purified as described. 8 To construct the CardioChip, we took 113 of these microplates containing 10,848 nonredundant PCR products (Table 1) ▶ , spotted them onto CMT-GAPS-II amino-silane-coated glass microarray slides (Corning) using the GMS 417 arrayer (Affymetrix, Santa Clara, CA) and postprocessed the arrays. 15
Table 1.
Categorical Distribution of the 10,848 Expressed Sequence Tags on the CardioChip
| Category of EST | Number of clones on array (% of total) |
|---|---|
| Known, matched gene* | 2774 (25.6%) |
| Matched other EST in dbEST† | 4091 (36.8%) |
| Unmatched, putative novel EST‡ | 3887 (35.8%) |
| Bacterial clones (negative controls) | 192 (1.8%) |
*Includes matches to full-length hypothetical proteins in the “nr” database of GenBank.
‡Includes matches to human genome sequences in the “nr” database of GenBank.
‡Includes matches to sequences in the “htgs” database of GenBank.
Fluorescent Probe Labeling and Hybridization
Total RNA was extracted from the left ventricular free wall of five nonfailing human adult hearts (rejected as donors because of infection) and seven explanted dilated cardiomyopathic hearts (from patients undergoing transplantation) using Trizol reagent (Life Technologies, Inc., Grand Island, NY), according to the manufacturer’s protocol. Integrity of the RNA was verified with a 1% agarose-formaldehyde minigel and concentrations were obtained by measuring absorbance at 260 nm. For the labeling process, two types of experiments were performed. To assess intersample variation, 10 μg of each RNA species (ie, 7 DCM and 5 nonfailing hearts for a total of 12 hybridizations) were oligo-dT primed and probe synthesis was performed in the presence of Cy5-dUTP (Amersham, Arlington Heights, IL) at 42°C. 7 A parallel labeling with Cy3-dUTP was performed with human universal reference RNA (Stratagene, La Jolla, CA). After purification of the labeled probe by gel exclusion chromatography (ProbeQuant G-50, Amersham), the two probes of interest were mixed (one with Cy3 and one with Cy5), reduced to a volume of 10 μl and combined with 30 μl of hybridization solution [stock solution containing 100 μl of DIG EasyHyb hybridization solution (Roche, Indianapolis, IN), 5 μl of yeast tRNA (10 mg/ml), 5 μl of human COT1 DNA, 5 μl of poly-dA, and 5 μl of salmon sperm DNA (10 mg/ml) as blocking agents]. The probe solution was then hybridized to the arrayed slide at 37°C overnight. The next day, slides were washed first with 1× standard saline citrate to remove the coverslip (Hybri-Slips; Sigma, St. Louis, MO), then three successive washes of 1× standard saline citrate/0.1% sodium dodecyl sulfate for 15 minutes each at 50°C, followed by a rinse with 1× standard saline citrate. The slides were dried in individual 50-ml conical tubes and centrifuged at 500 rpm to remove excess fluid. Scanning of the slides was performed using the GMS 418 scanner (Affymetrix) at 532 nm (Cy3) and 635 nm (Cy5).
To identify interesting clusters of genes showing differential expression, three individual DCM RNA samples were labeled and hybridized against a pool of nonfailing heart tissue RNA; subsequently, the labeling was reversed (ie, the RNA species labeled with Cy3 in the first experiment was labeled with Cy5 in a subsequent experiment). Altogether, six individual reverse-dye-labeling hybridization experiments were performed (three tissues times two experiments each).
Data Processing
Raw scanned images of Cy3 and Cy5 fluorescence were processed using ScanAlyze 2.44 microarray image analysis software (Eisen, MB http://rana.lbl.gov). Local background was subtracted from the fluorescent value of each spot to obtain a net value. Spots whose net fluorescent value did not exceed the mean value obtained from the 192 bacterial-negative control spots were excluded. To account for incomplete hybridization on each spot, we only included in our analysis those spots in which at least 50% of pixels (within the defined area of the spot) displayed fluorescence at least 1.5 times greater than local background in all experiments.
Cluster Analysis
To determine congruence of the DCM and nonfailing heart tissue samples, expression data from the 12 samples were submitted to hierarchical clustering analysis (GeneSpring version 4.1.1; Silicon Genetics, Redwood City, CA). The median ratio for each spot (MRAT; calculated as the ratio of the median fluorescence from each pixel, minus background, in Cy5 to that in Cy3) was entered. Spots that did not meet the background-filtering criteria stated above were omitted. Hybridization results from reverse-dye-labeling were also clustered (Cluster, Eisen, MB http://rana.lbl.gov) using the same criteria.
Verification of Candidate Gene Expression Using Quantitative Real-Time Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
To confirm the expression patterns of certain candidate genes, expression of eight transcripts [atrial natriuretic peptide (ANP), elongation initiation factor 1A-Y isoform (EIF1AY), Ca2+/calmodulin kinase kinase (CaCKK), inositol 1,4,5-triphosphate-3-kinase (InosK), inositol 1,4,5-triphosphate receptor (InosTR), cardiac Ca2+ ATPase (CCaATPase), and putative heart-specific expressed sequence tags cnn3870 and ha6788] were analyzed using quantitative real-time RT-PCR (QRT-PCR) in 96-well format (Applied Biosystems, Foster City, CA). For each gene of interest, single QRT-PCR reactions were performed on each individual DCM heart (n = 7) and nonfailing adult heart (n = 5) mRNA samples. As an internal control, primers for GAPDH were also designed and amplified in parallel with the genes of interest. One-step QRT-PCR reactions were performed using 100 ng of total RNA per reaction. Primers were designed online (http://alces.med.umn.edu/rawprimer.html), verified for complementarity (http://www.basic.nwu.edu/biotools/oligocalc.html), and searched against the public database to confirm unique amplification products (http://www.ncbi.nlm.nih.gov). The primer sequences were: 1) ANP forward: 5′ GGA TTT CAA GAA TTT GCT GG3′, reverse: 5′ GCA GAT CGA TCA GAG GAG TC3′; 2) EIF1A forward: 5′ CAT CCT TTG TGC CTC GGT TA3′, reverse: 5′ TGG TTG CCA CTT ATT GTT CG3′; 3) cnn3870 forward: 5′ GCC ACT TTG GAA AAG GTG AG3′, reverse: 5′ CGA CTG GGT TTG GAT GTG AT3′; 4) ha6788 forward: 5′ AGA CAA CCT AGT GGC CGT GAC3′, reverse: 5′ TCC ATA GAT GTT CAC AGC GTT3′; 5) InosTR forward: 5′ TTG CCT TCT CTG GGA TTC AG 3′, reverse: 5′ TTT CCT GGG GCT TAC TGT TG 3′; 6) InosK forward: 5′ GCC ACC ATC AGG TTA ATT GG 3′, reverse: 5′ ACT TTT CCA CGT TGG TCT CG 3′; 7) CaCKK forward: 5′ ACC TCC TGG TCG GAG AAG AT 3′, reverse: 5′ AGA AGA TCT TGC GGG TCT CA 3′; 8) CCaATPase forward: 5′ CCT TTT CAT CTG TCG CTG TTG 3′, reverse: 5′ CGA ACA CCC TTA CAT TTC TGC 3′; 9) GAPDH forward: 5′ TGG GTG TGA ACC ATG AGA AG 3′, reverse: 5′ GTG TCG CTG TTG AAG TCA GA 3′. All reactions were carried in 50-μl volumes containing 1× SYBR Green PCR Master Mix, 0.25 U/μl of Multiscribe Reverse Transcriptase (Applied Biosystems), 0.4 U/μl of RNase inhibitor, and 10 pmol of each forward and reverse primer. Reactions in 96-well format were performed in the Applied Biosystems ABI Prism 7700 Sequence Detection System. The cycling parameters were 30 minutes at 48°C (reverse transcription), followed by PCR: 40 cycles of 30 seconds at 94°C and 1 minute at 60°C.
Results
Individual DCM (n = 7) and nonfailing heart (n = 5) RNA samples were labeled and hybridized against a human universal RNA sample; in addition, three of these DCM samples were reverse-dye labeled and hybridized against a pool of nonfailing heart RNA, for a grand total of 18 experiments. Each array was then normalized in GeneSpring to account for fluorescent dye incorporation bias by taking the median distribution of the signal intensities.
To determine which clones were differentially expressed, processed data were submitted to GeneSpring. The three slides representing the three patient samples were grouped together and treated as replicates; those in which DCM RNA was labeled with Cy3 were pooled into one set, whereas those in which DCM RNA was labeled with Cy5 were pooled into a second set. This generated an average MRAT for each spot (n = 3) in each group. Differential expression was determined as the average MRAT of each spot from three experiments, unless derived from a slide on which DCM was labeled with Cy3; for these, the inverse MRAT was included in the average. A clone was considered to be significantly up- or down-regulated if the average MRAT was significantly different from a ratio of 1.0 (P < 0.05) in at least one of the two sets.
Table 2 ▶ lists 145 selected known transcripts from statistical analysis determined to be differentially expressed (of which 111 genes are at a level of at least 1.5-fold) in end-stage heart failure consequent to DCM. Atrial natriuretic peptide (19.15-fold up-regulated) was the most differentially expressed transcript detected in this array. In general, genes encoding numerous sarcomeric and cytoskeletal proteins were consistently up-regulated, including cardiac troponin, α- and β-cardiac actin, tropomyosin, and β-myosin heavy chain. An increase in the expression of 40-kd, 70-kd, and 90-kd heat shock proteins were also observed. Several isoforms of collagen were identified as being expressed up-regulated, including α-1 type 1, type XVII, and type XVIII, whereas type XV was down-regulated. Genes encoding proteins regulating transcription and translation were noticeably altered. Elongation initiation factor 1 (eIF-1A) was most prominently up-regulated, as well as elongation factors 1-α and EF-2, but not eIF-2.
Table 2.
Selected Differentially-Expressed Transcripts in DCM (P < 0.05 versus MRAT of 1.0)
| Fold | Gene name | Acc. number |
|---|---|---|
| Up-regulated | ||
| 19.15 | Atrial natriuretic peptide (ANP) (=cardiodilatin) | M30262 |
| 12.45 | EST | T60005 |
| 9.62 | EST (yb56d02.r1) | T57506 |
| 6.14 | CCAAT-box binding factor (cbf) | M37197 |
| 3.78 | 3-Phosphoglycerate dehydrogenase | AF006043 |
| 3.61 | Eukaryotic translation elongation factor 1 alpha | NM001403 |
| 2.94 | EST-HS (cl0054.seq.F) | AA247158 |
| 2.75 | EST(qh14e06.x1) | R55392 |
| 2.69 | Hypothetical protein (KIAA0013) | D13638 |
| 2.61 | EST (EST65893) | T34317 |
| 2.58 | EST (ys10a01.r1) | H73017 |
| 2.45 | EST (zl51g02.r1) | AA147583 |
| 2.45 | EST (zd97e04.r1) | W93909 |
| 2.41 | cAMP phosphodiesterase | L12052 |
| 2.33 | Actin, alpha 2 | NM001613 |
| 2.28 | EST(ZL33G06.S1) | AA326222 |
| 2.25 | P2x purinoceptor | AF000234 |
| 2.23 | EST(EST112922) | AA297388 |
| 2.18 | EST(yr54g07.r1) | H63932 |
| 2.15 | Hypothetical protein (KIAA0164) | D79986 |
| 2.09 | Cyclin I | D50310 |
| 2.09 | EST(yj18g12.r1) | R82468 |
| 2.09 | Clathrin assembly protein 50 (AP50) | D63475 |
| 2.07 | EST(zl06e11.r1) | AA115552 |
| 2.05 | Ankyrin 1 (ANK1) | AF005213 |
| 2.05 | Cardiac troponin I | M64247 |
| 2.03 | EST(yh48g03.r1) | R23872 |
| 2.01 | EST(yi77c07.r1) | AA910337 |
| 2.01 | 20-kd myosin light chain (MLC-2) | J02854 |
| 1.99 | Natural killer cell enhancing factor (nkefb) | L19185 |
| 1.99 | SCID complementing gene 2 | D78188 |
| 1.96 | Osteoblast specific factor 2 (OSF-2p1) | D13665 |
| 1.96 | Supervillin | AF051850 |
| 1.93 | Eukaryotic translation elongation factor 1 gamma (EEF1G) | NM001404 |
| 1.93 | Alpha-cardiac actin | NM_005159 |
| 1.92 | Eomesodermin | P79944 |
| 1.90 | Four and a half LIM domains 1 (FHL1) | NM001449 |
| 1.89 | Fibronectin | K00799 |
| 1.79 | Ribosomal protein L39 homologue | L05096 |
| 1.78 | eIF-1A, Y isoform (EIF1AY) | AF000987 |
| 1.69 | Ribosomal protein L12 | L06505 |
| 1.69 | 90-kd heat-shock protein | M16660 |
| 1.67 | Mida1 | D63784 |
| 1.61 | N-acylsphingosine amidohydrolase (ASAH) | 4757785 |
| 1.50 | Heat-shock protein 40 | D49547 |
| 1.45 | Cytoskeletal tropomyosin TM30 | 37423 |
| 1.37 | OS-9 | AB002806 |
| 1.35 | Ubiquitin isopeptidase T | U35116 |
| 1.35 | Heat shock protein HSP70 (HSPA7) | 4139180 |
| 1.34 | Heat shock cognate 70 | P11142 |
| 1.32 | Microfibril-associated glycoprotein 4 (MFAP4) | L38486 |
| 1.32 | ATP synthase beta subunit (ATPSB) | M27132 |
| 1.31 | Beta myosin heavy chain (MYH7) | M58018 |
| 1.31 | Dysferlin, limb girdle muscular dystrophy 2B | XM_010780 |
| 1.31 | PKU-deta | AB004885 |
| 1.24 | Collagen alpha-1 type I | X06269 |
| 1.24 | Transcriptional regulator homolog RPD3 | U31814 |
| 1.24 | Collagen type XVIII | AF018081 |
| 1.20 | Zinc finger protein clone L3–4 | AF024706 |
| 1.19 | Factor X | K01886 |
| 1.12 | Prothymosin alpha | M26708 |
| 1.12 | ATP-dependent RNA-helicase | L15441 |
| 1.10 | Proton pump polypeptide | M58758 |
| 1.09 | Dynactin subunit (p22) | AF082513 |
| Down-regulated | ||
| 3.19 | EST(EST177676) | AA306721 |
| 2.96 | EST(zb02d03.r1) | W07662 |
| 2.61 | Hypothetical protein (R27090_2) | AC002985 |
| 2.48 | EST(yg11g03.r1) | R17208 |
| 2.45 | Transforming growth factor-beta (tgf-beta) | M60316 |
| 2.45 | Fos proto-oncogene (c-fos) | K00650 |
| 2.32 | Motor protein | D21094 |
| (Table continues) | ||
| 2.29 | EST(zh78e02.r1) | W90263 |
| 2.28 | Tob family | D64110 |
| 2.25 | EST(EST40980) | AA336265 |
| 2.25 | EST(zl33c05.r1 clone 503720 5′) | AA131632 |
| 2.24 | Membrane cofactor protein | X59405 |
| 2.23 | EST(EST108171 5′) | H32755 |
| 2.21 | Hypothetical protein (KIAA0642) | AB014542 |
| 2.20 | EST (nc80h02.r1) | AA580765 |
| 2.18 | EST(EST35620) | AA331759 |
| 2.17 | EST(zk05d07.r1) | AA027894 |
| 2.16 | EST-HS (j0145.seq.F) | AA249323 |
| 2.16 | EST(yh59e02.r1) | R30903 |
| 2.14 | Cytotoxic granule-associated RNA-binding protein p40-TIA-1 | S70114 |
| 2.12 | Sialyltransferase, putative | AJ007310 |
| 2.10 | Embryonic lung protein (HUEL) | AF006621 |
| 2.07 | EST(EST64412) | AA355938 |
| 2.06 | EST(zb66c05.r1) | W24921 |
| 2.06 | EST(EST91651) | AA378879 |
| 2.05 | Hepatitis C-associated microtubular aggregate protein p44 | D28910 |
| 2.05 | Putative transmembrane protein precursor (B5) | L38961 |
| 2.02 | EST(zw34c09.r1) | AA443492 |
| 2.02 | E1-like protein | AF094516 |
| 2.00 | EST(yz62a12.r1) | N75168 |
| 2.00 | Calcium-ATPase | M23115 |
| 2.00 | Hypothetical protein (KIAA0084) | D42043 |
| 1.99 | Thioredoxin reductase | D88687 |
| 1.96 | YL-1 | D43642 |
| 1.94 | HM89 | D10924 |
| 1.94 | Hypothetical protein (KIAA0675) | AB014575 |
| 1.91 | Tristetraproline (TTP) | M63625 |
| 1.86 | Chromosome 1 specific transcript KIAA0493 | AB007962 |
| 1.86 | Inositol 1,4,5-trisphosphate receptor type-2 | D26350 |
| 1.86 | Inositol 1,4,5-trisphosphate 3-kinase | D38169 |
| 1.85 | Hypothetical protein (KIAA0027) | D25217 |
| 1.83 | Bone morphogenetic protein-4 (hBMP-4) | U43842 |
| 1.83 | Ribosomal protein L34 (RPL34) | L38941 |
| 1.82 | N-acetylglucosamide-(beta 1–4)-galactosyltransferase | M13701 |
| 1.80 | Blue cone photoreceptor pigment | M13299 |
| 1.80 | Secretory carrier membrane protein (SCAMP3) | AF005039 |
| 1.79 | Transfer valyl-tRNA synthetase | M98326 |
| 1.79 | Thrombospondin 3 (THBS3) | L38969 |
| 1.76 | RAS-related protein RAB-18 | P35293 |
| 1.76 | N-acetylgalactosamine 6-sulfate sulfatase | D17629 |
| 1.75 | Hypothetical protein (KIAA0292) | AB006630 |
| 1.75 | Hypothetical protein (KIAA0041) | D26069 |
| 1.75 | Arylhydrocarbon receptor | D38417 |
| 1.75 | Leukocyte adhesion protein and activation antigen (BLAST-1) (CD48) | M63911 |
| 1.74 | Sarco-/endoplasmic reticulum Ca-ATPase 3 (SERCA3) | AF068221 |
| 1.73 | Hypothetical protein (KIAA0393) | AB002391 |
| 1.73 | Glycoprotein GP-39 | M80927 |
| 1.72 | CD39L2 | AF039916 |
| 1.72 | Thymidylate synthase | D00596 |
| 1.70 | Hypothetical protein (KIAA0697) | AB014597 |
| 1.70 | Immunoglobulin-like transcript 3 protein variant 1 | AF072099 |
| 1.70 | GTP-binding protein (low match) | Z49068 |
| 1.70 | Hypothetical protein (KIAA0010) | D13635 |
| 1.68 | G protein-coupled receptor (EBI 1) | L31581 |
| 1.54 | MHC class I antigen HLA-B7 variant (HLA-B) | U29057 |
| 1.53 | Calcium-ATPase (hk2), cardiac | M23278 |
| 1.36 | RO PROTEIN SS-A | P19474 |
| 1.30 | EWS/FLI1 activated transcript 2 homologue (EAT-2) | AF020264 |
| 1.23 | Complement H factor | M17517 |
| 1.22 | b (2)gcn homologue (=integrin beta 4 binding protein) | AF047433 |
| 1.19 | Vacuolar H+ATPase, isoform 2 | L35249 |
| 1.18 | S100 calcium-binding protein A4 (S100A4) | M80563 |
| 1.17 | Lysosomal membrane glycoprotein CD63 | M58485 |
| 1.16 | IL-13 receptor alpha-1 chain | Y09328 |
| 1.14 | Cellular adhesion regulatory molecule | S54769 |
| 1.10 | Neurofibromatosis 2 (nf2) | L27065 |
| 1.09 | Ca2+/calmodulin-dependent kinase kinase | AF101264 |
| 1.09 | Cyclin D3 (CCND3) | M92287 |
| 1.08 | Legumain | Y09862 |
| 1.07 | Putative Cu++-transporting P-type ATPase | L06133 |
| 1.03 | APC | M74088 |
“Fold” represents average difference over 6 dye-swap experiments. ‘HS’ = heart specific EST; found in heart cDNA library only.
The most striking constituency of transcripts down-regulated were those involved in Ca2+ signaling and homeostasis. Inositol 1,4,5-triphosphate (ITP) receptor, ITP 3-kinase, sarcoendoplasmic reticulum Ca2+-ATPase 3, and Ca2+/calmodulin-dependent protein kinase kinase 2 showed lower levels of expression across all DCM samples.
From hierarchical cluster analysis, the six of seven DCM samples and four of five nonfailing heart samples clustered together (Figure 1) ▶ . Reverse-dye labeling identified two distinct cluster regions that were consistently expressed (Figure 2) ▶ . A cluster of up-regulated genes was enriched in sarcomeric and cytoskeletal proteins, including β-myosin heavy chain (β-MyHC), cardiac troponin I, tropomyosin, consistent with the statistical analysis. Another cluster was identified containing predominantly signal transducers (eg, GTP binding protein rho A, ras-like TC25) and energy metabolism modifiers (eg, aldose reductase, electron transfer flavoprotein). Many cardiovascular-enriched expressed sequence tag transcripts were interspersed within both of these clusters.
Figure 1.
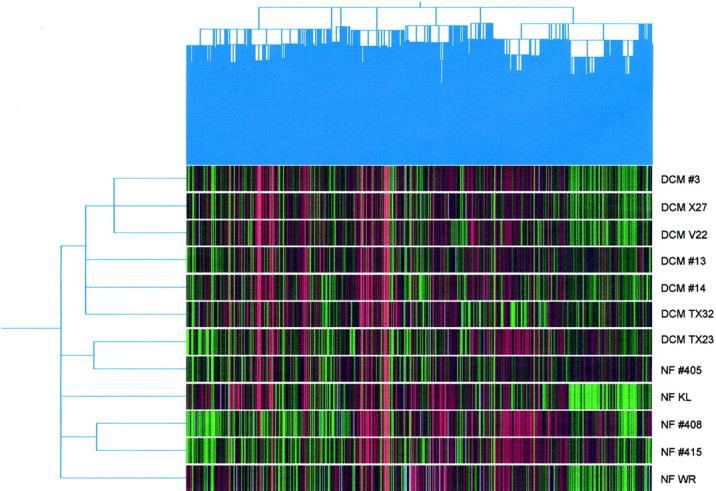
Hierarchical cluster analysis of seven DCM and five nonfailing (NF) heart patient samples against human universal reference RNA. Six of seven DCM patient samples and four of the five nonfailing heart clustered together.
Figure 2.
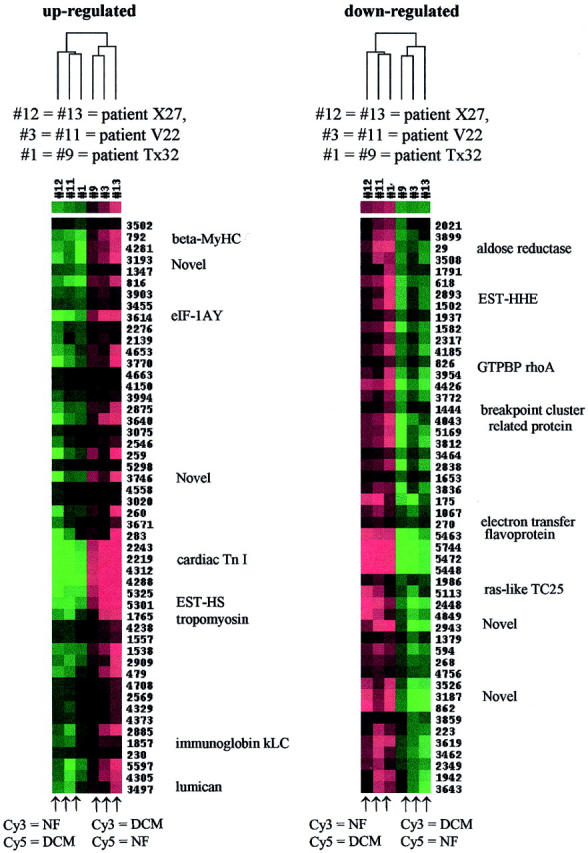
Hierarchical cluster analysis of DCM versus pooled nonfailing (NF) adult heart samples. Six slides representing three individual DCM patients (three reverse-labeled) were clustered, ie, slides 1, 11, and 12 were hybridized with DCM-Cy5-dUTP, whereas slides 3, 9, and 13 were hybridized with DCM-Cy3-dUTP. Colors represent Cy3/Cy5 ratio; red = positive ratio, green = negative ratio. A selection of genes corresponding to regions of the cluster are shown.
Verification Using Real-Time RT-PCR
Real-time RT-PCR was used to confirm the relative expression patterns of eight transcripts identified by microarray analysis. Threshold values were assigned where the normalized reporter signal Δ(Rn)-to-noise ratio exceed 1.0 for all genes in the plot. The midpoint of the linear phase of exponential amplification was determined to be the threshold cycle (Ct) for each gene. To determine relative expression levels in each mRNA population, a standard curve was plotted based on expressions of GAPDH in dilutions of nonfailing adult heart RNA (100%, 75%, 50%, 25%, and 12.5% of total stock RNA). For all experimental samples, the amount of product was determined from the standard curve, averaged, and divided by the average amount of GAPDH product to achieve a normalized value. Using a two-tailed Student’s t-test, significance was calculated by comparing the relative amounts of product between individual DCM and nonfailing adult heart samples. Fold difference was calculated by dividing each sample by the amount of product generated in the mean nonfailing adult heart samples (Figure 3) ▶ . Expression of ANP, cnn3870, EIF1A (up-regulated in DCM), CaCKK, InosK, InosTR CCaATPase, and ha6788 (down-regulated in DCM,) paralleled the results obtained with microarray.
Figure 3.

Real-time RT-PCR results of selected genes identified by the microarray analysis. a: Fold difference expression of each gene in DCM heart samples relative to nonfailing (NF) samples. ANP, atrial natriuretic peptide (positive control); CNN3870 and HA6788, novel heart transcripts; EIF1AY, elongation initiation factor 1A, Y isoform; CaCKK, Ca2+/calmodulin kinase kinase; InosK, inositol 1,4,5-triphosphate-3-kinase; InosTR, inositol 1,4,5-triphosphate receptor; CCaATPase, cardiac Ca2+ ATPase. b: Mean relative mRNA population of each gene from a versus GAPDH in DCM (n = 7) and NF (n = 5) samples. †, P < 0.01; *, P < 0.05 versus NF.
Discussion
Despite the recent advances contributing to our knowledge of DCM, its complex pathophysiological events especially at the molecular and genetic levels remain to be fully elucidated. The CardioChip, our 10,848-element cardiovascular-based cDNA microarray, has helped to answer some of the questions surrounding this complexity. This report reveals a profile of previously-suspected candidates involved in molecular events surrounding the pathology of heart failure; more importantly, it identifies novel candidates that may, with further verification at the functional level, be responsible for contributing to the demise of myocardial function.
As expected, ANP showed an intense level of up-regulation across the DCM patient samples, confirming at the microarray level its pivotal role as a circulating marker of cardiac muscle stress. 16 Indeed, its presence in our analysis lends a degree of credence to the validity of our study. Despite its significant up-regulation versus nonfailing samples, the level of ANP was highly variable among the patients.
In addition, we observed a consistent up-regulation of selected sarcomeric and extracellular matrix proteins (ie, β-myosin heavy chain, α-actinin, α-cardac actin, troponin I, tropomyosin, collagen, etc). Evidence in knockout mice and human studies has offered insight into the putative role of these proteins in maintaining sarcomeric integrity. 1,17-22 Mutations of proteins associated with α-actinin, namely MLP, cardiac α-actin, desmin and titin, have been shown to be present in certain forms of human DCM. 23-27 Ambiguities exist in the literature regarding the expression of collagen and other members of the extracellular matrix; nonetheless, regulation of the extracellular matrix is important in the formation of fibrosis and impaired contractile function. 28-30
Calcium signaling has recently become an important area of interest in the investigation of heart failure. 31 Decrease in calcium cycling genes has been shown to result in reduced contractility in mice whose β-adrenergic stimulation is blunted leading to decreased phospholamban phosphorylation. 32 Ca2+ATPase is key in regulating contractility, and its approximately twofold average down-regulation in our DCM samples lends credence to its involvement. This is supported by a recent study in which the transfer of the Ca2+ATPase gene into the rat myocardium prevents certain features of heart failure. 33 The presence of Ca2+/calmodulin-dependent kinase kinase in our analysis, despite showing only ∼ 1.1-fold down-regulation, is particularly intriguing, as it is known to phosphorylate phospholamban. 34 In addition, inositol 1,4,5-triphosphate receptor (a member of the calcium channel family) that may be responsible for calcium release from intracellular stores, 35 was also significantly down-regulated (1.86-fold). Inositol 1,4,5-trisphosphate 3-kinase was recently cloned 36 and may be another key component in this regulation (1.86-fold). Our findings suggest that the role of Ca2+ signaling down-regulation may be of crucial significance in the evolution of heart failure and would warrant further investigation.
A number of novel expressed sequence tags were also identified from our study to be differentially regulated. Verification with quantitative real-time RT-PCR confirmed this expression (Figure 3) ▶ . It is an intriguing prospect that these among other transcripts, after full-length sequencing, represent novel cardiac-specific genes encoding proteins that are potentially key to solving the puzzle of the molecular pathophysiology of heart failure. Indeed, our microarray analysis not only serves as a genomic model for a more complete understanding of DCM, but also as a focused target for possible therapeutic interventions specific to the cardiac tissue. Investigations are currently underway to elucidate the function of these candidates.
This report describes the most informative cDNA microarray-based analysis of end-stage heart failure derived from DCM currently available. Although we believe we have effectively demonstrated reproducibility and reliability of our technology (both for the entire array and for a selection of genes located on it), a larger n from our population would enhance the validity of our conclusions. Certainly, there exists no homogeneous heart failure genotype, especially among only seven DCM patients. Nonetheless, we have demonstrated a common expression pattern among our set of samples, from both microarray and QRT-PCR analysis. We are also limited by the genes (both in number and identity) present on this array. Although we are currently unable to spot every gene and gene cluster on our CardioChip, we have tried to draw from a diverse assortment of genes and gene pathways, both known and unknown. It must be emphasized that this investigation is not exhaustive; by no means does it attempt to fully characterize the molecular basis of heart failure. Its intention is to provide a preliminary portrait of global gene expression in complex cardiovascular disease using cDNA microarray and QRT-PCR technology, and to highlight the effectiveness of our ever-evolving platform for gene discovery. With even more patient samples and a CardioChip toward completeness, we will be in a better position to reap the important benefits from this initial work and expand our body of knowledge.
Acknowledgments
We thank Mr. James Ip, and Drs. Richard E. Pratt, Leonard Anderson, and Juey-Jen Hwang for their insight and helpful discussions on the microarray data.
Footnotes
Address reprint requests to Professor C. C. Liew, The Cardiovascular Genome Unit, Department of Medicine, Thorn 1334, Brigham and Women’s Hospital, 75 Francis St., Boston, MA 02115. E-mail: cliew@rics.bwh.harvard.edu.
Supported in part by the Heart and Stroke Foundation of Ontario, the Medical Research Council of Canada, the Canadian Genome Analysis and Technology Program, the National Institutes of Health [grants 5RO1-HL5851603, 5P5O-HL5931603, and 5RO1HL6166102; and a merit award (5R37-HL3561016 to V. J. D.], and the University of Toronto (open fellowship to J. D. B.).
References
- 1.Towbin JA, Bowles NE: Genetic abnormalities responsible for dilated cardiomyopathy. Curr Cardiol Rep 2000, 2:475-480 [DOI] [PubMed] [Google Scholar]
- 2.Chien KR: Stress pathways and heart failure. Cell 1999, 98:555-558 [DOI] [PubMed] [Google Scholar]
- 3.Hwang JJ, Dzau VJ, Liew CC: Genomics and the pathophysiology of heart failure. Curr Cardiol Rep 2001, 3:198-207 [DOI] [PubMed] [Google Scholar]
- 4.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever M, Byrd JC, Botstein D, Brown PO, Staudt LM: Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403:503-511 [DOI] [PubMed] [Google Scholar]
- 5.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO: Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 2000, 11:4241-4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor LA, Carthy CM, Yang D, Saad K, Wong D, Schreiner G, Stanton LW, McManus BM: Host gene regulation during coxsackievirus B3 infection in mice: assessment by microarrays. Circ Res 2000, 87:328-334 [DOI] [PubMed] [Google Scholar]
- 7.Yang J, Moravec CS, Sussman MA, DiPaola NR, Fu D, Hawthorn L, Mitchell CA, Young JB, Francis GS, McCarthy PM, Bond M: Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation 2000, 102:3046-3052 [DOI] [PubMed] [Google Scholar]
- 8.Barrans JD, Stamatiou D, Liew CC: Construction of a human cardiovascular cDNA microarray: portrait of the failing heart. Biochem Biophys Res Commun 2001, 280:964-969 [DOI] [PubMed] [Google Scholar]
- 9.Hwang DM, Hwang WS, Liew CC: Single pass sequencing of a unidirectional human fetal heart cDNA library to discover novel genes of the cardiovascular system. J Mol Cell Cardiol 1994, 26:1329-1333 [DOI] [PubMed] [Google Scholar]
- 10.Hwang DM, Dempsey AA, Lee CY, Liew CC: Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics 2000, 66:1-14 [DOI] [PubMed] [Google Scholar]
- 11.Hwang DM, Dempsey AA, Wang RX, Rezvani M, Barrans JD, Dai KS, Wang HY, Ma H, Cukerman E, Liu YQ, Gu JR, Zhang JH, Tsui SK, Waye MM, Fung KP, Lee CY, Liew CC: A genome-based resource for molecular cardiovascular medicine: toward a compendium of cardiovascular genes. Circulation 1997, 96:4146-4203 [DOI] [PubMed] [Google Scholar]
- 12.Liew CC, Hwang DM, Fung YW, Laurenssen C, Cukerman E, Tsui S, Lee CY: A catalogue of genes in the cardiovascular system as identified by expressed sequence tags. Proc Natl Acad Sci USA 1994, 91:10645-10649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liew CC, Hwang DM, Wang RX, Ng SH, Dempsey A, Wen DH, Ma H, Cukerman E, Zhao XG, Liu YQ, Qiu XK, Zhou XM, Gu JR, Tsui S, Fung KP, Waye MM, Lee CY: Construction of a human heart cDNA library and identification of cardiovascular based genes (CVBest). Mol Cell Biochem 1997, 172:81-87 [PubMed] [Google Scholar]
- 14.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215:403-410 [DOI] [PubMed] [Google Scholar]
- 15.DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM: Use of a cDNA microarray to analyze gene expression patterns in human cancer. Nat Genet 1996, 14:457-460 [DOI] [PubMed] [Google Scholar]
- 16.Vikstrom KL, Bohlmeyer T, Factor SM, Leinwand LA: Hypertrophy, pathology, and molecular markers of cardiac pathogenesis. Circ Res 1998, 82:773-778 [DOI] [PubMed] [Google Scholar]
- 17.Towbin JA: The role of cytoskeletal proteins in cardiomyopathies. Curr Opin Cell Biol 1998, 10:131-139 [DOI] [PubMed] [Google Scholar]
- 18.Elliott K, Watkins H, Redwood CS: Altered regulatory properties of human cardiac troponin I mutants that cause hypertrophic cardiomyopathy. J Biol Chem 2000, 275:22069-22074 [DOI] [PubMed] [Google Scholar]
- 19.Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T: Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 1997, 16:379-382 [DOI] [PubMed] [Google Scholar]
- 20.Redwood C, Lohmann K, Bing W, Esposito GM, Elliott K, Abdulrazzak H, Knott A, Purcell I, Marston S, Watkins H: Investigation of a truncated cardiac troponin T that causes familial hypertrophic cardiomyopathy: Ca(2+) regulatory properties of reconstituted thin filaments depend on the ratio of mutant to wild-type protein. Circ Res 2000, 86:1146-1152 [DOI] [PubMed] [Google Scholar]
- 21.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG: A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62:999-1006 [DOI] [PubMed] [Google Scholar]
- 22.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE: Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 1994, 77:701-712 [DOI] [PubMed] [Google Scholar]
- 23.Arber S, Hunter JJ, Ross J, Jr, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P: MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 1997, 88:393-403 [DOI] [PubMed] [Google Scholar]
- 24.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG: Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000, 342:770-780 [DOI] [PubMed] [Google Scholar]
- 25.Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A: Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun 1999, 262:411-417 [DOI] [PubMed] [Google Scholar]
- 26.Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT: Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280:750-752 [DOI] [PubMed] [Google Scholar]
- 27.Schonberger J, Seidman CE: Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet 2001, 69:249-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dempsey AA, Ton C, Liew CC: A cardiovascular EST repertoire: progress and promise for understanding cardiovascular disease. Mol Med Today 2000, 6:231-237 [DOI] [PubMed] [Google Scholar]
- 29.Francis GS: Changing the remodeling process in heart failure: basic mechanisms and laboratory results. Curr Opin Cardiol 1998, 13:156-161 [PubMed] [Google Scholar]
- 30.Rao VU, Spinale FG: Controlling myocardial matrix remodeling: implications for heart failure. Cardiol Rev 1999, 7:136-143 [DOI] [PubMed] [Google Scholar]
- 31.McKinsey TA, Olson EN: Cardiac hypertrophy: sorting out the circuitry. Curr Opin Genet Dev 1999, 9:267-274 [DOI] [PubMed] [Google Scholar]
- 32.Chien KR: Genomic circuits and the integrative biology of cardiac diseases. Nature 2000, 407:227-232 [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ: Adenoviral gene transfer of SERCA2a improves left ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci USA 2000, 97:793-798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tada M, Yabuki M, Toyofuku T: Molecular regulation of phospholamban function and gene expression. Ann NY Acad Sci 1998, 853:116-129 [DOI] [PubMed] [Google Scholar]
- 35.Marks AR: Cardiac intracellular calcium release channels: role in heart failure. Circ Res 2000, 87:8-11 [DOI] [PubMed] [Google Scholar]
- 36.Dewaste V, Pouillon V, Moreau C, Shears S, Takazawa K, Erneux C: Cloning and expression of a cDNA encoding human inositol 1,4,5-trisphosphate 3-kinase C. Biochem J 2000, 352:343-351 [PMC free article] [PubMed] [Google Scholar]