

Abstract
Gastrointestinal stromal tumors (GISTs) are mesenchymal neoplasms of the gut wall that express the receptor tyrosine kinase KIT. Somatic mutations that result in constitutive activation of KIT kinase have been identified in a number of studies of GISTs, although the reported frequency of these mutations has varied over a wide range (20 to 92%). Several reports have suggested that KIT gene mutations are more common in malignant GISTs than in benign lesions, and it has been proposed that mutations in exon 11 of KIT are a negative prognostic factor. To maximize sensitivity for KIT mutations we have adapted denaturing high-pressure liquid chromatography as a method for screening polymerase chain reaction amplimers of exons 9, 11, 13, and 17 from GIST genomic DNA. This approach was used to assess the frequency of KIT mutations in 13 morphologically benign, incidentally discovered, GISTs identified at autopsy, endoscopy, or laparotomy for unrelated disease. Representing the smallest pathologically recognizable GISTs, these lesions ranged in size from 4 to 10 mm in diameter and were all immunohistochemically positive for KIT. Eleven of the 13 tumors had sequence-confirmed mutations in KIT, including 10 mutations in exon 11 (77%) and one mutation in exon 9 (7.7%). The remaining two tumors were wild type for exons 9, 11, and 17; one of these was also analyzed for exon 13 and was wild type in this exon as well. The mutations found in the incidental GISTs were identical to those that have been documented in larger GISTs. In addition, the overall frequency of mutations in the incidental tumors (85%) did not differ significantly from that we previously reported in a series of 72 advanced/metastatic GISTs (86%), strongly supporting the view that activating mutations in KIT are acquired very early in the development of most GISTs. The findings suggest that KIT mutations per se are of little prognostic importance in GISTs.
Gastrointestinal stromal tumors (GISTs) are relatively uncommon mesenchymal neoplasms that arise in the wall of the stomach, small intestine, colon, and other sites within the abdominal cavity. 1 Classification of these neoplasms was a source of controversy for many years, but two key observations published by Hirota and colleagues 2 in 1998 helped to clarify the nature of GISTs. The first observation was that GISTs strongly express the receptor tyrosine kinase KIT (CD117). This marker, which is rarely present on other spindle cell tumors occurring in the abdominal cavity, is now accepted as the most specific immunohistochemical identifier for GISTs. 1,3 The second observation was that mutations were present in the juxtamembrane domain (exon 11) of the KIT gene (five of six examined GISTs; 83%), and that these mutations caused constitutive activation of the KIT kinase when expressed in COS cells. 2
From a number of follow-up studies it is clear that mutations of KIT exon 11 are the most common type present in GISTs, but that mutations also occur in exons 9, 13, and 17. 4-14 The mutations vary from single base pair substitutions to complex deletions/insertions, but they are invariably in-frame and in many cases have been documented to cause activation of the KIT kinase independent of its natural ligand, stem cell factor. Correspondingly, phosphorylated (activated) KIT is detectable in extracts of GISTs (M Heinrich, C Corless, and J Fletcher, manuscript submitted). 12 Activation of KIT seems important to the growth of GISTs, because the proliferation of GIST cells in culture is inhibited by the tyrosine kinase inhibitor STI571, a potent blocker of KIT kinase activity. 15 Moreover, in recent clinical trials the majority of patients with malignant GIST have shown a benefit to treatment with STI571. 16-18
Although the importance of KIT mutations in the biology of GISTs is well established, it remains unclear at what point in the development of these tumors the mutations are acquired. Several studies have suggested that mutations in KIT exon 11 are more common in malignant than in benign GISTs. 5,6,14,19,20 In a large series published by Taniguchi and colleagues 7 (124 patients), exon 11 mutations were identified in 57% of tumors and seemed to correlate with disease recurrence and shortened survival (86% versus 49% 5-year survival). In contrast, Rubin and colleagues 12 did not observe a correlation between KIT mutation status and tumor grade.
In this report we examine the frequency of KIT gene mutations in a series of tumors that were small (10 mm or less), clinically incidental, and morphologically benign. The majority of these lesions, which represent the earliest pathologically recognizable GISTs, were found to harbor mutations of the type frequently identified in larger, malignant lesions. The results favor the view that activating mutations in KIT occur early in the development of GISTs.
Materials and Methods
One hundred twenty GISTs were identified in the pathology archives of the Oregon Health and Science University Hospital, the Portland VA Medical Center, and the Northwest Kaiser Permanente Regional Laboratory by searching for cases coded as “leiomyoma,” “leiomyosarcoma,” or “sarcoma, NOS” in association with abdominal organs (excepting uterus). Fifteen tumors were incidentally discovered lesions; 13 of these were 1.0 cm or less in size and these were selected for further study, following the institutional review board regulations of all three source institutions.
Immunohistochemistry for KIT (CD117) was performed as follows. Sections from the original paraffin blocks were heated in Citra buffer (Biogenex, San Ramon, CA) for 20 minutes in a vegetable steamer (Sunbeam-Oster Household Products, Schaumburg, IL) and then placed on a DAKO automated immunostainer (DAKO Corp., Carpinteria, CA). A standard avidin-biotin staining protocol was performed with the DAKO polyclonal rabbit antibody (DAKO A4502), used at 1:400 dilution, goat biotinylated anti-rabbit secondary (Vector Laboratories, Burlingame, CA), and the Vectastain Elite kit (Vector Laboratories). Endogenous mast cells served as internal-positive controls in all cases; the antibody did not stain other tissue elements (eg, epithelial cells) at the selected titer.
Tumor tissue was identified on unstained, 5 μm sections by comparison with hematoxylin and eosin (H&E)-stained slides and was carefully collected using a clean, sterile scalpel blade into a microfuge tube. Because the tumors were round discrete masses, dissection by this approach was straightforward and there was minimal contamination from adjacent normal muscularis cells (note that such contamination would serve to bias the results toward an apparent wild-type genotype). The dissected tissue was deparaffinized by serial extraction with xylenes and ethanol and allowed to air-dry. DNA was extracted using the Qiagen minikit (no. 51304; Qiagen, Valencia, CA) in accordance with the manufacturer’s recommendations.
Purified tumor DNA (0.5 μg) was subjected to 45 cycles of polymerase chain reaction (PCR) using the High-Fidelity PCR System (no. 1732078; Roche, Indianapolis, IN). Primer pairs for each exon analyzed are listed in Table 1 ▶ . Negative controls were included in every set of amplifications. In a minority of cases there was insufficient amplified DNA for screening by high-pressure liquid chromatography (HPLC) after single step amplification and therefore a second round of amplification was performed using nested primers (Table 1) ▶ .
Table 1.
PCR Primers Used to Analyze KIT Exons
| Single step PCR | Forward primer | Reverse primer |
|---|---|---|
| Exon 9 | ATGCTCTGCTTCTGTACTGCC | CAGAGCCTAAACATCCCCTTA |
| Exon 11 | CCAGAGTGCTCTAATGACTG | ACCCAAAAAGGTGACATGGA |
| Exon 13 | CATCAGTTTGCCAGTTGTGC | ACACGGCTTTACCTCCAATG |
| Exon 17 | TGTATTCACAGAGACTTGGC | GGATTTACATTATGAAAGTCACAGG |
| Nested PCR | Forward primer | Reverse primer | Second forward primer | Second reverse primer |
|---|---|---|---|---|
| Exon 9 | AGCCAGGGCTTTTGTTTTCT | CAGAGCCTAAACATCCCCTTA | ATGCTCTGCTTCTGTACTGCC | CCTTTGTTGTTACCTTTAAATGC |
| Exon 11 | CCTTTGCTGATTGGTTTCGT | AAACAAAGGAAGCCACTGGA | CCAGAGTGCTCTAATGACTG | ACCCAAAAAGGTGACATGGA |
| Exon 13 | GTTCCTGTATGGTACTGCATGCG | CAGTTTATAATCTAGCATTGCC | CATCAGTTTGCCAGTTGTGC | ACACGGCTTTACCTCCAATG |
| Exon 17 | GGTTTTCTTTTCTCCTCCAACC | GGATTTACATTATGAAAGTCACAGG | TGTATTCACAGAGACTTGGC | GAAACTAAAAATCCTTTGCAGGAC |
Aliquots (5 to 20 μl) of the final PCR reaction were screened for mutations on a Transgenomic WAVE HPLC system (D-HPLC; Transgenomic, Inc., Omaha, NE) by running at nondenaturing (50°C) or partially denaturing temperature (exon 11, 56°C; exon 13, 59°C; exon 17, 58°C). D-HPLC-detected mutations were confirmed by two methods: 1) re-amplification of the exon and repeat D-HPLC analysis on a different day; 2) bi-directional sequence analysis on an ABI 377 sequencer using the BigDye terminator kit (Applied Biosystems, Inc., Foster City, CA).
Case 9 was heavily infiltrated by lymphocytes and yielded only a small mutant peak at the nondenaturing temperature when its exon 11 amplimer was analyzed by D-HPLC. Although this peak suggested the presence of a deletion, the quantity of mutant DNA was too small to be confirmed by direct sequencing. Therefore the amplification products were cloned into pCR4-TOPO using the TOPO TA cloning kit (version H; Invitrogen, Carlsbad, CA) and the ligated plasmids were used to transform competent Escherichia coli (OneShot TOP10, Invitrogen). Isolated plasmids were screened for the mutant exon insert by PCR and D-HPLC. Direct sequence analysis of cloned mutant DNA confirmed the presence of an in-frame exon 11 deletion in this case.
Results and Discussion
In a series of 120 GISTs identified from the pathology archives of three Portland area hospitals, there were 15 tumors that were clinically inapparent and were discovered incidentally at the time of surgery, endoscopy, or autopsy. Two of these tumors were >1 cm in greatest diameter (3.2 cm and 2.7 cm) and had negative prognostic features (>5 mitoses per 50 high-power fields, necrosis); these were excluded from the study. The remaining 13 tumors ranged in size from 4 to 10 mm, were morphologically benign, and lacked mitoses on routine H&E-stained sections (Figure 1 ▶ ; A to C). Two of the tumors represented incidental findings at autopsy, two were discovered during endoscopy, and the remaining nine were identified at laparotomy performed for unrelated disease (Table 2) ▶ . In all cases, the tumor cells stained positively for KIT by immunohistochemistry (Figure 1D) ▶ .
Figure 1.
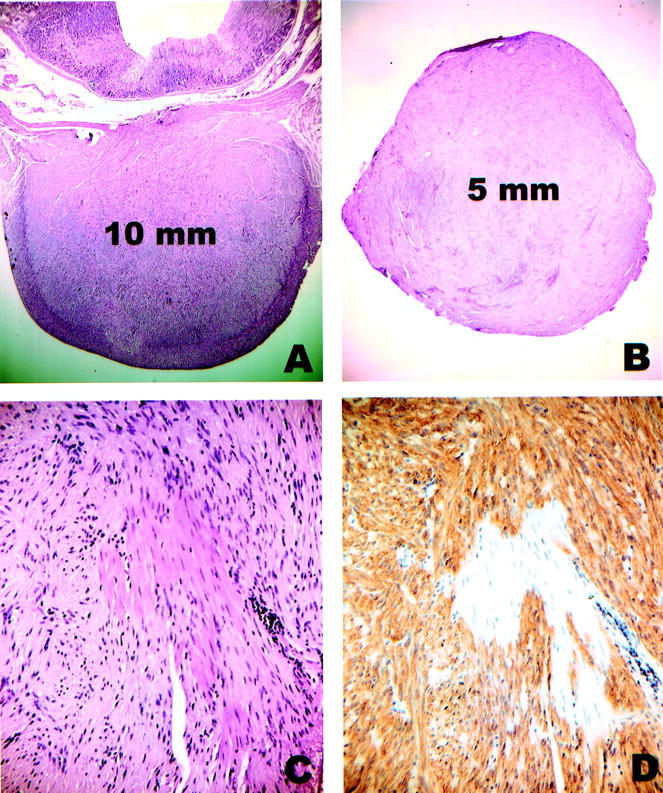
Incidental GISTs. A: H&E-stained section of case 6 showing a 10-mm tumor centered in the muscularis propria of the stomach. The lesion was discovered during a gastroesophagectomy for a large esophageal leiomyoma. B: H&E-stained section of case 11 showing a well-circumscribed 5-mm nodule that was removed from the serosal aspect of the small bowel during surgery for endometrial adenocarcinoma. C: Close-up of case 11. Note that entrapped smooth muscle cells of the muscularis propria are morphologically similar to the tumor cells. D: Same area of case 11 as in C, immunostained for KIT. The tumor cells are strongly positive whereas the smooth muscle cells are negative.
Table 2.
Incidental GISTs
| Case | Sex/age | Size | Location | How discovered | KIT mutation |
|---|---|---|---|---|---|
| 1 | F/88 | 9 mm | Stomach | Surgery for ovarian cystadenoma | Exon 11 |
| 2 | M/53 | 10 mm | Small intestine | Exploratory laparotomy (lymphoma) | Exon 11 |
| 3 | M/56 | <10 mm | Stomach | Autopsy (massive pulmonary emboli) | Exon 11 |
| 4 | M/45 | 6 mm | Rectum | Incidental finding at endoscopy | Exon 11 |
| 5 | M/76 | 4 mm | Stomach | Surgery for esophageal adenocarcinoma | Exon 11 |
| 6 | F/48 | 10 mm | Stomach | Surgery for large esophageal leiomyoma | Exon 11 |
| 7 | M/65 | 7 mm | Stomach | Surgery for gastric adenocarcinoma | Exon 11 |
| 8 | F/78 | 10 mm | Stomach | Vagotomy for unrelated gastric ulcer disease | Exon 11 |
| 9 | F/65 | 8 mm | Small intestine | Surgery for villous adenoma of colon | Exon 11 |
| 10 | M/60 | 6 mm | Stomach | Incidental finding at endoscopy | Exon 11 |
| 11 | F/62 | 5 mm | Small intestine | Surgery for endometrial adenocarcinoma | Exon 9 |
| 12 | M/64 | 5 mm | Stomach | Incidental to gastric polyp resection | None detected |
| 13 | F/80 | 5 mm | Stomach | Autopsy (myelodysplastic syndrome) | None detected |
Genomic DNA prepared from each of the 13 incidental tumors was subjected to PCR amplification of exon 11 of the KIT gene, yielding a 236-bp product that was screened for the presence of a mutation on a modified HPLC system (D-HPLC) run at nondenaturing and partially denaturing temperatures. Ten tumors (77%) demonstrated aberrant D-HPLC profiles for exon 11, as illustrated in Figure 2, B and C ▶ . The corresponding mutations found by direct sequencing are listed in Table 3 ▶ . Three were point mutations and the remainder consisted of deletions with or without small substitutions. Each of these mutations was confirmed by D-HPLC analysis of a repeat amplification from the original tumor DNA; in some cases, the DNA sequence analysis was also repeated. All of these mutations have been observed in larger, clinically significant GISTs (M Heinrich et al, manuscript in preparation). 17 Figure 2C ▶ illustrates a tumor (case 7) in which the deletion mutation was detected but the wild-type allele was not. The locus or chromosome bearing the wild-type allele in this case, as well as that in case 4, was apparently lost. Detection of such events in subclinical GISTs suggests that there may be a selective advantage to down-regulating wild-type KIT expression in the presence of a KIT mutation.
Figure 2.

D-HPLC elution profiles of KIT exon amplimers. A: Wild-type exon 11 amplimer (case 12) run at denaturing and nondenaturing temperatures. B: Exon 11 amplimer containing a point mutation (case 6) is resolved from wild-type amplimer at the denaturing temperature. C: Comparison of the elution profiles for exon 11 amplimers representing pure wild-type sequence (case 12), pure deletion mutation (case 7), and a mixture of wild-type and deletion mutations (case 8). In the latter case there are four separate peaks that represent homotypic and heterotypic combinations of mutant and wild-type strands after re-annealing. D: The exon 9 insertion mutation discovered in case 11 has a distinctive elution pattern in comparison with the wild-type amplimer from case 12.
Table 3.
Mutations Detected in Incidental GISTs
| Case | Exon 11 | Exon 9 | Exon 13 | Exon 17 |
|---|---|---|---|---|
| 1 | Deletion KVVEEING 558–565 R* | − | − | − |
| 2 | Point mutation V559D* | − | − | − |
| 3 | Deletion KPMYEVQWK 550–558* | − | − | − |
| 4 | Deletion D570 (homozygous)* | − | − | − |
| 5 | Deletion NYVYIDPTQL 567–576 KV* | − | − | − |
| 6 | Point mutation V560D* | WTDHPLC | − | − |
| 7 | Deletion QKPMYEVQWK 549–558Q (homozygous)* | WTDHPLC | WTDHPLC | WTDHPLC |
| 8 | Deletion KPMYEVQWK 550–558* | WTDHPLC | − | − |
| 9 | Deletion YIDPTQLPY 570–578* | WTDHPLC | − | − |
| 10 | Point mutation V559D* | WTDHPLC | − | − |
| 11 | WTDHPLC | Insertion AY 502–503* | − | − |
| 12 | WT* | WTDHPLC | WTDHPLC | WTDHPLC |
| 13 | WT* | WTDHPLC | −† | WTDHPLC |
*Sequence confirmed.
†Insufficient DNA available DHPLC denaturing HPLC.
−, Not done.
Tumors that were negative for exon 11 mutations were further analyzed for mutations in exon 9 of KIT by the combination of D-HPLC and direct sequencing. Case 11 reproducibly yielded an AY duplication/insertion in this exon (Figure 2C ▶ ; Table 3 ▶ ). This mutation has been observed in several recent studies of GISTs and has been shown to cause constitutive activation of KIT. 8,11,13,21 The other two tumors showed wild-type profiles for all exons examined (insufficient DNA was available to allow exon 13 analysis on one of these tumors). It remains possible that both of these tumors harbor a mutation elsewhere in the KIT gene.
The issue of whether KIT mutations are related to the oncological progression of GISTs has been raised in several studies, with differing results. Lasota and co-workers 6 found only one exon 11 mutation in 19 benign GISTs (5.2%), whereas 12 of 24 malignant GISTs (50%) harbored detectable mutations. Similarly, Li and colleagues 20 reported exon 11 mutations in 3 of 4 borderline tumors and 6 of 10 malignant tumors, but found no mutations in the 2 benign lesions examined. This apparent difference between benign and malignant GISTs was not as pronounced in the study by Debiec-Rychter and colleagues 14 that included sequencing of exons 9 and 13. They observed KIT mutations in 3 of 9 (33%) benign tumors and 8 of 14 (57%) malignant tumors.
Using a reverse transcriptase-PCR approach, Rubin and colleagues 12 recently documented KIT mutations in 10 of 10 benign GISTs (100%), paralleling the results reported here. Indeed, there was no difference in the KIT mutation frequency among the benign, borderline, and malignant tumors examined by Rubin and colleagues, 12 (total 48 cases), and the overall incidence of mutations was strikingly high (92%). The data from this study suggest that technical issues may influence the detection of KIT mutations in genomic DNA, reducing the apparent mutation frequency. In a recent analysis of genomic DNA from 72 malignant GISTs, we used D-HPLC to screen PCR amplimers of the KIT gene and found mutations (sequence-confirmed) in 86% of cases, which is quite close to the frequency observed by Rubin and colleagues 17 using reverse transcriptase-PCR. Based on our experience, there are two advantages of using D-HPLC to screen PCR amplimers. First, D-HPLC screening predicts the type of mutation to be expected during DNA sequence analysis. Second, there is increased sensitivity for mutations (<10% mutant DNA can be detected). This is illustrated by case 9, in which a minor peak representing an exon 11 deletion was reproducibly detected but could not be confirmed by direct sequence analysis. Contaminating wild-type DNA from infiltrating lymphocytes likely interfered with the sequence analysis, although it is also possible that only a minority of the tumor cells harbored the mutation. Subcloning of the mutant peak yielded sufficient mutant amplimer to document the in-frame deletion in this case.
In applying the D-HPLC approach to the incidental tumors in this report, we detected exon 11 mutations in 77% (10 of 13), matching the 70% frequency that we observed among the 72 malignant GISTs. Moreover, the overall percentage of KIT mutations in the group of incidental tumors (85%) was essentially identical to that of the malignant GISTs (86%). It has previously been suggested that GISTs harboring an exon 11 mutation have a worse prognosis than tumors without detected mutations. 7,19 However, the relatively low overall frequency of KIT mutations observed in these studies (37 to 57%) complicates the interpretation of their findings.
In the course of selecting incidental GISTs for this study, we identified several small leiomyomas of the type recently reviewed by Miettinen and colleagues. 22 These lesions, which are often associated with the muscularis mucosae, may have areas of hyalinization and other morphological features that closely resemble low-grade GISTs on H&E stain. Nevertheless, they are immunohistochemically negative for KIT and are usually positive for desmin. We examined one small leiomyoma for KIT mutations and found none (data not shown).
The presence of activating KIT mutations in a high percentage of early, benign GISTs is entirely consistent with the proposal that these mutations represent a gatekeeper alteration that plays a fundamental role in GIST development. It is also consistent with recent studies of four different kindreds in which heritable mutations of KIT exon 11 or 13 have been demonstrated. 23-26 In all four kindreds, affected individuals develop multiple GISTs, and in some of these patients there is demonstrable hyperplasia of KIT-positive cells in the area of Auerbach’s plexus, consistent with interstitial cells of Cajal.
In summary, we have examined 13 clinically incidental, morphologically benign GISTs that were 10 mm or less in size and found an 85% incidence of mutations in the KIT gene. This high frequency of mutations is strikingly similar to that observed by the same methodology in a larger series of clinically significant GISTs, suggesting that these mutations occur very early in the course of GIST development. Our findings do not support a negative prognostic impact of KIT mutations, as has been proposed in other studies.
Acknowledgments
We thank Drs. John Thompson, Karen Oyama, and Robert Krum for their assistance in identifying gastrointestinal stromal tumors in the archives of the Northwest Kaiser Regional Laboratory; and Linda Jauron-Mills and Carolyn Gendron for expert technical assistance.
Footnotes
Address reprint requests to Christopher L. Corless, M.D., Ph.D., Dept. of Pathology (L471), Oregon Health and Science University, 3181 SW Sam Jackson Park Rd., Portland, OR 97201. E-mail: corlessc@ohsu.edu.
Supported in part by grants from the Veteran’s Administration Merit Review Program, the Northwest Health Foundation (to M. C. H.), and the Oregon Cancer Institute (to C. L. C.).
References
- 1.Miettinen M, Lasota J: Gastrointestinal stromal tumors—definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001, 438:1-12 [DOI] [PubMed] [Google Scholar]
- 2.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279:577-580 [DOI] [PubMed] [Google Scholar]
- 3.Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M: CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol 1998, 11:728-734 [PubMed] [Google Scholar]
- 4.Nakahara M, Isozaki K, Hirota S, Miyagawa J, Hase-Sawada N, Taniguchi M, Nishida T, Kanayama S, Kitamura Y, Shinomura Y, Matsuzawa Y: A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology 1998, 115:1090-1095 [DOI] [PubMed] [Google Scholar]
- 5.Moskaluk CA, Tian Q, Marshall CR, Rumpel CA, Franquemont DW, Frierson HF, Jr: Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999, 18:1897-1902 [DOI] [PubMed] [Google Scholar]
- 6.Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M: Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999, 154:53-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, Nomura T, Matsuda H, Kitamura Y: Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999, 59:4297-4300 [PubMed] [Google Scholar]
- 8.Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, Xiao S, Singer S, Fletcher CD, Fletcher JA: KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000, 156:791-795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rader AE, Avery A, Wait CL, McGreevey LS, Faigel D, Heinrich MC: Fine-needle aspiration biopsy diagnosis of gastrointestinal stromal tumors using morphology, immunocytochemistry, and mutational analysis of c-kit. Cancer 2001, 93:269-275 [DOI] [PubMed] [Google Scholar]
- 10.Fukuda R, Hamamoto N, Uchida Y, Furuta K, Katsube T, Kazumori H, Ishihara S, Amano K, Adachi K, Watanabe M, Kinoshita Y: Gastrointestinal stromal tumor with a novel mutation of KIT proto-oncogene. Intern Med 2001, 40:301-303 [DOI] [PubMed] [Google Scholar]
- 11.Hirota S, Nishida T, Isozaki K, Taniguchi M, Nakamura J, Okazaki T, Kitamura Y: Gain-of-function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol 2001, 193:505-510 [DOI] [PubMed] [Google Scholar]
- 12.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA: KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001, 61:8118-8121 [PubMed] [Google Scholar]
- 13.Sakurai S, Oguni S, Hironaka M, Fukayama M, Morinaga S, Saito K: Mutations in c-kit gene exons 9 and 13 in gastrointestinal stromal tumors among Japanese. Jpn J Cancer Res 2001, 92:494-498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debiec-Rychter M, Lasota J, Sarlomo-Rikala M, Kordek R, Miettinen M: Chromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutation. Cancer Genet Cytogenet 2001, 128:24-30 [DOI] [PubMed] [Google Scholar]
- 15.Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, Fletcher JA, Demetri GD: STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene 2001, 20:5054-5058 [DOI] [PubMed] [Google Scholar]
- 16.Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, Silberman S, Capdeville R, Dimitrijevic S, Druker B, Demetri GD: Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001, 344:1052-1056 [DOI] [PubMed] [Google Scholar]
- 17.Blanke CD, von Mehren M, Joensuu H, Roberts PJ, Esienberg B, Heinrich M, Druker B, Tuveson D, Dimitrijevic S, Demetri GD: Evaluation of the safety and efficacy of an oral molecularly-targeted therapy, STI571, in patients with unresectable or metastatic gastrointestinal stromal tumors (GISTs) expressing c-kit (CD117). Proc Am Soc Clin Oncol 2001, 20:1a(Abstract) [Google Scholar]
- 18.van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, Martens M, Webb A, Sciot R, Van Glabbeke M, Silberman S, Nielsen OS: Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet 2001, 358:1421-1423 [DOI] [PubMed] [Google Scholar]
- 19.Ernst SI, Hubbs AE, Przygodzki RM, Emory TS, Sobin LH, O’Leary TJ: KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998, 78:1633-1636 [PubMed] [Google Scholar]
- 20.Li SQ, O’Leary TJ, Sobin LH, Erozan YS, Rosenthal DL, Przygodzki RM: Analysis of KIT mutation and protein expression in fine needle aspirates of gastrointestinal stromal/smooth muscle tumors. Acta Cytol 2000, 44:981-986 [DOI] [PubMed] [Google Scholar]
- 21.Lasota J, Wozniak A, Sarlomo-Rikala M, Rys J, Kordek R, Nassar A, Sobin LH, Miettinen M: Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol 2000, 157:1091-1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miettinen M, Sarlomo-Rikala M, Sobin LH: Mesenchymal tumors of muscularis mucosae of colon and rectum are benign leiomyomas that should be separated from gastrointestinal stromal tumors—a clinicopathologic and immunohistochemical study of eighty-eight cases. Mod Pathol 2001, 14:950-956 [DOI] [PubMed] [Google Scholar]
- 23.Maeyama H, Hidaka E, Ota H, Minami S, Kajiyama M, Kuraishi A, Mori H, Matsuda Y, Wada S, Sodeyama H, Nakata S, Kawamura N, Hata S, Watanabe M, Iijima Y, Katsuyama T: Familial gastrointestinal stromal tumor with hyperpigmentation: association with a germline mutation of the c-kit gene. Gastroenterology 2001, 120:210-215 [DOI] [PubMed] [Google Scholar]
- 24.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, Kanakura Y, Tanaka T, Takabayashi A, Matsuda H, Kitamura Y: Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet 1998, 19:323-324 [DOI] [PubMed] [Google Scholar]
- 25.Isozaki K, Terris B, Belghiti J, Schiffmann S, Hirota S, Vanderwinden JM: Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol 2000, 157:1581-1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirota S, Okazaki T, Kitamura Y, O’Brien P, Kapusta L, Dardick I: Cause of familial and multiple gastrointestinal autonomic nerve tumors with hyperplasia of interstitial cells of Cajal is germline mutation of the c-kit gene. Am J Surg Pathol 2000, 24:326-327 [DOI] [PubMed] [Google Scholar]