

Abstract
Sepsis and trauma are the two most common causes of disseminated intravascular coagulation and multiple organ dysfunction syndrome. Both disseminated intravascular coagulation and the systemic inflammatory response syndrome often lead to multiple organ dysfunction syndrome. The current studies have evaluated the relationship between the anaphylatoxin, C5a, and changes in the coagulation/fibrinolytic systems during the cecal ligation and puncture (CLP) model of sepsis in rats. CLP animals treated with anti-C5a had a much improved number of survivors (63%) compared to rats treated with pre-immune IgG (31%). In CLP rats treated with pre-immune IgG there was clearly increased procoagulant activity with prolongation of the activated partial thromboplastin time and prothrombin time, reduced platelet counts, and increased levels of plasma fibrinogen. Evidence for thrombin formation was indicated by early consumption of factor VII:C, subsequent consumption of factors XI:C and IX:C and anti-thrombin and increased levels of the thrombin-anti-thrombin complex and D-dimer. Limited activation of fibrinolysis was indicated by reduced plasma levels of plasminogen and increased levels of tissue plasminogen activator and plasminogen activator inhibitor. Most of these parameters were reversed in CLP rats that had been treated with anti-C5a. Production of C5a during sepsis may directly or indirectly cause hemostatic defects that can be reduced by blockade of C5a.
Multiple organ dysfunction syndrome is the leading cause of death in patients in surgical intensive care units and is accompanied by consumptive hemostatic changes often leading to disseminated intravascular coagulation (DIC), progressive complement activation, and unregulated release of pro-inflammatory mediators. 1-4 Activation in plasma of the coagulation/fibrinolytic and complement systems in sepsis may be related to release of cell wall components from gram-negative or gram-positive bacteria. 5 In sepsis-induced DIC, thrombin formation leads to diffuse fibrin formation in the microvascular system, which is considered to be an important pathogenic factor for tissue hypoxia and organ damage. 6 Recently, therapeutic interventions aimed at restoring the hemostatic balance with anti-thrombin (AT) or with activated protein C concentrates have been used in animal and human sepsis associated with DIC. 7-11 There is ample evidence for an interrelationship between the coagulation system and the inflammatory response. 3,12 Inhibition of inflammatory mediators by monoclonal antibodies in experimental sepsis has resulted in reduced disturbances in the coagulation system. 13,14 Administration of pro-inflammatory cytokines 15,16 or endotoxin 17,18 into human volunteers has induced alterations in the coagulation system, similar to those observed changes in sepsis.
Interactions between the coagulation and complement systems have been controversial. Complement activation products, especially the anaphylatoxins, C3a, C4a and C5a, appear during sepsis. Elevated anaphylotoxin plasma levels highly correlate with the development of multiorgan failure. 19,20 In sepsis, complement may directly promote procoagulant activity or indirectly induce cytokine production. 21-23 In vitro C5a and the terminal complex of complement, C5b-9, induce tissue factor expression on endothelial cells and monocytes. 24-27 Furthermore, assembly of C5b-9 on the surface of platelets has been shown to stimulate prothrombinase activity. 26 C4b-binding protein, a regulator of the classical pathway of complement activation, also modulates the anti-coagulant effects of the protein S system. 28,29
Previously, we have shown that antibody blockade of C5a improves survival in cecal ligation and puncture (CLP)-induced sepsis in rats. 30,31 In the present investigation we determined changes of pro- and anti-coagulant activities in the plasma proteins of rats after CLP-induced sepsis, and we determined if anti-C5a would ameliorate these changes. Our results indicate that anti-C5a treatment significantly reduces changes in sepsis-induced coagulation/fibrinolytic proteins of plasma, leading to improved survival in this animal model.
Materials and Methods
Preparation and Characterization of Rabbit Anti-Rat C5a
A peptide of rat C5a, 17KHRVPKKCCYDGARENKYET36, was coupled to keyhole limpet hemocyanin for rabbit immunization. After several immunizations, anti-C5a immunoglobulin was affinity-purified from rabbit serum using the synthetic peptide coupled to Sepharose beads (Amersham Pharmacia, Piscataway, NJ). Preparation of the antibody was performed by Research Genetics (Huntsville, AL). This polyclonal antibody immunoprecipitated a 14-kd protein from activated rat serum, consistent with the molecular mass of glycosylated C5a. 30
Experimental Sepsis Induced by CLP
Male Long-Evans, specific pathogen-free rats (Harlan Breeders, Indianapolis, IN) weighing 275 to 300 g were used in all experiments. Anesthesia was induced by intraperitoneal administration of ketamine (20 mg/100 g body wt). After shaving the abdomen and using a 2-cm midline abdominal incision, the cecum was identified and ligated below the ileocecal valve. The cecum was then subjected to a single through-and-through perforation with a 21-gauge needle and was gently squeezed to ensure patency of the perforation sites. The bowel was returned to its usual position and the abdominal incision closed in layers with plain gut surgical suture 4-0 and skin clips (Ethicon, Somerville, NJ). Within 5 minutes after induction of sepsis, each CLP animal received either 400 μg of affinity purified rabbit anti-rat C5a (anti-C5a) or 400 μg of pre-immune rabbit IgG by intravenous infusion. Sham-operated rats underwent the same procedure of laparotomy except for ligation and puncture of the cecum. For each group, the sample or group size is indicated in the text. Before and after surgery, animals had unrestricted access to food and water.
Blood Sample Collection
CLP-treated or sham-operated rats were sacrificed by exsanguination by venipuncture at 12-hour intervals for up to 36 hours. Blood was obtained from the inferior vena cava from seven or more animals at 12, 24, and 36 hours from each of the three groups of animals. Rats were anesthetized by ketamine. A 5-ml syringe containing 0.5 ml of 3.8 g% sodium citrate was used to withdraw a total volume of 4.5 ml of blood (1:9 ratio of citrate:blood) from each animal by venipuncture of the inferior vena cava. Unless otherwise stated, whole blood samples were immediately centrifuged at 2400 rpm (Beckman GRP Instruments, Schaumberg, IL) for 10 minutes at room temperature. Platelet-poor plasma was obtained and frozen in 50- to 100-μl aliquots at −70°C until assay.
Assays
All samples of rat plasma were thawed once from the time of blood collection at the time of testing. The prothrombin time (PT) was performed by incubating 50 μl of plasma for 5 minutes at 37°C and then adding 100 μl of an equal volume mixture of Simplastin (Organon Teknika, Durham, NC) and 30 mmol/L of CaCl2. The activated partial thromboplastin time (APTT) was performed by mixing 50 μl of APTT reagent (Organon Teknika) and 50 μl of rat plasma followed by incubation for 5 minutes at 37°C and recalcification with 50 μl of 30 mmol/L CaCl2. Both assays were performed in an Amelung KC4 coagulation instrument (Sigma Chemical Co., St. Louis, MO). The factor V:C (FV) and VII:C (FVII) coagulant assays were performed with human factor V- and VII-deficient plasmas (George King, Inc., Overland Park, KS) in PT-based assays using Simplastin (Organon Teknika). Factors XII:C (FXII), XI:C (FXI), IX:C (FIX), and VIII:C (FVIII) were measured using human factor-deficient plasmas (George King, Inc.), in APTT-based assays were measured using APTT Reagent (Organon Teknika). Pooled normal human plasma was purchased from George King, Inc. Rat clottable plasma fibrinogen was determined using human α-thrombin (50 U/ml) and the values compared to a human plasma fibrinogen standard (Sigma Chemical Co.).
Platelet counts were measured in platelet-rich plasma using a Coulter Z analyzer (Coulter Electronics, Hialeah, FL). After centrifugation of 500 μl of citrated whole blood for 10 minutes at room temperature at 1000 rpm in a microcentrifuge (Beckman Instruments, Palo Alto, CA), the platelet-rich plasma supernatant was removed and the platelet counts obtained according to the manufacturer’s instructions.
The AT amidolytic assay was performed by a modified method of Odegard and colleagues. 32 Human or rat plasma was diluted in 0.05 mol/L Tris, 0.0075 mol/L ethylenediaminetetraacetic acid, pH 8.4, containing 3 U/ml standard heparin, after which human α-thrombin (Hematological Technologies, Essex Junction, VT) at 1.5 U/ml (final concentration) was added and the mixture was incubated for 7 minutes at room temperature. The chromogenic substrate H-D-Phe-Pip-Arg-paranitroanilide (S2238) (Diapharma, Inc., West Chester, OH) was added at 0.17 mmol/L (final concentration). After 5 minutes of hydrolysis, the reaction was stopped by the addition of an equal volume of acetic acid. Rat plasma AT levels were expressed as percent activity of pooled normal human plasma. The plasma plasminogen assay was performed by a modified assay of Lijnen and colleagues 33 by activation of rat plasma with 2000 U/ml human urokinase (Abbott Laboratories, Chicago, IL) for 5 minutes. After incubation, the chromogenic substrate Pefachrom Pl (2 AcOH.H-D-Ala-Cht-Lys-paranitroanilide) (Centerchem, Inc., Stanford, CT) was added at a final concentration of 0.5 mmol/L, and the reaction continued for 37°C for 2 hours. Preliminary experiments determined that the hydrolysis of the substrate was linear throughout the period of time of the assay. Rat plasma plasminogen levels were expressed as percent activity of pooled normal human plasma. Plasma tissue plasminogen activator (t-PA) and plasminogen activator inhibitor (PAI) were assayed using Spectrolyse/fibrin chromogenic assay kit according to the manufacturer’s instructions (Biopool, Inc., Ventura, CA). To retain full t-PA activity, 500 μl of citrated blood samples for determination of t-PA were collected in an ice-cold centrifuge test tube and acidified within 30 seconds of collection with 250 μl of ice-cold acetate. After centrifugation at 1500 × g for 5 minutes, 10 μl of 20% acetic acid was added to 200-μl aliquots that were then frozen and stored at −70°C until assay. The plasma thrombin-anti-thrombin (TAT) complex was measured by using the enzyme immunoassay kit Enzygnost TAT micro (Dade Behring, Marburg, Germany) according to the manufacturer’s instructions. Plasma D-dimer levels were assayed using the enzyme immunoassay kit Asserachrom D-Di (Diagnostica Stago, Asnières, France) according to the manufacturer’s instructions. For the reagents to detect rat D-dimer, the rat samples were 20-fold less diluted than the human samples.
Statistical Analysis
Experimental data were analyzed by determining the means ±SEM. Data sets among groups were analyzed using one-factor analysis of variance or the Kruskal-Wallis one-way analysis of variance on ranks. Individual group means were then compared using the Tukey or the Dunn’s multiple comparison test. For analysis of survival curves, outcomes in different treatment groups were compared using chi-square and Fisher’s exact tests. Significance was established if the P value was <0.05.
Results
Effects of Anti-C5a on Mortality
A Kaplan-Meier survival curve for the total observation time of 36 hours was analyzed, including animals with laparotomy in the absence (sham animals) or presence of CLP (Figure 1) ▶ . Blood was taken from surviving animals 12, 24, or 36 hours after the operation. All sham animals survived the time period of 36 hours. Ten animals were sacrificed at 12 hours, 10 were sacrificed after 24 hours, and 10 were sacrificed at 36 hours. Only 22 of 69 animals subjected to CLP and simultaneous infusion of pre-immune IgG survived to the planned times for sacrifice (eight pre-immune IgG-treated CLP rats were sacrificed after 12 hours, seven animals after 24 hours, and seven animals at 36 hours). These animals exhibited piloerection, huddling, and reduced motor activity with a mean survival rate of 87% at 12 hours, 40% at 24 hours, and 31% at 36 hours, respectively. Thirty of 47 animals (63%) in the CLP group treated with anti-C5a survived by 36 hours. In the anti-C5a group of animals, 7 rats were sacrificed after 12 hours, 12 at 24 hours, and 11 at 36 hours. Anti-C5a treatment in CLP animals resulted in increased survival rates, 92% at 12 hours, 72% at 24 hours, and 63% at 36 hours. When comparing the outcomes of the anti-C5a to pre-immune IgG treatment, survivals were significantly different (P = 0.016 by chi-square test).
Figure 1.

Effects of anti-C5a on survival rates in CLP rats. Animals received either 400 μg of pre-immune IgG (CLP + IgG) or 400 μg of anti-C5a IgG (CLP + anti-C5a) intravenously immediately after the procedure of CLP. Sham animals underwent laparotomy without CLP. The data are presented as percentage of survival (n = total number of animals in each group).
Effects of Anti-C5a Treatment on APTT and PT
When compared to the sham groups, APTT values in both groups of the CLP animals had significantly prolonged times (P < 0.05) at 24 hours, suggesting depletion/activation of clotting factors associated with the sepsis (Figure 2A) ▶ . By 36 hours after CLP, APTT values returned to the same levels found in the sham group. Similar to the APTT values at 24 hours, there was an early prolongation of the PT in both the pre-immune IgG-treated and anti-C5a-treated CLP animals when compared to sham animals at 12 hours (P < 0.05) (Figure 2B) ▶ . However the anti-C5a-treated animals had a significantly less (P < 0.05) prolongation at 12 hours than the pre-immune IgG-treated group. At 24 hours, these values had returned to levels found in sham animals.
Figure 2.
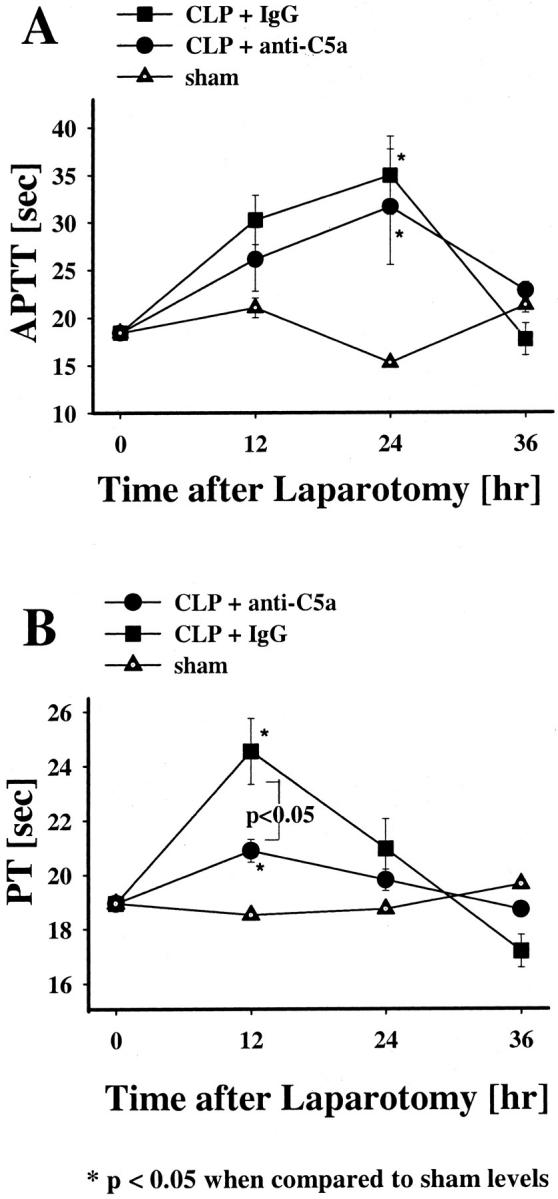
Effects of anti-C5a or pre-immune IgG on APTT or PT in CLP rats. APTT (A) and PT (B) levels were measured in plasma collected from the various groups at 12, 24, or 36 hours after laparotomy. The data represent means ±SEM from seven or more individual animals at each time point.
Effects of Anti-C5a on Platelet Counts and Fibrinogen Levels in CLP Animals
Blood platelet counts and fibrinogen levels were examined in the plasma of animals before and 12, 24, and 36 hours after laparotomy (Figure 3) ▶ . Compared to sham animals, platelet counts in the pre-immune IgG-treated CLP animals declined significantly at 12 hours and remained reduced during the entire study time (P < 0.001, Figure 3A ▶ ). The anti-C5a-treated CLP animals also showed significantly reduced platelet levels (when compared to sham animals) at 12 and 36 hours after CLP (P < 0.001, Figure 3A ▶ ). However, at both 12 and 36 hours after CLP, rats receiving anti-C5a had platelet values that showed significantly less reduction when compared to platelet levels found in pre-immune IgG-treated CLP animals (P < 0.05, respectively).
Figure 3.
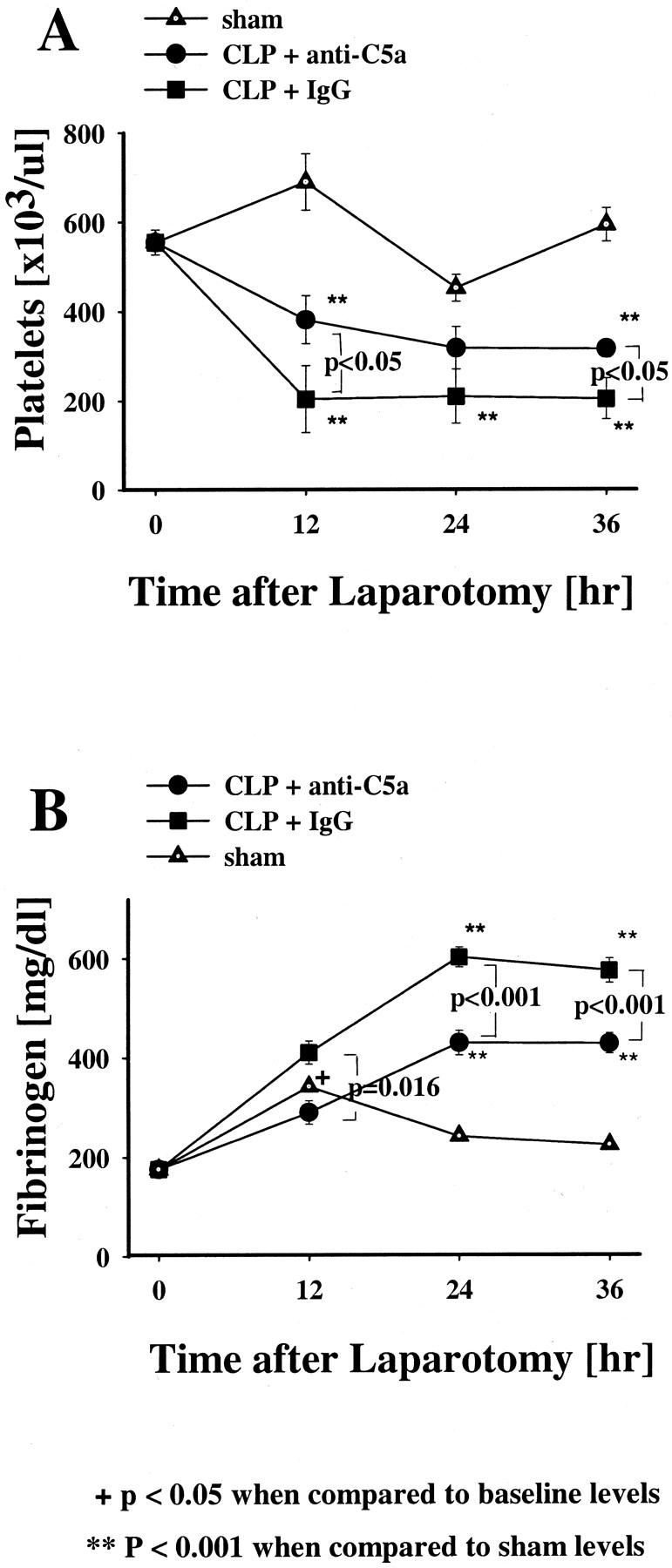
Effects of anti-C5a and pre-immune IgG on plasma platelet or fibrinogen levels in the indicated groups. Platelet counts were done in platelet-rich plasma (A) and plasma clottable fibrinogen levels (B) were measured in CLP rats and sham rats at 12, 24, or 36 hours after laparotomy. The data represent means ±SEM from six or more individual animals at each time point.
Plasma fibrinogen levels initially rose significantly (P < 0.05) at 12 hours in the sham group when compared to baseline values and then fell back into the normal range (Figure 3B) ▶ . Fibrinogen levels in the pre-immune IgG and anti-C5a-treated animals at 12 hours were not significantly different from the sham group, but in the pre-immune IgG and anti-C5a-streated CLP groups, the fibrinogen continued to rise and remained at significantly higher levels at 24 and 36 hours (P < 0.001). When compared to the pre-immune IgG-treated CLP animals, the anti-C5a-treated CLP animals showed significantly reduced elevations of fibrinogen (P = 0.016, P < 0.001) throughout the total observation time.
Effects of Anti-C5a on Levels of Factors (F) VII:C, XI:C, IX:C, XII:C, VIII:C, and V:C
To evaluate indirectly whether elevation of tissue factor (TF) contributed to the hemostatic changes during CLP-induced sepsis, studies were performed to determine whether there was a change in the plasma FVII:C levels in these animals (Figure 4) ▶ . When compared to the sham-operated animals, there was an initial and significant decrease in plasma FVII:C levels at 12 hours (P = 0.007) in the pre-immune IgG-treated CLP group, returning to baseline values after 24 and 36 hours. In contrast, FVII:C levels of the anti-C5a-treated CLP animals did not significantly drop after 12 hours when compared to the sham group. These results suggested that the FVII:C-tissue factor pathway initially was activated, prolonging the PT, during CLP-induced sepsis but not in CLP animals treated with anti-C5a. However, it is also possible that sepsis caused suppressed hepatic synthesis of factor VII:C, resulting in reduced plasma levels of this factor.
Figure 4.

Effects of anti-C5a and pre-immune IgG on coagulation factor VII:C (FVII) in CLP in rats. FVII levels were measured at 12, 24, or 36 hours after laparotomy. The data represent means ±SEM from seven or more individual animals.
FIX:C levels significantly fell in the preimmune IgG-treated animals at 12 hours and both groups at 24 hours (Figure 5A) ▶ . FXI:C levels in the CLP animals showed a decline in activity after 24 and 36 hours after CLP when compared to the sham group (P < 0.001) (Figure 5B) ▶ . There was no significant difference in the degree of FXI:C reduction between the pre-immune IgG and anti-C5a-treated CLP animals. The fall in the FIX:C and FXI:C values both contributed to the prolongation of the APTT at 24 hours. Last, there were no significant change in the plasma FXII:C, FVIII:C, or FV:C levels among the three groups (data not shown).
Figure 5.
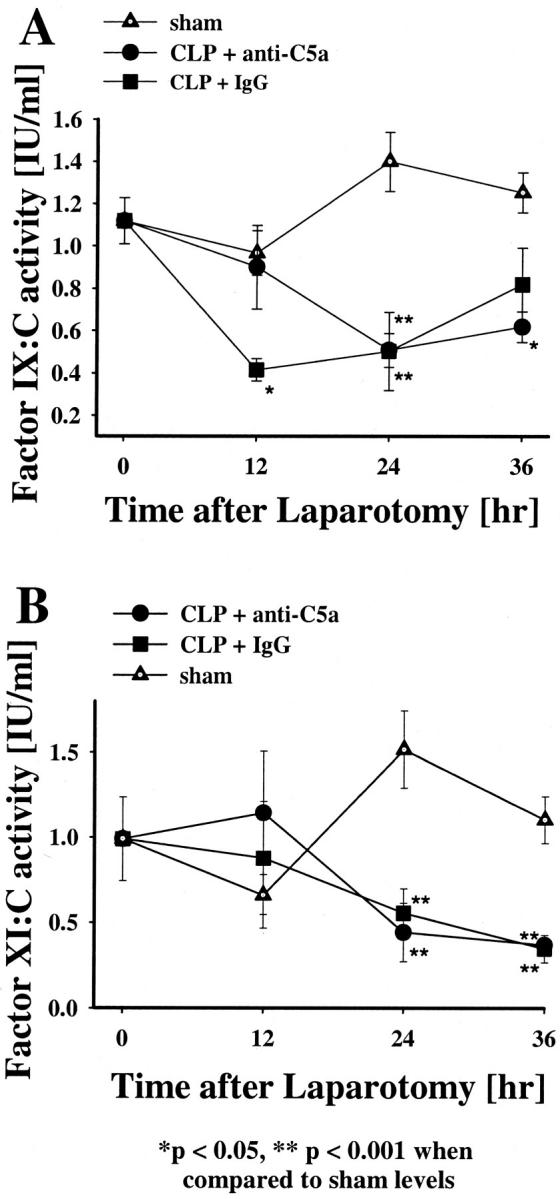
Effects of anti-C5a and pre-immune IgG on coagulation factors IX:C (FIX) and XI:C (FXI) in CLP in rats. FIX (A) and FXI (B) levels were measured at 12, 24, or 36 hours after laparotomy. The data represent means ±SEM from seven or more individual animals.
Effect of Anti-C5a on AT and Fibrinolytic Systems
Activation of the coagulation system was further demonstrated by changes in AT levels. AT consumption occurred in the pre-immune IgG-tested CLP animals, indirectly indicating thrombin formation (Figure 6A) ▶ . When compared to the sham group, AT levels in the pre-immune IgG-treated CLP animals progressively declined after 12 hours, by 50% at 24 hours and 36 hours (P < 0.001, Figure 6A ▶ ). In contrast, AT levels of anti-C5a-treated CLP animals did not significantly decrease throughout the total observation time when compared to sham groups. The difference in AT levels between the pre-immune IgG or anti-C5a-treated animals was significantly higher at the 24- and 36-hour time points (P < 0.001).
Figure 6.
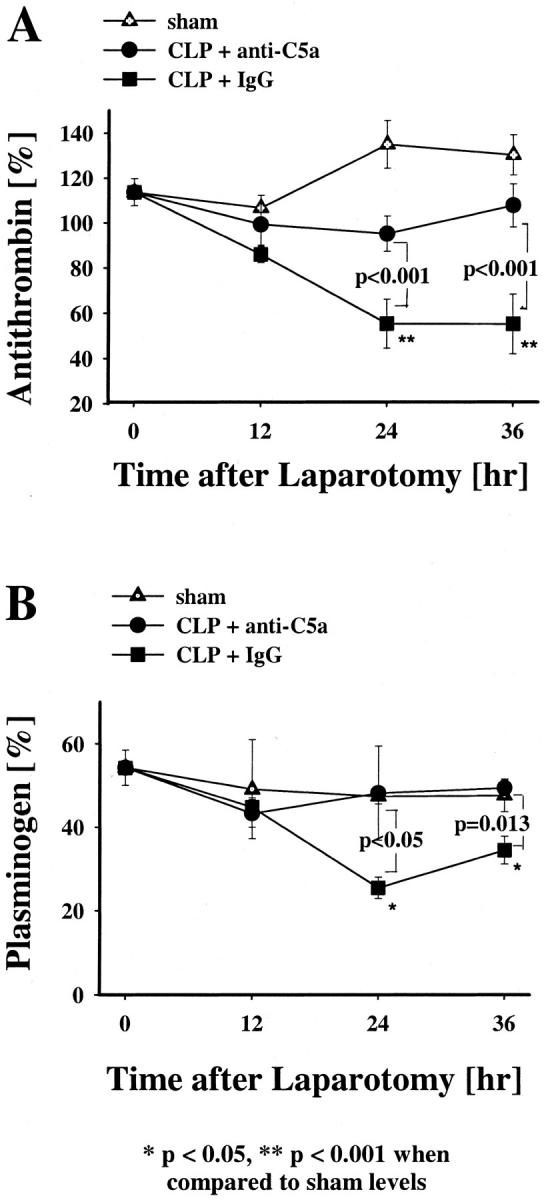
Effects of anti-C5a and pre-immune IgG on plasma AT or plasminogen levels in CLP-induced sepsis. AT (A) and plasminogen plasma (B) activities were measured in the CLP groups and in the sham group at 12, 24, or 36 hours after laparotomy. The data represent means ±SEM from six or more individual animals at each time point.
Sepsis-induced changes in the fibrinolytic system was examined by measurements of plasma plasminogen. When compared to sham animals, there was no significant change in the plasma plasminogen levels of the sham animals and anti-C5a-treated CLP rats throughout the total period of observation (Figure 6B) ▶ . However, the pre-immune IgG-treated group showed significant decreases (P < 0.05, P = 0.013) in plasma plasminogen levels 24 and 36 hours after CLP when compared to sham animals or anti-C5a-treated CLP animals.
The pre-immune IgG-treated CLP rats further showed an early increase in plasma t-PA, which peaked after 24 hours (P < 0.001, Figure 7A ▶ ) when compared to sham-treated animals. The plasma t-PA levels of anti-C5a-treated CLP rats were not significantly different from the values of the sham animals during the total observation period. The difference in t-PA levels between pre-immune IgG and anti-C5a-treated animals was especially evident at the 24-hour time point (P = 0.006). Plasma PAI levels also increased in CLP animals. The pre-immune IgG-treated CLP animals had progressively increasing levels of PAI throughout the total observation time (P < 0.001, Figure 7B ▶ ). It is possible that the small (but statistically significant) increases in PAI in the two CLP groups could also be because of an acute phase response that is not C5a related. PAI levels of CLP animals treated with anti-C5a also increased when compared to the sham group (P < 0.001), but not to the same extent as seen in the IgG-treated animal group (P < 0.001, Figure 7B ▶ ).
Figure 7.
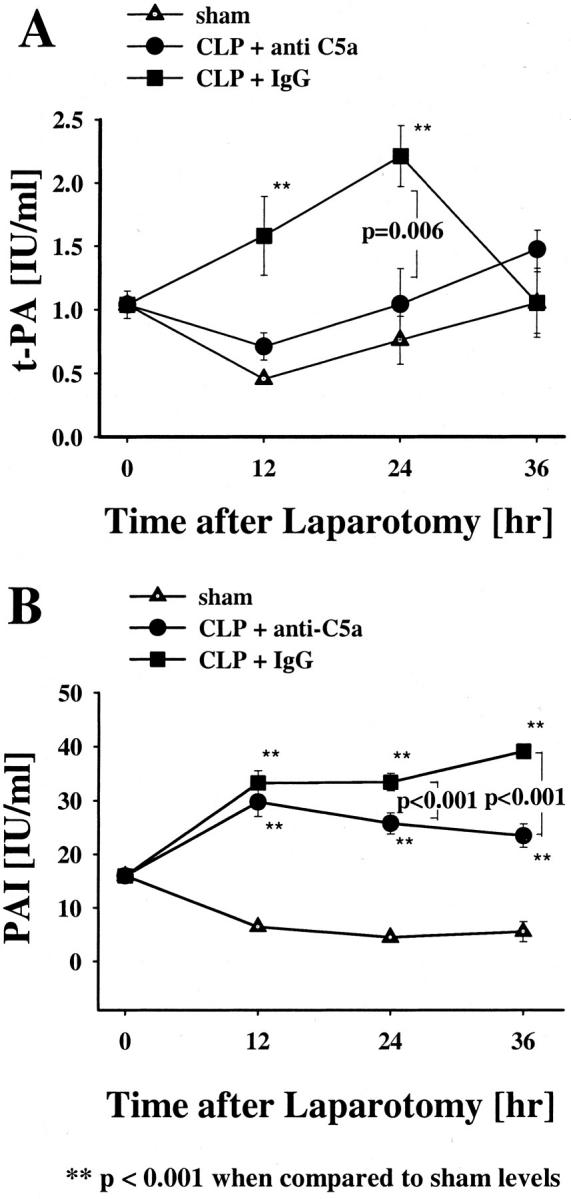
Effects of anti-C5a and pre-immune IgG on t-PA or PAI in CLP and sham rats. t-PA (A) and PAI (B) plasma activities were measured at 12, 24, and 36 hours after the operation. Data represent means ±SEM from six or more animals at each time point.
Effects of C5a Blockade on Plasma TAT and D-Dimer Levels
Plasma TAT complex levels in the anti-C5a-treated CLP rats did not change significantly at any time point when compared to sham controls (Figure 8A) ▶ . Alternatively, in the CLP group treated with pre-immune IgG direct evidence for thrombin activation was indicated by a dramatic increase in TAT levels at 12 and 24 hours after CLP (Figure 8A) ▶ . The difference in TAT complex levels between pre-immune IgG-treated and anti-C5a-treated animals was especially evident at the 12- and 24-hour time points (P = 0.012 and P = 0.01, respectively). D-dimer levels did also not significantly change in sham controls and in CLP animals treated with anti-C5a (Figure 8B) ▶ throughout the total observation time. However, the D-dimer levels of the pre-immune IgG-treated CLP animals showed an early significant increase at 12 hours (P = 0.039) when compared to sham group (Figure 8B) ▶ .
Figure 8.
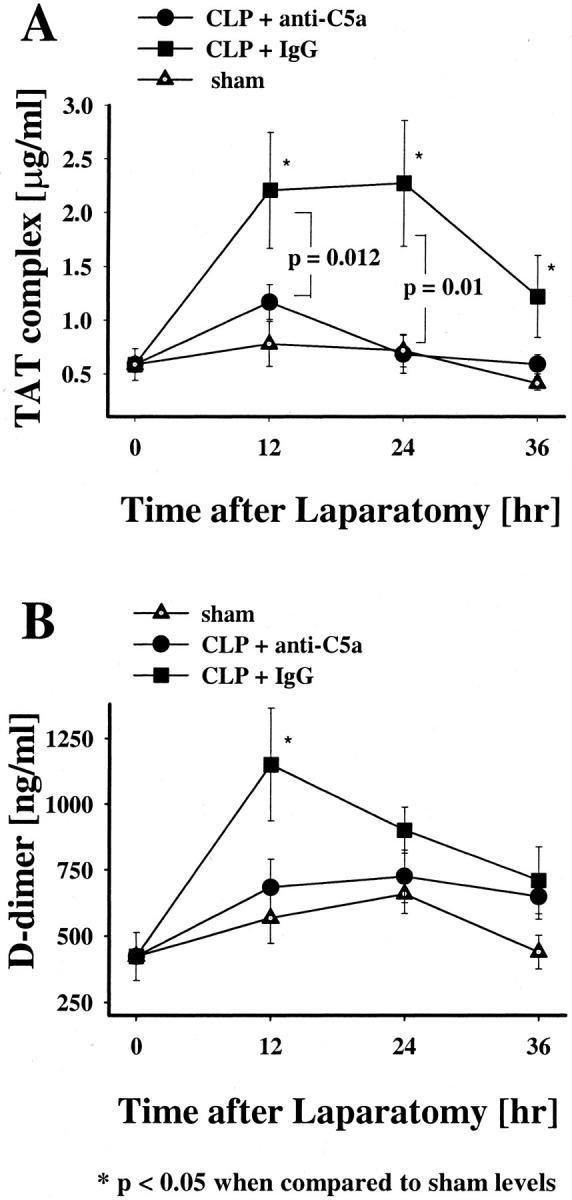
Effects of anti-C5a and pre-immune IgG on plasma TAT complex or D-dimer levels in CLP and sham rats. TAT complex (A) and D-dimer (B) levels were measured at 12, 24, or 36 hours after laparotomy. The data represent means ±SEM from six or more animals at each time point.
Discussion
It is well established in humans that sepsis results in complement activation/consumption as defined by loss of total hemolytic activity of serum complement (CH50) and the presence of complement activation products in serum. 5,19,20,34-37 C5a is a powerful anaphylatoxin, as reflected by the intravenous administration of C5a to animals leading to severe hypotension. 38 Pretreatment with anti-C5a in a primate model of sepsis (intravenous injection of live bacteria) results in reduced mean arterial pressure. 39 C5a is known to induce TF activity in vitro in endothelial cells and leukocytes. 24,25 To determine the effects of rabbit-rat polyclonal anti-C5a IgG on changes in the coagulation/fibrinolytic responses in experimental sepsis, a rat CLP model of septic shock was used. This model closely mimics the pathophysiology of sepsis in humans. 40,41 Anti-C5a therapy reduced the frequency of death in CLP-treated animals (Figure 1) ▶ , confirming our recent publication that anti-C5a-treated CLP animals show improved survival rates (31% versus 63%) at 36 hours when lethality is very pronounced. 30,31 C5a induction of tissue factor activity as reflected by a fall in factor VII and IX levels leads to thrombin generation. Thrombin formation activates factor XI that is not ameliorated by anti-C5a, and, in turn, leads to increased fibrinolytic activity as reflected by the fall in plasminogen and rise in D-dimer concentrations secondary to the release of t-PA from the endothelium by thrombin. The data suggest that C5a contributes significantly to this sequence, although it is not likely to be the sole responsible factor. The reduction in the t-PA response in the anti-C5a-treated group (compared to the group receiving pre-immune IgG) is very clear and coincides closely with the reduction in peak TAT levels and the attenuation of the subsequent consumption of plasminogen and D-dimer levels. Those findings suggest a link between thrombin production and thrombin-induced release of t-PA from the vascular endothelium.
CLP animals treated with the pre-immune IgG had more significant changes characteristic of early activation of the coagulation/fibrinolytic systems. CLP animals treated with pre-immune IgG developed an acute phase reaction with high levels of fibrinogen. Increased plasma fibrinogen levels are usually not found in severe sepsis-induced DIC, which is characterized by consumption of fibrinogen. 28,42 However, in early sepsis fibrinogen will increase as an acute phase reactant. Taylor and colleagues 43 demonstrated in a baboon model of Escherichia coli-induced sepsis that increased levels of fibrinogen mask evidence of DIC. 43 Although thrombocytopenia is not specific for the development of sepsis-related DIC, it is a reliable sign of hemostatic activation. The differences in the degree of thrombocytopenia in the CLP groups (treated with pre-immune IgG versus anti-C5a IgG) were somewhat modest (Figure 3A) ▶ . It is also possible that other events could be occurring, such as reduced platelet production in the marrow and splenic sequestration of platelets. FVII:C is known to be activated by tissue factor. The overall small, but significant, early decline in FVII:C may reflect early generation of tissue factor during CLP-induced sepsis. This finding agrees with in vitro observations that incubation of endothelial cells with C5a enhances tissue factor production. 24,25 Thrombin generation in pre-immune IgG-treated CLP animals is reflected by early prolongation of APTT and PT, early consumption of FIX:C and FVII:C, decrease of AT levels, increase in plasma TAT complexes, and the transient increase of D-dimer levels. Such consumptive changes may have been initiated by bacterial proteases or cell wall (lipopolysaccharide) products. Decreased AT and increased D-dimer plasma levels, both of which are known to be sensitive markers for DIC, 44 are closely correlated with high mortality in septic shock in other animal and human sepsis studies. 7,8,45-47 The overall low levels of fibrinolytic activity in the pre-immune IgG-treated animals were offset by elevated PAI and a fall in t-PA. The increased PAI levels in other animal experimental sepsis studies highly correlate with poor prognostic outcome. 48
Collectively, these data indicate that there is evidence for DIC in the pre-immune IgG-treated CLP animals, whereas treatment of CLP rats with anti-C5a attenuates the development of DIC. Despite the observation that hemostatic disturbances in CLP rats do not manifest florid DIC, as defined by elevated levels of D-dimer, TAT complexes, and PAI as well as reductions in AT and plasminogen levels, the data are consistent with this diagnosis of DIC. 43 We demonstrate that anti-C5a significantly ameliorates coagulation/fibrinolytic changes and DIC in septic rats. The changes in survival, platelet counts, fibrinogen, FVII:C, AT, plasminogen, t-PA, and PAI as well as TAT complexes and D-dimer were all markedly attenuated in CLP rats treated with anti-C5a.
In recent years, it has become apparent that mediators of inflammation have critical roles in the hemostatic response, and vice versa. 49 Pro- and anti-inflammatory cytokines modulate coagulation reactions in sepsis. 50 Likewise, numerous coagulation factors have been clearly shown to impact on the inflammatory response. Replacement therapies using recombinant tissue factor plasma inhibitor, AT, or activated protein C concentrates are promising in preclinical and early clinical investigations, seeming to improve clinical outcomes in sepsis because of their combined anti-inflammatory and anti-coagulant activities. 49
Our data suggest that the anaphylatoxin, C5a, modulates the hemostatic response in vivo, supporting the interpretation that coagulation and inflammation are coupled systems, interactive, and mutually reinforcing in sepsis. Administration of anti-C5a to septic rats ameliorates thrombin formation and lowers the mortality rate.
These findings support the hypothesis that interventions directed against complement activation products may result in amelioration of DIC in septic patients, thereby improving survival rates. We suggest that, in an effort to develop new anti-sepsis strategies, the combination of anti-coagulant and anti-inflammatory properties of two or more substances (eg, anti-C5a treatment combined with activated protein C concentrates) deserve consideration.
Footnotes
Address reprint requests to Peter A. Ward, M.D., Department of Pathology, University of Michigan Medical School, 1301 Catherine Rd., Ann Arbor, MI 48109-0602. E-mail: pward@umich.edu.
Supported by National Institutes of Health grants GM61656 (to P. A. W.), GM29507 (to P. A. W.), HL65194 (to A. H. S.), and HL57346 (to A. H. S.).
References
- 1.Deitch EA: Multiple organ failure. Pathophysiology and potential future therapy. Ann Surg 1992, 216:117-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bone RC: Toward an epidemiology and natural history of SIRS (systemic inflammatory response syndrome). JAMA 1992, 268:3452-3455 [PubMed] [Google Scholar]
- 3.Levi M, ten Cate H, van der Poll T, van Deventer SJ: Pathogenesis of disseminated intravascular coagulation in sepsis. JAMA 1993, 270:975-979 [PubMed] [Google Scholar]
- 4.Gando S, Kameue T, Nanzaki S, Nakanishi Y: Cytokines, soluble thrombomodulin and disseminated intravascular coagulation in patients with systemic inflammatory response syndrome. Thromb Res 1995, 80:519-526 [DOI] [PubMed] [Google Scholar]
- 5.Morrison DC, Kline LF: Activation of classical and properdin pathways of complement by bacterial lipopolysaccharides (LPS). J Immunol 1977, 118:362-368 [PubMed] [Google Scholar]
- 6.Gando S, Kameue T, Nanzaki S, Nakanishi Y: Disseminated intravascular coagulation is a frequent complication of systemic inflammatory response syndrome. Thromb Haemost 1996, 75:224-228 [PubMed] [Google Scholar]
- 7.Nielsen JD: The effect of antithrombin on the systemic inflammatory response in disseminated intravascular coagulation. Blood Coagul Fibrinolysis 1998, 9:S11-S15 [PubMed] [Google Scholar]
- 8.Mammen EF: Antithrombin: its physiological importance and role in DIC. Semin Thromb Hemost 1998, 24:19-25 [DOI] [PubMed] [Google Scholar]
- 9.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fischer CJ, Jr: Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001, 344:699-670 [DOI] [PubMed] [Google Scholar]
- 10.Colman RW: The role of plasma proteases in septic shock. N Engl J Med 1989, 320:1207-1209 [DOI] [PubMed] [Google Scholar]
- 11.Esmon CT: Regulation of blood coagulation. Biochim Biophys Acta 2000, 1477:349-360 [DOI] [PubMed] [Google Scholar]
- 12.Gando S, Kameue T, Nanzaki S, Hayakawa T, Nakanishi Y: Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome. Crit Care Med 1997, 25:1820-1826 [DOI] [PubMed] [Google Scholar]
- 13.Gray E, Thomas S, Mistry Y, Poole S: Inhibition of tissue factor and cytokine release. Haemostasis 1996, 26:92-95 [DOI] [PubMed] [Google Scholar]
- 14.ten Cate H, Levi M, van der Poll T, Biemond BJ, van Deventer SJ, Buller HR, ten Cate JW: Inhibition of extrinsic coagulation activation in endotoxemia; therapeutic implications. Prog Clin Biol Res 1994, 388:215-219 [PubMed] [Google Scholar]
- 15.Bevilacqua MP, Pober JS, Majeau GR, Cotran RS, Gimbrone MA, Jr: Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J Exp Med 1984, 160:618-623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tijburg PN, Ryan J, Stern DM, Wollityky B, Rimon A, Handley D, Nawroth P, Sixma JJ, de Groot PG: Activation of the coagulation mechanism on tumor necrosis factor-stimulated cultured endothelial cells and their extracellular matrix. The role of flow and factor IX/IXa. J Biol Chem 1991, 266:12067-12074 [PubMed] [Google Scholar]
- 17.Moore KL, Andreoli SP, Esmon NL, Esmon CT, Bang NU: Endotoxin enhances tissue factor and suppresses thrombomodulin expression of human vascular endothelium in vitro. J Clin Invest 1987, 79:124-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colucci M, Balconi G, Lorenzet R, Pietra A, Locati D, Donati MB, Semeraro N: Cultured human endothelial cells generate tissue factor in response to endotoxin. J Clin Invest 1983, 71:1893-1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hack CE, Nuijens JH, Felt-Bersma RJF, Schreuder WO: Elevated plasma levels of the anaphylatoxins C3a and C4a are associated with a fatal outcome in sepsis. Am J Med 1989, 86:86-20 [DOI] [PubMed] [Google Scholar]
- 20.McCabe WR: Serum complement levels in bacteremia due to gram-negative organisms. N Engl J Med 1973, 288:21-23 [DOI] [PubMed] [Google Scholar]
- 21.Goodman MG, Chenoweth DE, Weigle WO: Induction of interleukin 1 secretion and enhancement of humoral immunity by binding of human C5a to macrophage surface C5a receptors. J Exp Med 1982, 156:912-917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okusawa S, Yancey KB, van der Meer JW, Endres S, Lonnemann G, Hefter K, Frank MM, Burke JF, Dinarello CA, Gelfand JA: C5a stimulates secretion of tumor necrosis factor from human mononuclear cells in vitro. Comparison with secretion of interleukin 1 beta and interleukin 1 alpha. J Exp Med 1988, 168:443-448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scholz W, McClurg MR, Cardenas GJ, Smith M, Noonan DJ, Hugli TE, Morgan EL: C5a-mediated release of interleukin 6 by human monocytes. Clin Immunol Immunopathol 1990, 57:297-307 [DOI] [PubMed] [Google Scholar]
- 24.Ikeda K, Nagasawa K, Horiuchi T, Nishizaka H, Niho Y: C5a induces tissue factor activity on endothelial cells. Thromb Haemost 1997, 77:394-398 [PubMed] [Google Scholar]
- 25.Muhlfelder TW, Niemetz J, Kreutzer D, Beebe D, Ward PA, Rosenfeld SI: C5 chemotactic fragment induces leukocyte production of tissue factor activity: a link between complement and coagulation. J Clin Invest 1979, 63:147-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sims PJ, Faioni EM, Wiedmer T, Shattil SJ: Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 1988, 263:18205-18212 [PubMed] [Google Scholar]
- 27.Carson SD, Johnson DR: Consecutive enzyme cascades: complement activation at the cell surface triggers increased tissue factor activity. Blood 1990, 76:361-367 [PubMed] [Google Scholar]
- 28.Taylor FB, Jr: Studies on the inflammatory-coagulant axis in the baboon response to E. coli: regulatory roles of proteins C, S, C4bBP and of inhibitors of tissue factor. Prog Clin Biol Res 1994, 388:175-194 [PubMed] [Google Scholar]
- 29.Hesselvik JF, Malm J, Dahlback B, Blomback M: Protein C, protein S and C4b-binding protein in severe infection and septic shock. Thromb Haemost 1991, 65:126-129 [PubMed] [Google Scholar]
- 30.Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, Schmal H, Friedl HP, Ward PA: Protective effects of C5a blockade in sepsis. Nat Med 1999, 5:788-792 [DOI] [PubMed] [Google Scholar]
- 31.Huber-Lang M, Sarma VJ, Lu KT, McGuire SR, Padgaonkar VA, Guo RF, Younkin EM, Kunkel RG, Ding J, Erickson R, Curnutte JT, Ward PA: Role of C5a in multiorgan failure during sepsis. J Immunol 2001, 166:1193-1199 [DOI] [PubMed] [Google Scholar]
- 32.Odegard OR, Lie M, Abildgaard U: Heparin cofactor activity measured with an amidolytic method. Thromb Res 1975, 6:287-294 [DOI] [PubMed] [Google Scholar]
- 33.Lijnen HR, van Hoef B, Beelen V, Collen D: Characterization of the murine plasma fibrinolytic system. Eur J Biochem 1994, 224:863-871 [DOI] [PubMed] [Google Scholar]
- 34.Morrison DC, Ulevitch RJ: The effect of bacterial endotoxins on host mediator systems. Am J Pathol 1978, 2:526-617 [PMC free article] [PubMed] [Google Scholar]
- 35.Nakae H, Endo S, Inada K, Yoshida M: Chronological changes in the complement system in sepsis. Surg Today 1996, 26:225-229 [DOI] [PubMed] [Google Scholar]
- 36.Solomkin JS, Jenkins MK, Nelson RD, Chenoweth D, Simmons RL: Neutrophil dysfunction in sepsis. II. Evidence for the role of complement activation products in cellular deactivation. Surgery 1981, 90:319-327 [PubMed] [Google Scholar]
- 37.Alexander JW, Stinnett JD, Ogle CK, Ogle JD, Morris MJ: A comparison of immunologic profiles and their influence on bacteremia in surgical patients with a high risk of infection. Surgery 1979, 86:94-104 [PubMed] [Google Scholar]
- 38.Lundberg C, Marceau F, Hugli TE: C5a-induced hemodynamic and hematologic changes in the rabbit. Role of cyclooxygenase products and polymorphonuclear leukocytes. Am J Pathol 1987, 128:471-483 [PMC free article] [PubMed] [Google Scholar]
- 39.Steven JH, O’Hanley P, Shapiro JM, Mihm FG, Satoh PS, Collins JA, Raffin TA: Effects of anti-C5a antibodies on the adult respiratory distress syndrome in septic primates. J Clin Invest 1986, 77:1812-1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deitch EA: Animal models of sepsis and shock: a review and lessons learned. Shock 1997, 9:1-11 [DOI] [PubMed] [Google Scholar]
- 41.Heumann D, Glauser MP: Anticytokine strategies for the treatment of septic shock: relevance of animal models. Curr Top Microbiol Immunol 1996, 216:299-311 [DOI] [PubMed] [Google Scholar]
- 42.Owen CA, Bowie EJ: Induced chronic intravascular coagulation in dogs. Bibl Haematol 1977, 44:169-173 [DOI] [PubMed] [Google Scholar]
- 43.Taylor FB, Jr, Wada H, Kinasewitz G: Description of compensated and uncompensated disseminated intravascular coagulation (DIC) responses (nonovert and overt DIC) in baboon models of intravenous and intraperitoneal Escherichia coli sepsis and in the human model of endotoxemia: toward a better definition of DIC. Crit Care Med 2000, 28:S12-S19 [DOI] [PubMed] [Google Scholar]
- 44.Carey MJ, Rodgers GM: Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998, 59:65-73 [DOI] [PubMed] [Google Scholar]
- 45.Baudo F, Arlati S, Casella GP, Caimi TM, de Cataldo F, Giudici D, D’Angelo A, Petrini F, Ridolfi L, Palareti G, Legnani C: DIC in the multiple organ dysfunction syndrome in patients in intensive care unit: prognostic index. Faist E eds. Fifth World Congress on Trauma, Shock, Inflammation and Sepsis. 2000, :pp 835-839 Monduzzi Editone, (Munich). Bologna, Italy [Google Scholar]
- 46.Wada H, Mori Y, Shimura M, Hiyoyama K, Ioka M, Nakasaki T, Nishikawa M, Nakano M, Kumeda K, Kaneko T, Nakamura S, Shiku H: Poor outcome in disseminated intravascular coagulation or thrombotic thrombocytopenic purpura patients with severe vascular endothelial cell injuries. Am J Hematol 1998, 58:189-194 [DOI] [PubMed] [Google Scholar]
- 47.Carey MJ, Rodgers GM: Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998, 59:65-73 [DOI] [PubMed] [Google Scholar]
- 48.Schmaier AH, Srikanth S, Elghetany MT, Normolle D, Gokhale S, Feng H-M, Walker DH: Hemostatic/fibrinolytic proteins changes in C3H/HeN mice infected with Rickettsia conorii. Thromb Haemost 2001, 86:871-879 [PubMed] [Google Scholar]
- 49.Esmon CT: Role of coagulation inhibitors in inflammation. Thromb Haemost 2001, 86:51-56 [PubMed] [Google Scholar]
- 50.van der Poll T, Levi M, van Deventer SJ, ten Cate H, Haagmans BL, Biemond BJ, Buller HR, Hack CE, ten Cate JW: Differential effects of anti-tumor necrosis factor monoclonal antibodies on systemic inflammatory responses in experimental endotoxemia in chimpanzees. Blood 1994, 83:446-451 [PubMed] [Google Scholar]