

Abstract
Diabetic retinopathy remains a leading cause of irreversible blindness. A critical early pathology in the disease is the adhesion of leukocytes to the retinal vasculature, a process that occurs, in part, via intercellular adhesion molecule-1. Once leukocyte adhesion occurs, endothelial cell injury ensues, as does blood-retinal barrier breakdown. Here we show that angiopoietin-1 can prevent and reverse these diabetic retinal vascular changes in both new and established diabetes. Angiopoietin-1, when given intravitreally to newly diabetic rats, normalized retinal vascular endothelial growth factor (VEGF) and intercellular adhesion molecule-1 mRNA and protein levels, leading to reductions in leukocyte adhesion, endothelial cell injury, and blood-retinal barrier breakdown. When an adenovirus coding for angiopoietin-1 was given systemically to mice with established diabetes, it similarly inhibited leukocyte adhesion and endothelial cell injury and blood-retinal barrier breakdown. These changes coincided with reductions in retinal eNOS, nitric oxide, Akt (protein kinase B), and MAP kinase activity, known mediators of VEGF bioactivity and leukocyte adhesion. When endogenous VEGF bioactivity was inhibited with a soluble Flt-1/Fc chimera, retinal Akt kinase activity was significantly reduced in vivo. Taken together, these data document new vascular and anti-inflammatory bioactivities for angiopoietin-1 and identify it as the first naturally occurring protein that directly protects the retinal vasculature in diabetes.
Diabetic retinopathy in rodents recapitulates much of the pathology of human diabetic retinopathy. In both species, the diabetic retinal vasculature up-regulates intercellular adhesion molecule-1 (ICAM-1) and contains increased numbers of leukocytes. 1-4 In rodents, leukocytes adhere to the retinal vasculature via ICAM-1 and trigger capillary occlusion, endothelial cell injury and death, and blood-retinal barrier breakdown—hallmarks of human diabetic retinopathy. 1,2
Recent findings with angiopoietin-1 (Ang-1) suggested that it might be worth evaluating in models of diabetic retinopathy. Ang-1 promotes endothelial cell survival in vitro without causing endothelial cell proliferation, 5-9 blocks the leak-inducing action of vascular endothelial growth factor (VEGF) in vivo, 10,11 and stabilizes endothelial interactions with surrounding support cells. 10 Ang-1 is a 70-kd protein that binds to the tyrosine kinase with immunoglobulin and endothelial growth factor homology domains (Tie-2) receptor, which is selectively expressed on endothelial cells. 5 Ang-1 is highly conserved, with the human and mouse Ang-1 amino acid sequences displaying 94% homology. Whereas VEGF has a requisite role in early endothelial cell differentiation, proliferation, and vessel formation; Ang-1 seems to function later in vascular development, promoting vascular remodeling and maturation. 12 Ang-1 is expressed primarily in nonendothelial cells, including peri-endothelial cells, 13-15 however relatively little is known about the regulation of its expression. In cultured cells, platelet-derived growth factor, epidermal growth factor, transforming growth factor-β, and hypoxia all down-regulate Ang-1 mRNA levels. 16 In the current study, we explored the therapeutic effects of Ang-1 and its related mechanisms in two relevant models of diabetic retinopathy.
Materials and Methods
Cell Culture
Bovine retinal endothelial cells were isolated as previously described 17 and cultured in EBM 131 (Clonetics, San Diego, CA), 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT), 1× gentamicin, penicillin, streptomycin, and 2 ng/ml of basic fibroblast growth factor in 5% CO2. The endothelial phenotype was confirmed by CD31 staining [fluorescein isothiocyanate (FITC)-coupled anti-human CD31 mAb; Pharmingen, San Diego, CA]. All cells were cultured at 37°C in 5% CO2, 95% air, and media were changed every 2 to 3 days.
Animals and Experimental Diabetes
All animal experiments followed the guidelines of the Association for Research in Vision and Ophthalmology and were approved by the Animal Care and Use Committees of the Children’s Hospital and the Massachusetts Eye and Ear Infirmary. Male Long-Evans rats weighing ∼200 g (Charles River Laboratories, Wilmington, MA) received a single 60-mg/kg injection of streptozotocin (Sigma Chemical Co., St. Louis, MO) in 10 mmol/L of citrate buffer, pH 4.5, after an overnight fast. Control nondiabetic rats received injections of citrate buffer alone. Rats with blood glucose levels >250 mg/dl at 24 hours later were deemed diabetic. All rat experiments were performed 1 week after the induction of diabetes. Male C57/BL6 mice (Charles River Laboratories), weighing 20 to 25 g received daily 60-mg/kg intraperitoneal injections of streptozotocin for consecutive 5 days. A urine volume >15 ml/24 hours 1 week after the final injection correlated to a blood glucose level >300 mg/dl. The mice were analyzed 16 weeks after the induction of diabetes. The diabetic state of the rats and mice, defined as a blood glucose level >250 mg/dl, was confirmed 24 hours before the onset of each experiment and once again before sacrifice. All animals were fed standard laboratory chow and were allowed free access to food and water in an air-conditioned room with a 12-hour light/dark cycle.
Treatment of Retinal Endothelial Cells with Ang-1
Bovine retinal endothelial cells were maintained in basal medium with growth supplements, serum-starved for 24 hours, and then challenged with Ang-1 (250 ng/ml) for 30 minutes. The cells were subsequently washed, lysed in buffer containing 20 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L glycerophosphate, 1 mmol/L Na3O4, 1 μg/ml leupeptin, and 1 mmol/L phenylmethyl sulfonyl fluoride. Akt activity was then measured as described below.
Intraocular Ang-1 Therapy
Recombinant human Ang-1 was produced and purified at Regeneron Pharmaceuticals (Tarrytown, NY). Ang-1 was genetically modified at the NH2 terminus and contained a mutated Cys245 for facilitated purification. The material retained all of the properties of native Ang-1 in all assays (data not shown). All concentrations of recombinant human Ang-1 were formulated in vehicle containing 10 mmol/L of phosphate, 150 mmol/L of NaCl and 5% glycerol, pH 7.2. The protein was >95% pure, as judged by reducing and nonreducing silver-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (data not shown). Each rat eye received a single intravitreous injection. After anesthesia with xylazine hydrochloride (4 mg/kg; Phoenix Pharmaceutical, St. Joseph, MO) and ketamine hydrochloride (25 mg/kg; Parke-Davis, Morris Plains, NJ), the pupils of both eyes were dilated with 1% tropicamide (Alcon, Humancao, PR). Doses of 800, 200, and 50 ng of Ang-1 each were diluted in 5 μl of the above-listed buffer to make concentrations of 160 μg/ml, 40 μg/ml, and 10 μg/ml, respectively. A fourth condition, vehicle alone, was also tested. The animals were randomly assigned to the various treatment groups and the experimenters were masked to the identity of the injections. Under direct visualization, the material was injected into the center of the vitreous, 0.5-mm posterior to the limbus, using a 32-gauge needle. The contralateral eye of each rat received 5 μl of phosphate-buffered saline (PBS) alone as a control. Any eyes with evidence of injury to the lens or retina were discarded. The eyes were studied 24 hours later as described below.
Systemic Ang-1 Therapy
Ad-Ang1 was constructed using the Adeno-Quest system (Quantum Biotechnology, Montreal, Canada). Full-length cDNA encoding a genetically engineered version of human Ang-1 was cloned into shuttle vector pQBI-AdCMV5GFP. Linearized plasmid and viral long-arm DNA were co-transfected into 293 cells (CRL-1573; American Type Culture Collection, Rockville, MD). Viral plaques were assayed for protein expression and further purified by two more rounds of plaque purification. Viral particles were precipitated with polyethylene glycol, further purified by cesium chloride density centrifugation, and stored at −80°C in buffer. The viral titer was determined using the Tissue Culture Infection Dose50 method as outlined in the manual for the Adeno-Quest system (Quantum Biotechnology). The amount of replication competent adenovirus was determined by applying a test dose of each adenoviral preparation to HeLa cells. After a 4-day culture period, a lysate from the HeLa cells was applied to A459 cells and the cytopathic effect was assessed. All adenoviral preparations tested had less than one replication competent adenoviral particle in 108 PFU replication-deficient adenovirus. Anesthetized diabetic mice received an intravenous tail vein injection of 1 × 109 PFU Ad-Ang1. Control diabetic mice received an equal amount of an adenovirus expressing green fluorescent protein (Ad-GFP). In this system, serum Ang-1 levels are 5 to 15 μg/ml at 72 hours and decline slowly thereafter. 11 The mice retinae were examined for leukocyte adhesion and endothelial cell death 72 hours after virus injection. The animals receiving the Ad-Ang1 and Ad-GFP viruses remained healthy throughout the experiment.
Systemic VEGF TrapA40 and Interleukin (IL)-6 Trap Therapy
VEGF TrapA40 and IL-6R Trap were synthesized at Regeneron Pharmaceuticals Inc. (Tarrytown, NY). VEGF TrapA40 consisted of immunoglobulin repeats 1 to 3 of the extracellular domain of human Flt-1 fused to the Fc portion of human IgG1. The protein was expressed in Chinese hamster ovary cells and purified via protein A affinity chromatography and size exclusion chromatography. The recombinant Flt-Fc chimera was then chemically modified to improve the pharmacokinetic profile of the parent molecule, without affecting its ability to bind VEGF with high affinity (Rudge and colleagues, unpublished data). The purity of the modified recombinant protein was determined to be >95% by Coomassie-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The protein was filter sterilized and stored in PBS, pH 7.2, containing 5% glycerol at −20°C. VEGF TrapA40 bioactivity was confirmed in endothelial cell proliferation assays before its use (data not shown). IL-6R Trap was made from the extracellular domain of human IL-6Rα (the low-affinity IL-6 receptor) fused to the Fc domain of human IgG1. IL-6R Trap binds only human IL-6 with low affinity, and not mouse or rat IL-6. IL-6R Trap was Chinese hamster ovary cell-derived, purified via protein A and size exclusion chromatography, and was >95% pure on Coomassie-stained gels. VEGF TrapA40 and IL-6R Trap were dissolved in sterile Tris-BisTris-Cl-sodium acetate buffer (15 mg/ml). On day 7 of diabetes, diabetic rats were randomized to receive a single 25-mg/kg intraperitoneal injection of either VEGF TrapA40 or IL-6R Trap.
Ex Vivo Retinal Leukostasis Quantitation in Rats and Mice
After the induction of deep anesthesia, the chest cavity was opened and a 14-gauge perfusion cannula was introduced into the left ventricle. Drainage was achieved using a 16-gauge needle placed in the right atrium. The animals were perfused with 250 ml of PBS per kg body weight (BW) to remove erythrocytes and nonadherent leukocytes. After PBS perfusion, fixation with 1% paraformaldehyde and 0.5% glutaraldehyde was achieved using 200 ml/kg BW of perfusate for ∼3 minutes. The PBS perfusion was performed at a physiological pressure because the pumping heart provided the motive force. All subsequent perfusions were post mortem and were performed at 100 mmHg pressure. Nonspecific binding was blocked with 1% albumin in PBS (total volume 100 ml/kg BW) followed by perfusion with FITC-coupled concanavalin A lectin (20 μg/ml in PBS, pH 7.4, 5 mg/kg BW) (Vector Laboratories, Burlingame, CA). Concanavalin A was used to label adherent leukocytes and vascular endothelial cells. Residual unbound lectin was removed with a 1% albumin in PBS perfusion for 1 minute followed by a PBS perfusion for 4 minutes. The retinae were carefully removed and flat mounts prepared using a fluorescence anti-fading medium (Southern Biotechnology, Birmingham, AL). The retinae were then imaged using a fluorescence microscope (FITC filter; Zeiss Axiovert, Oberkochen, Germany). Retinae in which the peripheral collecting vessels of the ora serrata were not visible were discarded. The total number of leukocytes in the retinal arterioles, venules, and capillaries was then determined.
Propidium Iodide (PI) Labeling
Injured and/or dying endothelial cells were labeled in vivo using PI (Molecular Probes, Eugene, OR), a molecule that is excluded from uninjured viable cells. PI fluoresces when it leaks through injured cell membranes and binds to DNA and RNA, identifying transiently injured viable cells or cells undergoing either necrosis or apoptosis. 18 After the induction of deep anesthesia with 50 mg/kg of intraperitoneal sodium pentobarbital, PI (1 mg/ml in PBS) was injected intravenously via the tail vein at a concentration of 5 μmol/kg (0.668 ml/200 mg BW). The solution was allowed to circulate for 20 minutes, after which it was followed by body perfusion, fixation, and lectin labeling as described above. Retinal flat mounts were examined via fluorescence microscopy as described above. Labeled endothelial cells were distinguished from surrounding cells, especially pericytes, by focusing through the tissue to discern the distinct cellular outline and nuclear shape of the endothelial cells.
Measurement of Blood-Retinal Barrier Breakdown Using Evans Blue
Blood retinal barrier breakdown was quantitated using Evans blue dye, which noncovalently binds to plasma albumin in the blood stream. 19 Evans blue dye (Sigma Chemical Co.) was dissolved in normal saline (30 mg/ml), sonicated for 5 minutes, and filtered through a 5-μm filter. A heparinized PE50 catheter was inserted in the femoral artery. Under deep anesthesia, Evans blue was injected through a jugular vein catheter for 10 seconds at a dose of 30 mg/kg. Blood was withdrawn through the femoral catheter into a heparinized syringe every 15 minutes, for a total of 120 minutes. After the dye circulated for 120 minutes, the chest cavity was opened and left heart ventricle cannulated. Each rat was perfused with citrate-buffered 1% paraformaldehyde (37°C) for 2 minutes to clear the dye, keeping the physiological pressure of 100 mmHg. Immediately after perfusion, the eyes were enucleated and the retinae were carefully dissected away under an operating microscope. The weight of each retina was measured after thorough drying in a Speed-Vac. Albumin leakage into the retinal tissue was estimated via the measurement of extravasated Evans blue dye. Evans blue was extracted by incubating each retina in 0.3 ml of formamide for 18 hours at 70°C. The extract was filtered through a 30,000 MW filter at a speed of 70,000 rpm for 45 minutes at 4°C. The absorbance of the filtrate was measured with a spectrophotometer at 620 nm, the absorption maximum for Evans blue in formamide. The arterial blood samples were centrifuged at 12,000 rpm for 20 minutes at 4°C. The concentration of dye in the extracts was calculated from a standard curve of Evans blue in formamide and normalized to the dry retinal weight and the time-averaged concentration of Evans Blue in the plasma. 20
Ribonuclease Protection Assay
Rat retinae were gently dissected free, cut at the optic disk after enucleation, and immediately frozen in liquid nitrogen. Total RNA was isolated according to the acid guanidinium thiocyanate-phenol-chloroform extraction method. A 425-bp EcoRI/BamHI fragment of rat ICAM-1 cDNA and a segment of rat VEGF cDNA containing exons 4 through 7 and a portion of exon 8 was prepared by reverse transcription-polymerase chain reaction. Nucleotide antisense riboprobes were prepared by in vitro transcription (Promega, Madison, WI) of linearized plasmid DNA with T7 RNA polymerase in the presence of [32P]dUTP. The sequence of the cloned cDNA was verified by DNA sequencing. Twenty μg of total cellular RNA was used for the ribonuclease protection assay. All samples were simultaneously hybridized with an 18S riboprobe (Ambion, Austin, TX) to normalize for variations in loading and recovery of RNA. Protected fragments were separated on a gel of 5% acrylamide, 8 mol/L urea, 1× Tris-borate-EDTA, and quantified with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA). All signals were within the linear range of quantitation.
Enzyme-Linked Immunosorbent Assay (ELISA) for ICAM-1
Retinal ICAM-1 protein was quantified after intravitreal injection of either Ang-1 (800 ng) or PBS. Each retina was homogenized in 100 μl of a solution consisting of 20 mmol/L imidazole hydrochloride, 100 mmol/L KCl, 1 mmol/L MgCl, 1 mmol/L EGTA, 1% Triton, 10 mmol/L NaF, 1 mmol/L sodium molybdenate, 1 mmol/L EDTA, and supplemented with a protease inhibitor cocktail (Complete; Roche, Basel, Switzerland). Samples were cleared by centrifugation for 10 minutes at 13,000 rpm and assessed for protein concentration with the bicinchoninic acid (BCA) assay (Mini BCA kit; Pierce Scientific, Rockford, IL). Flat-bottomed 96-well microtiter plates (Immuno-Plate I 96-F; Nunc, Naperville, IL) were coated with 50 μl/well (1 ng/ml) of a specific rabbit anti-ICAM-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) in coating buffer (0.6 mol/L NaCl, 0.26 mol/L H3PO4, and 0.08 N NaOH, pH 9.6) for 16 to 24 hours at 4°C. Nonspecific sites were blocked with 2% bovine serum albumin in PBS for 60 minutes at 37°C, followed by sample addition of a 50-μl aliquot in duplicate and incubated for 60 minutes at 37°C. After washing, 50 μl of a biotinylated anti-ICAM-1 rabbit polyclonal Ab (3.5 μg/ml in PBS, pH 7.5, 0.05% Tween-20, and 2% fetal calf serum) was added and incubated for 45 minutes at 37°C. The antibody was biotinylated with the MiniBiotin XX protein labeling kit (Molecular Probes). The plates were washed, streptavidin-peroxidase conjugate (1/1000; R&D Systems, Minneapolis, MN) was added, and the plates were incubated for 30 minutes at 37°C. The plates were washed again, the substrate TMB (3,3′,5,5′-tetramethylbenzidine; Kirkegaard & Perry, Gaithersburg, MD) was added for color development, and the reaction was quenched using 100 μl of 1 mol/L H2PO4. The plates were read at 450 nm with an automated microplate reader. A standard curve was generated by using serial dilutions of the raising peptide (Santa Cruz Biotechnology).
ELISA for VEGF
Retinal VEGF protein was quantified 24 hours after intravitreal injection of either Ang-1 (800 ng) or solvent. The samples were prepared as described above. Briefly, the VEGF protein levels were estimated with an ELISA kit (R&D Systems) according to the manufacturer’s instructions. All measurements were performed in duplicate. The tissue sample concentration was calculated from a standard curve and corrected for protein concentration.
Measurement of Akt-Kinase Activity
Retinal and cellular Akt kinase activity was quantified using a modified ELISA protocol. Briefly, each well of a 96-well microtiter plate was coated with 100 μl of 1 μmol/L of GSK-3αβ fusion protein, which serves as a substrate for the activated Akt kinase, overnight at 4°C in carbonate buffer (15 mmol/L Na2CO3, 35 mmol/L NaHCO3, 0.2 g/L NaN3, pH 9.6). After washing, the remaining active sites were blocked in 10 mg/ml of bovine serum albumin in PBS containing 0.05% Tween-20. Whole retinas were dissected from treated animals as described above, or cultured cells isolated, and homogenized in lysis buffer containing 20 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L glycerophosphate, 1 mmol/L Na3O4, 1 μg/ml leupeptin, 1 mmol/L phenylmethyl sulfonyl fluoride. The lysates were cleared by centrifugation and protein was quantified with the BCA assay. The Akt kinase was precipitated from 100 μl of lysate from each condition with the addition of 15 μl of immobilized Akt 1G1 monoclonal antibody (Cell Signaling, Beverly, MA) overnight at 4°C. After washing, the Akt kinase was dissociated from the immobilized antibody by high-speed centrifugation (14,000 rpm 10 minutes at 4°C) and incubated with the GSK-3αβ fusion protein in kinase buffer (25 mmol/L Tris, pH 7.5, 5 mmol/L glycerophosphate, 2 mmol/L dithiothreitol, 0.1 mmol/L Na3HO4, 10 mmol/L MgCl2) supplemented with 200 μmol/L of ATP for 1 hour at 30°C. After three washes with lysis buffer the plate was incubated for with the anti-phopsho-GSK-3αβ(Ser21/9) antibody against the phosphorylated fusion protein (1:1000, Cell Signaling) for 1 hour in blocking buffer [Tris-buffered saline (TBS) with 0.1% Tween 20, with 2% fetal bovine serum]. After three washes with TBS supplemented with 0.1% Tween 20, the plate was incubated with a secondary anti-rabbit antibody conjugated with horseradish peroxidase in blocking buffer (TBS with 0.1% Tween 20, with 2% fetal bovine serum) for 1 hour at room temperature. The plate was subsequently washed three times and the peroxidase reaction was developed and measured at 450 nmol/L with a reference wavelength at 620 nmol/L.
Measurement of Retinal Erk1 Kinase (MAPK p44/p42) Activity
MAP kinase activity was analyzed in whole retinal tissue using an ELISA-based technique. Briefly, each well of a 96-well microtiter plate was coated with 100 μl of 1 μmol/L Elk1 fusion protein, which serves as a substrate for the activated Erk kinase, overnight at 4°C in carbonate buffer (15 mmol/L Na2CO3, 35 mmol/L NaHCO3, 0.2 g/L NaN3, pH 9.6). After washing, the remaining active sites were blocked in 10 mg/ml of bovine serum albumin in PBS containing 0.05% Tween-20. Retinal tissue was homogenized in lysis buffer containing 20 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3O4, 1 μg/ml leupeptin, and 1 mmol/L phenylmethyl sulfonyl fluoride. The lysates were cleared by centrifugation and protein was quantified with the BCA assay. Active Erk kinase was precipitated from 100 μl of lysate from each condition with the addition of 15 μl of immobilized phospho-p44/42 MAPK monoclonal antibody (Cell Signaling) for 6 hours at 4°C. After washing, the Erk kinase was dissociated from the immobilized antibody by high-speed centrifugation (14,000 rpm for 10 minutes at 4°C) and incubated with the Elk fusion peptide in kinase buffer (25 mmol/L Tris, pH 7.5, 5 mmol/L glycerophosphate, 2 mmol/L dithiothreitol, 0.1 mmol/L Na3VO4, 10 mmol/L MgCl2) supplemented with 200 μmol/L of ATP for 1 hour at 30°C. After three washes with lysis buffer the plate was incubated for 1 hour with blocking buffer (TBS with 0.1% Tween 20, with 5% w/v nonfat dry milk) with an antibody against the phosphorylated Erk-1(1:1000, Cell Signaling). After three washes with TBS supplemented with 0.1% Tween 20, the plate was incubated with a secondary anti-rabbit antibody conjugated with horseradish peroxidase. The peroxidase reaction was developed and measured at 450 nmol/L with a reference wavelength at 620 nmol/L.
Measurement of Retinal Nitric Oxide
Retinal nitric oxide levels were measured with the Total Nitric Oxide Assay (R&D Systems), which quantifies the nitric oxide reaction product nitrite. Briefly, each retina was placed in 100 μl of 40 mmol/L Tris buffer, pH 7.8, supplemented with 3 mmol/L dithiothreitol, 1 mmol/L l-arginine, 1 mmol/L NADH, and 4 μmol/L each of FAD, FMN, and H4 biopterin, and homogenized mechanically. The samples were cleared by centrifugation and the protein levels were estimated using a commercial assay (BCA kit; Bio-Rad, Hercules, CA). Equal amounts of protein per sample were ultrafiltered through a 10,000-molecular weight cutoff filter to eliminate proteins. The samples were subsequently incubated with nitric reductase to convert nitric oxide to nitrite according to the manufacturer’s instructions and incubated with the Griess reagent (1% sulfonamide, 0.1% naphthylethylene diamine dihydrochloride, 2.5% H3PO4) at room temperature for 10 minutes. Nitrite was determined at 550 nm using a microplate reader and the concentration was calculated using sodium nitrite standards. Results are expressed as nitrite concentration in μmol/L.
ELISA for eNOS
Each retina was placed in 100 μl of solution (4°C) consisting of 20 mmol/L imidazole hydrochloride, 100 mmol/L KCl, 1 mmol/L MgCl, 1 mmol/L EGTA, 1% Triton, 10 mmol/L NaF, 1 mmol/L sodium molybdenate, and 1 mmol/L EDTA. Complete Mini Proteinase Inhibitor Cocktail (Roche) was added before use. Samples were centrifuged for 10 minutes at 13,000 rpm. Two μl of the supernatant was used for protein determination via the Mini BCA Assay (Pierce Scientific). A commercially available ELISA kit (R&D Systems) was used to quantitate eNOS levels according to the manufacturer’s instructions. All measurements were performed in duplicate. The tissue sample concentration was calculated from a standard curve and corrected for protein concentration.
Statistical Analysis
A nonparametric test for trend was used to analyze the different treatment groups for a dose response. 21 Unpaired groups of two were compared using ranks in one-criterion variance analysis according to Kruskal-Wallis. 22 Differences were considered statistically significant when P values were <0.05.
Results and Discussion
Ang-1 Dose-Dependently Reduces Diabetic Blood-Retinal Barrier Breakdown
Retinal edema is the single most important cause of vision loss in diabetes. 23 Edema results, in part, when the tight junction-possessing retinal vasculature becomes permeable to fluid and solutes. Because Ang-1 induces leakage resistance in certain blood vessels 10,11 the ability of Ang-1 to prevent diabetes-related blood-retinal barrier breakdown was tested. Seven days after the induction of experimental diabetes, adult rat eyes received a single, unilateral injection of either vehicle, or 50 ng, 200 ng, or 800 ng of Ang-1. The contralateral eyes served as controls and received an injection of PBS. Twenty-four hours later, blood-retinal barrier breakdown was quantified with Evans blue dye. 20 In agreement with previously published data, blood-retinal barrier breakdown was increased approximately twofold in the 1-week diabetic eyes and was not altered by the PBS injections (data not shown). 1,20,24 In marked contrast, the diabetes-related blood-retinal barrier breakdown in the Ang-1-treated eyes was dose-dependently suppressed, with the 800-ng dose suppressing permeability ∼50% (Figure 1A) ▶ .
Figure 1.
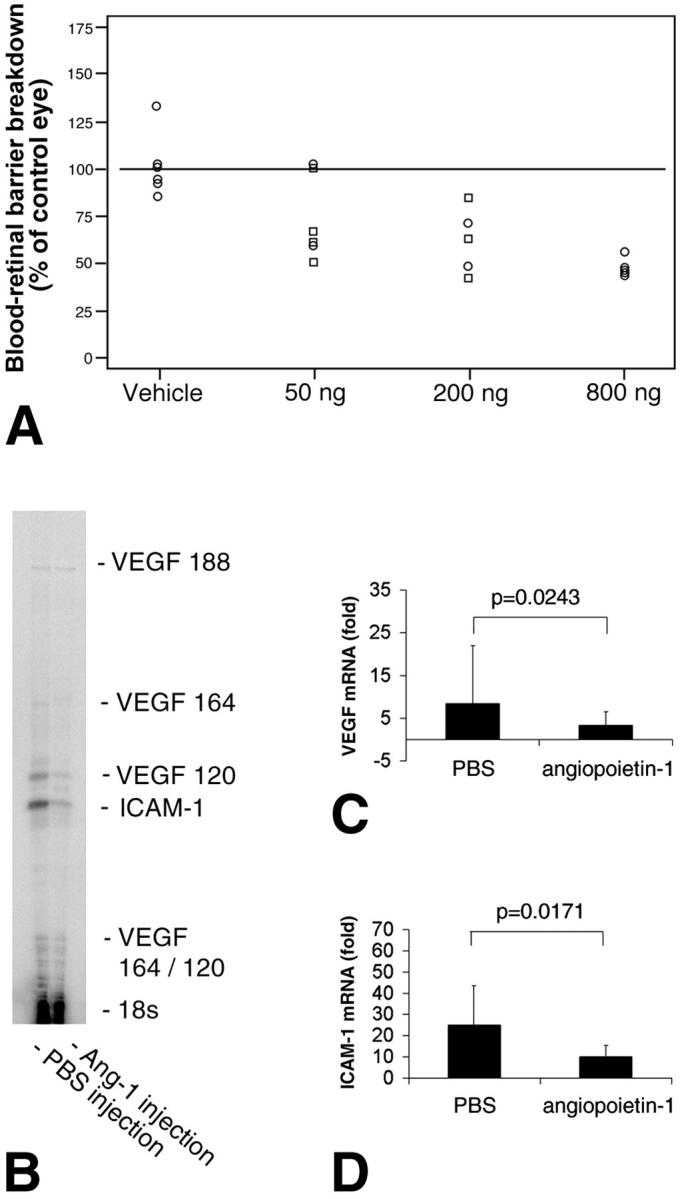
Ang-1 suppresses blood-retinal barrier breakdown and reduces VEGF and ICAM-1 expression in the retina. A: Locally administered Ang-1 suppresses diabetic retinal vascular permeability. Five μl of 160 μg/ml, 40 μg/ml, or 10 μg/ml of Ang-1 (800 ng, 200 ng, or 50 ng total) or vehicle was injected into one eye of 1-week diabetic rats in a masked manner. The contralateral eye of each rat received an equivalent volume of PBS alone as a control. The animals were studied 24 hours later. Evans blue dye was used to measure vascular permeability 24 hours after intra-ocular injection. Ang-1 dose-dependently reduced the retinal vascular permeability induced by experimental diabetes (P < 0.0001). B–D: Locally administered Ang-1 (200 ng) suppresses VEGF and ICAM-1 mRNA levels in diabetic retina. Retinae were harvested for RNA isolation 24 hours after intra-ocular injection with either Ang-1 or vehicle alone. B: A representative VEGF/ICAM-1 ribonuclease protection assay of eyes treated with PBS (left lane) or Ang-1 (right lane). C: When normalized to 18S RNA, diabetic retinal VEGF mRNA levels were reduced by 61.5% with Ang-1 (P < 0.03, n = 5). The graph shows the combined signal of the VEGF120, VEGF164, and VEGF188 transcripts, compared with diabetic eyes treated with PBS. D: When normalized to 18S RNA, retinal ICAM-1 mRNA levels were reduced by 59.5% with Ang-1 (P < 0.02, n = 5).
Ang-1 Decreases Retinal ICAM-1 and VEGF mRNA and Protein Levels
Retinal vascular permeability in early diabetes is primarily a function of ICAM-1-mediated leukocyte adhesion to the retinal vasculature. 1,2 Previous work has shown that VEGF increases retinal ICAM-1 expression and leukocyte adhesion, amplifying retinal vascular permeability. 1,25 Because Ang-1 suppresses VEGF-induced vascular permeability in the skin 10,11 the effect of Ang-1 on retinal ICAM-1 and VEGF mRNA and protein levels were quantified. A single unilateral injection of Ang-1 was given to randomly selected diabetic eyes. The contralateral eyes received PBS alone. In a similar manner to its effect on permeability, Ang-1 potently suppressed the expression of endogenous retinal VEGF and ICAM-1 mRNA by 61.5% (P = 0.024, n = 5) and 59.5%, (P = 0.017, n = 5), respectively, when compared to the PBS-treated contralateral eyes (Figure 1 ▶ ; B to D). Similarly, retinal VEGF and ICAM-1 protein levels were reduced by 39.6% (0.95 ± 0.14 pg/μg versus 1.57 ± 0.17 pg/μg; n = 4 to 5, P < 0.001) and 55.9% (0.41 ± 0.07 pg/μg versus 0.92 ± 0.12 pg/μg; n = 4 to 6, P < 0.001), respectively (Figure 2) ▶ .
Figure 2.

Ang-1 decreases retinal VEGF and ICAM-1 protein levels. VEGF and ICAM-1 protein levels in the retina were measured using an ELISA technique. A: Ang-1 reduced diabetic retinal VEGF protein levels by 39.6% (0.95 ± 0.14 pg/μg versus 1.57 ± 0.17 pg/μg; n = 4 to 6, P < 0.001). B: Ang-1 reduced diabetic retinal ICAM-1 levels by 55.9% (0.41 ± 0.07 pg/μg versus 0.92 ± 0.12 pg/μg; n = 4 to 5, P < 0.001).
Ang-1 Decreases Leukocyte Adhesion in the Diabetic Retina
One of the most important lesions in diabetic retinopathy is the obliterated or acellular microvessel. 26 The formation of acellular capillaries, via endothelial cell death, 27,28 contributes to retinal ischemia and the up-regulation of VEGF, a process that triggers intra-ocular neovascularization. 29,30 Initially, endothelial cell death precedes acellular microvessel formation. However, with time, acellular microvessels appear and become widespread. New data suggest that retinal endothelial cell injury and/or death are secondary, in part, to ICAM-1-mediated adhesion of leukocytes to the retinal vasculature. 2
To study the effect of Ang-1 on leukocyte adhesion, nonadherent leukocytes were removed via perfusion and the retinal vasculature and leukocytes were labeled with concanavalin A. 2,31,32 When selected diabetic eyes were treated with a single unilateral injection of Ang-1 ranging from 50 to 800 ng, as described above, a dose-dependent inhibition of diabetes-associated leukocyte adhesion in arterioles (P < 0.0001), venules (P < 0.0008), and capillaries (P < 0.02) was observed (Figure 3) ▶ .
Figure 3.

Ang-1 reduces leukocyte adhesion in the retina. A–D: Locally administered Ang-1 suppresses diabetic retinal leukocyte adhesion in rats with new-onset diabetes. Ang-1 or vehicle alone, at the indicated doses, was injected unilaterally into 1-week diabetic rat eyes. The contralateral eyes received an equal volume injection of PBS buffer alone. Endothelial cells and adherent leukocytes were labeled with FITC-linked concanavalin A lectin (green fluorescence). A: A representative microscopic image of diabetic flat-mounted retina stained with FITC-linked concanavalin A showing a cluster of adherent leukocytes. The number of adherent retinal leukocytes was dose-dependently decreased in the arterioles (P < 0.0001) (B), venules (P < 0.0008) (C), and capillaries (P < 0.02) (D). The data are presented as a percentage, comparing the Ang-1- or vehicle-treated eyes to the contralateral PBS-injected eyes.
The above results are in agreement with those of Kim and colleagues 33 showing that angiopoeitin-1 reduces VEGF-stimulated ICAM-1 expression and leukocyte adhesion in vitro. In addition, related experiments show that the inhibition of VEGF suppresses retinal leukostasis, as well as eNOS, NO, and ICAM-1 levels in the diabetic rat. 34 The administration of the NO inhibitor L-NAME has the same effect on retinal leukostasis. The combined evidence indicates that Ang-1 suppresses diabetes-induced Akt and NO indirectly, via its effects on VEGF expression.
Ang-1 Prevents Vascular Endothelial Cell Injury in the Diabetic Retina
Because Ang-1 suppresses retinal ICAM-1 expression and leukocyte adhesion, the ability of Ang-1 to prevent endothelial cell injury and/or death was assessed. To do this, damaged endothelial cells were labeled in situ with PI before perfusion with concanavalin A lectin. 2 When flat mounts of retinae prepared in this manner were studied, the diabetic eyes were notable for clusters of adherent leukocytes and PI-positive endothelial cells (Figure 4) ▶ . As previously shown, 2 the diabetic eyes exhibited a marked increase in adherent leukocytes and PI-labeled cells (data not shown). In contrast, the number of PI-labeled endothelial cells in the Ang-1-treated diabetic eyes was dose-dependently suppressed in the retinal arterioles (P < 0.04), venules (P < 0.008), and capillaries (P < 0.004) (Figure 4) ▶ .
Figure 4.

Ang-1 reduces endothelial cell damage in the retina. A–D: Locally administered Ang-1 suppresses endothelial cell injury and death in rats with new-onset diabetes. Injured and/or dead retinal endothelial cells were labeled in vivo using PI (orange fluorescence). A: Representative microscopic image of a diabetic flat-mounted retina stained with PI showing a cluster of injured and/or dying endothelial cells. Locally administered Ang-1 dose-dependently reduced the retinal vascular endothelial cell PI staining in arterioles (P < 0.004) (B), venules (P < 0.008) (C), and capillaries (P < 0.004) (D). The data are presented as a percentage, comparing the Ang-1- or vehicle-treated eyes to the contralateral PBS-injected eyes.
Ang-1 Activates Akt Kinase in Vitro, but Reduces Retinal Akt Kinase Activation in Vivo
The serine/threonine protein kinase Akt/PKB was recently shown to be activated by VEGF and to mediate the induction of eNOS, a cascade leading to NO increases, and in turn, ICAM-1 up-regulation. 35 Based on these data, we hypothesized that Ang-1, via the inhibition of endogenous VEGF, would lead to the down-regulation of retinal ICAM-1 through a reduction in Akt kinase activation and eNOS expression. The results obtained in this study are consistent with this hypothesis. Akt kinase activity increased by 54.3% in the diabetic retina compared to the nondiabetic controls (0.46 ± 0.01 versus 0.21 ± 0.03 [OD at 450 nm]; P < 0.001, n = 5 to 6) (Figure 5) ▶ . Treatment with 800 ng of Ang-1 decreased Akt kinase activity by 39.2% (0.28 ± 0.02; P < 0.005 versus diabetic control, n = 6) (Figure 5) ▶ . Furthermore, Akt activation was significantly suppressed in the diabetic animals receiving VEGF Trap A40 (0.4 ± 0.05 versus 0.25 ± 0.09 (IL-6 Trap) [OD at 450 nm], P < 0.005, n = 6) (Figure 6) ▶ . However, when retinal endothelial cells were exposed to Ang-1 in vitro, Akt activation was significantly increased (1.42 ± 0.22 versus 3.26 ± 0.34 09 [OD at 450 nm], P < 0.0001, n = 3) (Figure 6) ▶ . These disparate results argue that the indirect effects of Ang-1 on Akt kinase, via VEGF suppression, override the direct effects of Ang-1.
Figure 5.
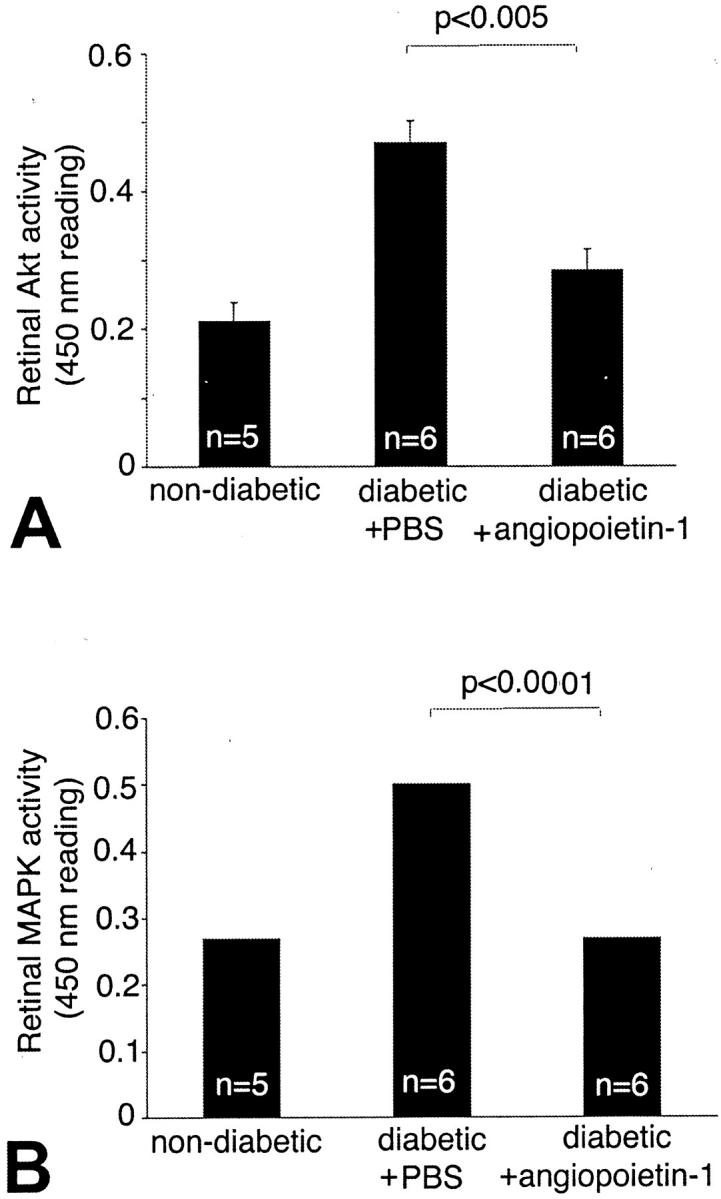
Ang-1 suppresses Akt kinase and Erk kinase activation in vivo. Akt kinase activity increased by 54.3% in the diabetic retina when compared to the nondiabetic controls [0.46 ± 0.01 versus 0.21 ± 0.003 (OD at 450 nm), P < 0.005, n = 5 to 6]. A: Treatment with Ang-1 resulted in a 39.2% reduction in Akt kinase activation.(0.28 ± 0.02 pg/mg; P < 0.005, n = 6). Erk kinase activity increased by 1.9-fold in the diabetic retina compared to the nondiabetic controls [0.26 ± 0.01 versus 0.5 ± 0.01 (OD at 450 nm); P < 0.0001, n = 5 to 6]. B: Treatment with Ang-1 resulted in a 55% reduction in retinal Erk activity compared to PBS-injected diabetic controls (0.26 ± 0.04; P < 0.0001, n = 6).
Figure 6.
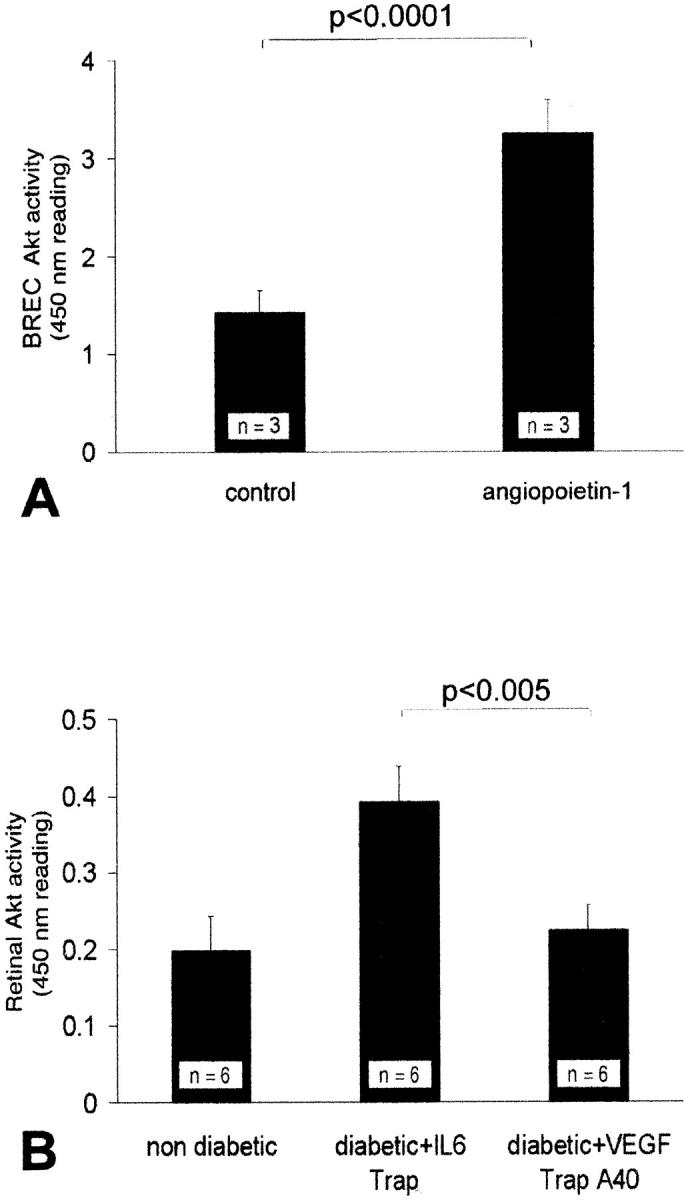
Ang-1 activates Akt kinase in vitro; VEGF inhibition suppresses Akt kinase activation in vivo. A: Akt kinase activity increased significantly in retinal endothelial cells treated with Ang-1 (250 ng/ml) in vitro [1.42 ± 0.22 versus 3.26 ± 0.34 09 (OD at 450 nm); P < 0.0001, n = 3]. However, the inhibition of endogenous VEGF, via the in vivo administration of the VEGF Trap A40, suppressed Akt kinase activity [0.4 ± 0.05 versus 0.25 ± 0.09 (IL-6 Trap); P < 0.005, n = 6]. B: Akt kinase activation increased in the diabetic retinae as compared to the nondiabetic controls [from 0.2 ± 0.045 to 0.4 ± 0.05 (OD at 450 nm); P < 0.005, n = 6].
Ang-1 Reduces MAP Kinase Activity in the Diabetic Retina
The mitogen-activated protein kinases p44Erk1 and p42Erk2 are serine/threonine kinases that play an important role in integrin-dependent neutrophil adhesion. 36 ICAM-1 cross-linking causes the activation of Erk1. 37 The VEGF-induced activation of MAPK/Erk and inhibition of SAPK/JNK activity also seems to be a key event in determining endothelial cell survival or apoptosis. 38 Further, MAPK is operative in the regulation of VEGF expression in certain systems. 39 In the current study, MAP kinase activity increased 1.9-fold in the diabetic retina (0.26 ± 0.01 versus 0.5 ± 0.01 (OD at 450 nm); P < 0.0001, n = 5), mirroring the diabetes-induced increases in retinal VEGF (Figure 5) ▶ . Treatment with 800 ng of Ang-1 resulted in a 55% reduction in the diabetes-related MAP kinase activity increases (0.26 ± 0.04; P < 0.0001, n = 6) (Figure 5) ▶ .
Ang-1 Reduces NO Levels and eNOS Expression in the Diabetic Retina
Because Ang-1 suppresses retinal VEGF and ICAM-1 expression, the effect of Ang-1 on retinal NO levels and eNOS expression was investigated. The effect of NO on the expression of inflammatory molecules is controversial and the effects may be tissue-dependent. 40,41 Following the demonstration by Radisavljevic and colleagues 35 of the actions of VEGF and NO on ICAM-1 up-regulation, a series of experiments was conducted showing that retinal VEGF and NO up-regulate ICAM-1 in the early diabetic rat retina. 34
In light of these data, retinal NO and eNOS levels were measured after treatment with Ang-1 (800 ng) or an equivalent volume of PBS. Retinal nitric oxide was converted to nitrite and quantified (Figure 7) ▶ . Compared to the retinae of nondiabetic animals, the diabetic animals exhibited a 2.8-fold increase in retinal nitrite levels (48.57 ± 6.05 μmol/L versus 135.89 ± 12.22 μmol/L; P < 0.001, n = 4 to 5) (Figure 6) ▶ . Treatment with Ang-1 reduced the retinal nitrite levels by 38.5% (83.36 ± 8.95 μmol/L; P < 0.001 versus PBS-injected diabetic eyes, n = 5). Using a sensitive ELISA, eNOS was quantified from retinal protein extracts. Treatment with Ang-1 reduced the diabetes-induced increases in retinal eNOS by 39.4% (0.95 ± 0.13 pg/mg versus 1.57 ± 0.16 pg/mg retinal weight; P < 0.001 versus PBS-injected eyes; n = 4 to 6) (Figure 7) ▶ .
Figure 7.
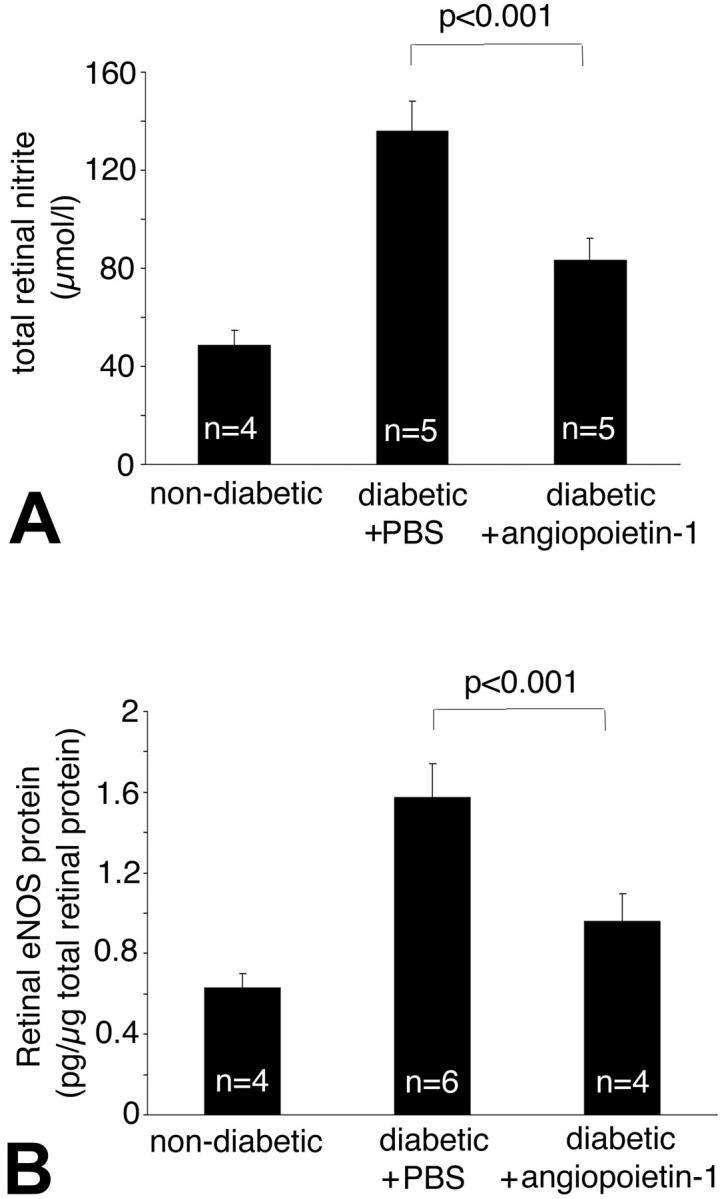
Ang-1 reduces retinal nitric oxide and eNOS expression. The total nitrite concentration was estimated from retinal tissue. Compared to the retinae of nondiabetic animals, control diabetic animals demonstrated a 2.8-fold increase in normalized NO levels (48.57 ± 6.05 μmol/L versus 135.89 ± 12.22 μmol/L; P < 0.0001, n = 4 to 6). A: Treatment with Ang-1 reduced retinal NO levels by 38.6% (83.36 ± 8.95 μmol/L; P < 0.001 versus nondiabetic controls, n = 4 to 5). The enzyme eNOS was quantified in retinal tissue by ELISA. B: Treatment with Ang-1 reduced retinal eNOS levels by 39.4% (0.95 ± 0.13 pg/mg versus 1.57 ± 0.16 pg/mg retinal weight; diabetic controls versus Ang-1-treated eyes; P < 0.001; n = 4 to 6).
Systemic Ang-1 Reduces Leukocyte Adhesion and Endothelial Cell Injury in Established Diabetes
Because diabetic retinopathy is a complex chronic disease, the operative mechanisms in early diabetes may not necessarily be relevant in established diabetes. To determine whether Ang-1 suppresses leukocyte adhesion and endothelial cell injury in more advanced diabetes, adult mice with diabetes of 16-weeks duration were studied. An Ang-1-containing adenovirus (Ad-Ang1) was administered systemically, resulting in circulating Ang-1 levels. The need for direct ocular manipulation was also obviated. As Thurston and colleagues 11 recently reported, intravenously administered Ad-Ang1 leads to specific viral uptake and expression in the mouse liver. The ensuing production of Ang-1 results in systemic and biologically active levels for at least 72 hours. A cohort of animals receiving a virus-encoding green fluorescent protein (Ad-GFP) served as controls. Using the same methods outlined above, leukocyte adhesion and endothelial cell damage were quantified with concanavalin A and PI, respectively. Seventy-two hours after intravenous Ad-Ang1 therapy, leukocyte adhesion was reduced by 62.8% in arterioles (P < 0.001), 68.7% in venules (P < 0.001), and 54.6% in capillaries (P < 0.001 versus Ad-GFP-treated mice) (Figure 8) ▶ . The numbers of PI-stained retinal endothelial cells were similarly reduced by 83.4% in arterioles (P < 0.001), 74.9% in venules (P < 0.001), and 87.2% in capillaries (P < 0.001) (Figure 8) ▶ . Ang-1 did not affect the diabetic state, as the blood glucose levels before sacrifice were 428.8 ± 5, 466.5 ± 57, and 453 ± 62 mg/dl in the Ad-GFP, Ad-Ang1 and control mice, respectively. Notably, the retinal vasculature of the Ad-Ang1 diabetic mice did not exhibit the larger, more numerous, and more highly branched vessels previously observed in transgenic mice chronically overexpressing Ang-1 in the skin 10 (data not shown).
Figure 8.

Systemic administration of Ang-1 in established diabetes. Systemically administered Ang-1 suppresses diabetic retinal leukocyte adhesion (A–C) and endothelial cell death and injury (D–F) in mice with established diabetes. Sixteen-week diabetic mice received a single tail vein injection of 1 × 109 PFU Ad-Ang-1. Age-matched control diabetic mice received an equivalent amount of Ad-GFP (control). Retinae were assayed for leukocyte adhesion and PI staining 72 hours after virus injection. The data are presented in a box and whiskers format. The middle line represents the median, with the box extending from the 25th to the 75th percentiles. Any data points beyond the percentile ranges are shown as individual data points (circles). The upper and lower values are the smallest and largest data points equal to, or greater than, the interquartile range. Twelve retinae per condition were analyzed. Systemic Ang-1 suppressed leukocyte adhesion in the retinal arterioles (P < 0.0001 versus diabetic Ad-GFP and diabetic control) (A), venules (P < 0.0001) (B), and capillaries (P < 0.0001) (C) of diabetic retinae. Similarly, systemic Ang-1 reduced vascular endothelial cell PI labeling in the retinal arterioles (P < 0.0001 versus diabetic Ad-GFP and diabetic control) (D), venules (P < 0.0001) (E), and capillaries (P < 0.0001) (F) of the diabetic retinae.
In summary, these data identify multiple new bioactivities for Ang-1. Ang-1 potently suppresses VEGF and ICAM-1 expression in the diabetic retina. These changes coincide with a reduction in retinal Akt kinase activity, a downstream mediator of VEGF action. The Akt kinase reductions also coincide with decreases in retinal eNOS and nitric oxide, known mediators of VEGF-induced ICAM-1 expression and leukocyte adhesion. Although Ang-1 activates Akt kinase in retinal endothelial cells in vitro, the situation in vivo seems to be more complex. The indirect effects of angiopoietin 1 on Akt, eNOS, and NO, via VEGF suppression, appear to outweigh the direct effects of Ang-1 on these molecules. Ang-1 also reduces retinal Erk kinase activity, a potential effector of both retinal leukocyte adhesion and endothelial cell death. At a more macroscopic level, Ang-1 suppresses diabetic blood-retinal barrier breakdown and protects the diabetic retinal vasculature against leukocyte-mediated endothelial cell injury and death. These data also indicate that the beneficial effects of Ang-1 are attainable in both new and established diabetes, and can be effected via either local or systemic therapy. Taken together, these observations identify Ang-1 as a potentially important therapeutic agent for the prevention and treatment of diabetic retinopathy.
Acknowledgments
We thank Nicholas Papadopoulos and Donna Hylton for reagent preparation and analysis, and Elizabeth Allred for statistical consultation.
Footnotes
Address reprint requests to Anthony P. Adamis, Massachusetts Eye and Ear Infirmary, 243 Charles St., Boston, MA 02114. E-mail: tony_adamis@meei.harvard.edu.
Supported by the Roberta W. Siegel Fund (to A. P. A.), the Deutsche Forschungsgemeinschaft (DFG Jo 324/4-1 to A. M. J.), the Juvenile Diabetes Foundation International (to A. M. J. and A. P. A.), the Zentrum für Molekulare Medizin Köln (to A. M. J.), the Falk Foundation (to A. P. A), the Iaccoca Foundation (to A. P. A.), the Grinnel Foundation (to A. P. A), the National Institutes of Health (EY11627 and EY12611 to A. P. A.), the Foundation Fighting Blindness (to V. P.), and the Massachusetts Lions (to A. P. A.).
A. M. J. and V. P. contributed equally to this work.
References
- 1.Miyamoto K, Khosrof S, Bursell S-E, Rohan R, Murata T, Clermont A, Aiello LP, Ogura Y, Adamis AP: Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci USA 1999, 96:10836-10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP: Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 2001, 158:147-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLeod DS, Lefer DJ, Merges C, Lutty GA: Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol 1995, 147:642-653 [PMC free article] [PubMed] [Google Scholar]
- 4.Schröder S, Palinski W, Schmid-Schönbein GW: Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol 1991, 139:81-100 [PMC free article] [PubMed] [Google Scholar]
- 5.Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD: Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87:1161-1169 [DOI] [PubMed] [Google Scholar]
- 6.Papapetropoulos A, Garcia-Cardena G, Dengler TJ, Maisonpierre PC, Yancopoulos GD, Sessa WC: Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab Invest 1999, 79:213-223 [PubMed] [Google Scholar]
- 7.Papapetropoulos A, Fulton D, Mahboubi K, Kalb RG, O’Connor DS, Li F, Altieri DC, Sessa WC: Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J Biol Chem 2000, 275:9102-9105 [DOI] [PubMed] [Google Scholar]
- 8.Kim I, Moon SO, Park SK, Chae SW, Koh GY: Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Circ Res 2000, 86:24-29 [DOI] [PubMed] [Google Scholar]
- 9.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ: Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284:1994-1998 [DOI] [PubMed] [Google Scholar]
- 10.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM: Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999, 286:2511-2514 [DOI] [PubMed] [Google Scholar]
- 11.Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD: Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med 2000, 6:460-463 [DOI] [PubMed] [Google Scholar]
- 12.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87:1171-1180 [DOI] [PubMed] [Google Scholar]
- 13.Moyon D, Pardanaud L, Yuan L, Breant C, Eichmann A: Selective expression of angiopoietin 1 and 2 in mesenchymal cells surrounding veins and arteries of the avian embryo. Mech Dev 2001, 106:133-136 [DOI] [PubMed] [Google Scholar]
- 14.Acker T, Beck H, Plate KH: Cell type specific expression of vascular endothelial growth factor and angiopoietin-1 and -2 suggests an important role of astrocytes in cerebellar vascularization. Mech Dev 2001, 108:45-57 [DOI] [PubMed] [Google Scholar]
- 15.Beck H, Acker T, Wiessner C, Allegrini PR, Plate KH: Expression of angiopoietin-1, angiopoietin-2, and tie receptors after middle cerebral artery occlusion in the rat. Am J Pathol 2000, 157:1473-1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enholm B, Paavonen K, Ristimaki A, Kumar V, Gunji Y, Klefstrom J, Kivinen L, Laiho M, Olofsson B, Joukov V, Eriksson U, Alitalo K: Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 1997, 14:2475-2483 [DOI] [PubMed] [Google Scholar]
- 17.Kräling BM, Bischoff J: A simplified method for growth of human microvascular endothelial cells results in decreased senescence and continued responsiveness to cytokines and growth factors. In Vitro Cell Dev Biol 1998, 33:308-315 [DOI] [PubMed] [Google Scholar]
- 18.Haugland RP: Handbook of Fluorescent Probes and Chemicals. 1996. Molecular Probes, Eugene
- 19.Rawson RA: The binding of T-1824 and structurally related diazo dyes by plasma proteins. Am J Physiol 1943, 138:708-717 [Google Scholar]
- 20.Xu Q, Qaum T, Adamis AP: Sensitive blood-retinal barrier breakdown quantitation using Evans blue. Invest Ophthalmol Vis Sci 2001, 42:789-794 [PubMed] [Google Scholar]
- 21.Cuzick J: A Wilcoxon-type test for trend. Stat Med 1985, 4:87-90 [DOI] [PubMed] [Google Scholar]
- 22.Kruskal WH, Wallis WA: Use of ranks in one-criterion variance analysis. J Am Statist Assoc 1952, 47:583-621 [Google Scholar]
- 23.Moss SE, Klein R, Klein BE: The incidence of vision loss in a diabetic population. Ophthalmology 1988, 95:1340-1348 [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto K, Khosrof S, Bursell S-E, Moromizato Y, Aiello LP, Ogura Y, Adamis AP: Vascular endothelial growth factor-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am J Pathol 2000, 156:1733-1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu M, Perez V, Ma N, Miyamoto K, Peng HB, Liao JK, Adamis AP: VEGF increases retinal vascular ICAM-1 expression in vivo. Invest Opthalmol Vis Sci 1999, 40:1808-1812 [PubMed] [Google Scholar]
- 26.Kohner EM, Porta M: Vascular abnormalities in diabetes and their treatment. Trans Ophthalmol Soc UK 1980, 100:440-444 [PubMed] [Google Scholar]
- 27.Kern TS, Tang J, Mizutani M, Kowluru RH, Nagaraj RH, Romeo G, Podesta F, Lorenzi M: Response of capillary cell death to animoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci 2000, 41:3972-3978 [PubMed] [Google Scholar]
- 28.Mizutani M, Kern TS, Lorenzi M: Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest 1996, 97:2883-2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adamis AP, Shima DT, Tolentino M, Gragoudas ES, Ferrara N, Folkman J, D’Amore PA, Miller JW: Inhibition of VEGF prevents retinal ischemia-associated iris neovascularization in a primate. Arch Ophthalmol 1996, 114:66-71 [DOI] [PubMed] [Google Scholar]
- 30.Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L, Ferrara N, King G, Smith LEH: Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA 1995, 92:10457-10461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolton PB, Lefevre P, McDonald DM: Salmeterol reduces early- and late-phase plasma leakage and leukocyte adhesion in rat airways. Am J Respir Crit Care Med 1997, 155:1428-1435 [DOI] [PubMed] [Google Scholar]
- 32.McDonald DM, Thurston G, Baluk P: Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation 1999, 6:7-22 [PubMed] [Google Scholar]
- 33.Kim I, Moon SO, Park SK, Chae SW, Koh GY: Angiopoietin-1 reduces VEGF-stimulated leukocyte adhesion to endothelial cells by reducing ICAM-1, VCAM-1, and E-selectin expression. Circ Res 2001, 14:477-479 [DOI] [PubMed] [Google Scholar]
- 34.Joussen AM, Poulaki V, Qin W, Kirchhof B, Mitsiades N, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP: Retinal VEGF induces ICAM-1 and eNOS expression and initiates early diabetic retinal leukocyte adhesion Am J Pathol 2002, 160:501-509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radisavljevic Z, Avraham H, Avraham S: Vascular endothelial growth factor up-regulates ICAM-1 expression via the phosphatidylinositol 3 OH-kinase/AKT/nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J Biol Chem 2000, 275:20770-20774 [DOI] [PubMed] [Google Scholar]
- 36.Lee SJ, Drabik K, Van Wagoner NJ, Lee S, Choi C, Dong Y, Benveniste EN: ICAM-1-induced expression of proinflammatory cytokines in astrocytes: involvement of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways. J Immunol 2000, 165:4658-4666 [DOI] [PubMed] [Google Scholar]
- 37.Lawson C, Ainsworth M, Yacoub M, Rose M: Ligation of ICAM-1 on endothelial cells leads to expression of VCAM-1 via a nuclear factor-kappaB-independent mechanism. J Immunol 1999, 162:2990-2996 [PubMed] [Google Scholar]
- 38.Gupta K, Kshirsagar S, Li W, Gui L, Ramakrishnan S, Gupta P, Law PY, Hebbel RP: VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp Cell Res 1999, 247:495-504 [DOI] [PubMed] [Google Scholar]
- 39.Tanaka T, Kanai H, Sekiguchi K, Aihara Y, Yokoyama T, Arai M, Kanda T, Nagai R, Kurabayashi M: Induction of VEGF gene transcription by IL-1 beta is mediated through stress-activated MAP kinases and Sp1 sites in cardiac myocytes. J Mol Cell Cardiol 2000, 32:1955-1967 [DOI] [PubMed] [Google Scholar]
- 40.Lindemann S, Sharafi M, Spiecker M, Buerke M, Fisch A, Grosser T, Veit K, Gierer C, Ibe W, Meyer J, Darius H: NO reduces PMN adhesion to human vascular endothelial cells due to downregulation of ICAM-1 mRNA and surface expression. Thromb Res 2000, 97:113-123 [DOI] [PubMed] [Google Scholar]
- 41.Liu P, Xu B, Hock CE, Nagele R, Sun FF, Wong PY: NO modulates P-selectin and ICAM-1 mRNA expression and hemodynamic alterations in hepatic I/R. Am J Physiol 1998, 275:H2191-H2198 [DOI] [PubMed] [Google Scholar]