

Abstract
Tumor development is thought to require both increased proliferation and inhibition of apoptosis. However, the relationship between cell replication and cell death in liver tumorigenesis is complex because both proliferation and apoptosis increase during hepatocarcinogenesis. To investigate the effect of the anti-apoptotic gene Bcl-2 in liver carcinogenesis, we established a line of double transgenic mice that express transforming growth factor-α (TGF-α), a liver mitogen, and Bcl-2. Double transgenic mice, TGF-α and Bcl-2 single transgenics, and wild type received an injection of diethylnitrosamine at 15 days of age. This alkylating agent induces liver carcinogenesis and its effect is greatly enhanced by TGF-α. We report that Bcl-2 expression inhibited diethylnitrosamine-induced liver carcinogenesis and counteracted the enhancing effect of TGF-α. Bcl-2 delayed the growth of proliferative foci at the early stages of carcinogenesis and inhibited cell proliferation in these foci. The effect of Bcl-2 on liver carcinogenesis is consistent with its reported ability to interfere with cell replication. The data demonstrate that the expression of an anti-apoptotic gene during liver carcinogenesis causes a delay rather than an increase in tumorigenesis.
The discovery that overexpression of Bcl-2 in follicular lymphomas, caused by a chromosomal translocation, inhibits apoptosis without increasing cell proliferation established a new mode of action for oncogenes. 1,2 It is now accepted that inhibition of apoptosis is a component of some stages of oncogenesis and that Bcl-2 itself and anti- and pro-apoptotic Bcl-2 family members may play important roles in the process.
Liver carcinogenesis has been extensively studied in experimental models. The best studied of these models involves the overexpression of oncogenes or growth factors, such as transforming growth factor-α (TGF-α), and the exposure to chemical carcinogens. A widely used method of inducing liver tumors in rats is the initiation/promotion protocol, in which the administration of an initiating agent is followed by application of a tumor promoter to expand the population of initiated cells. Studies with the tumor promoter phenobarbital, a non-genotoxic agent, demonstrated that its effects on rat liver carcinogenesis are more directly related to cell survival than cell proliferation. 3 Opposing relationships between cell proliferation and survival in tumorigenesis are easier to conceive in tissues that are normally proliferative, such as the mucosa of the gastrointestinal tract and the skin. However, the liver is not a proliferative organ in its normal state, and the rates of hepatocyte replication and apoptosis are very low. In a series of studies, Grasl-Kraupp et al 4,5 and Schulte-Hermann et al 6 showed that during liver carcinogenesis both cell replication and apoptosis are greatly increased over normal levels. As tumorigenesis develops, cell proliferation exceeds apoptosis, although cell replication and cell death are much higher than normal. These and other studies indicate that during hepatocarcinogenesis, the selective, proliferative advantage of altered hepatocytes is associated with enhanced apoptosis. 4,6,7
To analyze the relationships between hepatocyte proliferation and apoptosis in hepatocarcinogenesis, we established a line of double transgenic mice that overexpress TGF-α, a hepatocyte mitogen, and Bcl-2, an anti-apoptotic gene (TGF-α/Bcl-2 mice). Surprisingly, we found that Bcl-2 expression in these animals inhibited and delayed the development of TGF-α-induced hepatic tumors. 8 Further studies revealed that Bcl-2 can block hepatocyte replication in the regenerating liver through an effect on cell cycle progression, acting at a stage beyond cyclin D1 expression. 9 We concluded from these studies, as well as from the work of de La Coste et al, 10 that Bcl-2 expression inhibits liver carcinogenesis by slowing down the replication of altered cells. However, because of the lack of clear precursor lesions during TGF-α-induced hepatic tumorigenesis in the mice of hybrid genetic background used in the experiments, it was not possible to determine whether Bcl-2 expression would interfere with development and expansion of identifiable proliferative or early neoplastic foci. 8 Moreover, because the effects of Bcl-2 have so far been studied only in growth factor or oncogene-mediated tumorigenesis, 8,10 we wished to determine whether Bcl-2 would have similar effects in chemically induced hepatocarcinogenesis. To obtain answers to these questions, we injected newborn mice with the alkylating agent diethylnitrosamine (DEN), a liver carcinogen whose tumorigenic effect can be enhanced by TGF-α. An analysis of liver carcinogenesis in DEN-treated wild type (WT), Bcl-2 or TGF-α single transgenic, and TGF-α/Bcl-2 double transgenic mice demonstrated that Bcl-2 expression in single and double transgenic mice delayed tumorigenesis and inhibited the development of proliferative foci which precede tumor formation.
Materials and Methods
Transgenic Mice
We used male TGF-α/Bcl-2 double transgenic, TGF-α and Bcl-2 single transgenic, and WT mice, which were generated as reported previously. 8 In brief, female heterozygous TGF-α transgenic mice of the MT42 line (CD1 background) were mated to male heterozygous Bcl-2 mice (C57/B6C3H), yielding WT, TGF-α, and Bcl-2 single transgenics and TGF-α/Bcl-2 double transgenics, all in the same hybrid (CD1 × C57/B6C3H) background. Genotypes were determined by PCR amplification of genomic DNA obtained from mouse tails. Mice were maintained in specific pathogen-free housing and cared for in accordance with NIH guidelines for animal care and approval of protocols by the University of Washington Animal Care Committee.
DEN Treatment and Transgene Induction
Fifteen-day-old TGF-α/Bcl-2 double and TGF-α and Bcl-2 single transgenic and WT male mice received an intraperitoneal injection of 5 mg/kg DEN and were maintained on drinking water containing 25 mmol/L ZnSO4 (zinc water) to induce Bcl-2 transgene expression for the duration of the study. Animals were killed at 0, 1, 2, and 7 days for analysis of Bcl-2 and TGF-α expression and at 3, 4, 5, 6, 8–10, and 13 months for studies of tumorigenesis. At the time of death, livers were removed and examined, and portions were immediately fixed for histological analysis. The remaining liver was frozen in liquid nitrogen and stored at −80° for Western blot analysis.
Histology and Immunohistochemistry
Liver fragments were fixed in either neutral buffered formalin for 24 hours for routine histology and immunohistochemistry or methyl-Carnoy’s fluid (60% methanol, 30% chloroform, 10% acetic acid) for bromodeoxyuridine (BrdU) labeling. Sections (5 μm) were cut from paraffin-embedded blocks and stained with hematoxylin and eosin (H&E) for histological examination. PCNA immunohistochemistry was performed using a 1:200 dilution of mouse monoclonal antibody MO 879 (Dako, Carpinteria, CA) after antigen retrieval in Dako TRS. Endogenous peroxidase activity was blocked by 5 minutes of incubation in 3% hydrogen peroxide solution. The primary antibody was incubated with biotinylated sheep F(ab)2 fragment anti-mouse antibody (B-6774, Sigma, St. Louis, MO). Other studies of DNA replication were done in a small subset of animals by infusion of BrdU for 3 days using Alzet osmotic pumps (model 1003D, Alza Corporation, Palo Alto, CA). Detection of BrdU labeling was performed as described previously. 8
Analyses of Focal Lesions and Hepatocyte Proliferation
The identification of focal lesions was performed in H&E-stained liver sections. Images were captured with a digital camera (Spot Insight) mounted on an Olympus microscope. The images were imported into Adobe Photoshop (Seattle, WA) and foci highlighted for determination of number of foci/cm 2 and percentage of sections involved by foci using Image-ProPlus image analysis software (Media Cybernetics, Silver Spring, MD). PCNA staining of hepatocyte nuclei was analyzed with Image-ProPlus software. Fischer’s least significant difference analysis of variance was used for comparisons between means. For all tests, P < 0.05 was considered significant.
Western Blot Analysis
Total liver protein was isolated from snap-frozen tissue by homogenization in RIPA buffer (1.0% NP40, 0.5% sodium deoxycholate, 0.1% SDS in PBS) containing 1 mmol/L dithiothreitol, 0.5 mmol/L p-aminoethylbenzenesulfonyl fluoride, 2 μg/ml aprotinin, 2 μg/ml pepstatin A, 2 μg/ml leupeptin, and 10 μg/ml soybean trypsin inhibitor. Protein concentrations were determined with the Bradford protein assay (BioRad, Hercules, CA). Proteins (25 μg) were separated on a 10% SDS-PAGE gel, transferred to nylon membranes, and blocked in 5% milk containing 0.1% Tween-20 before incubation with antibody (rabbit anti-Bcl-2, clone N19, Santa Cruz Biotechnology). Mature TGF-α protein was detected using mouse anti TGF-α (Ab-1, Calbiochem) as previously described. 8 Enhanced chemiluminescence (ECL, Santa Cruz Biotechnology) was used for detection.
Results
We previously described the generation of TGF-α/Bcl-2 double transgenic mice and the ability of Bcl-2 to delay and reduce the frequency of liver tumor development induced by the TGF-α transgene. 8 The goal of the present study was to determine whether overexpression of Bcl-2 would delay the progression of liver proliferative foci and tumorigenesis initiated by a chemical carcinogen. We injected 15-day-old male TGF-α/Bcl-2 double transgenic (n = 29) and TGF-α (n = 23) and Bcl-2 (n = 24) single transgenic mice, as well as WT (n = 33) mice with 5 mg/kg of DEN. Groups of mice were killed at 3, 4, 5, and 6 months, between 8 and 10 months, and at 13 months. Both the TGF-α and the Bcl-2 transgenes are under the control of the MT-1 promoter and are inducible by zinc water. 8 When maintained on zinc water, Bcl-2 is very strongly expressed, and TGF-α transgene expression increases only approximately twofold. While Bcl-2 is uniformly expressed throughout the liver, TGF-α expression is more restricted. 8 In this study, we placed the mice on zinc water at the time of DEN injection at 15 days. Because the mice were still nursing at this time, we first needed to determine when the expression of the Bcl-2 transgene would start. We analyzed the induction of the Bcl-2 transgene by Western blot analysis (Figure 1) ▶ in livers of WT and Bcl-2 single transgenic mice maintained on zinc water and killed between 0 and 7 days after the start of the zinc water regimen. In the absence of zinc, there was very little detectable Bcl-2 protein in the liver lysates obtained from the Bcl-2 transgenic mice and none in lysates from WT mice. In Bcl-2 transgenic mice, Bcl-2 protein was low 1 day after exposure to zinc water, increased at 2 days, and was maximally induced by 7 days. In contrast, no Bcl-2 protein was detected in liver lysates from WT mice. Liver TGF-α expression was not detectable in the liver of WT mice, was high in 1 day old TGF-α transgenics and by 7 days, reached the levels found in adult liver (Figure 1) ▶ .
Figure 1.

Expression of Bcl-2 and TGF-α in livers of newborn mice. WT, Bcl-2, and TGF-α transgenic mice were injected with DEN at 15 days of age and were immediately placed on zinc water to induce the transgenes. Two Bcl-2 and two TGF-α transgenic mice were analyzed for BCL-2 protein and TGF-α, respectively by Western blot at 0, 1, 2, and 7 days after the start of zinc water treatment. WT littermates were analyzed either before zinc water treatment (WT, 0) or 7 days after the start of the treatment (WT, 7).
Development of Liver Tumors
Mice of all four genotypes injected with DEN and maintained in zinc water were examined for the presence of liver tumors at monthly intervals between 3 and 6 months (Figure 2A) ▶ . In addition, Bcl-2 single transgenics and WT mice were analyzed later, between 8 and 10 months and at 13 months. A minimum of 4 animals of each genotype per group was examined. In the 3- to 4-month group, no tumors were detected in any of the mice examined. It is known that TGF-α enhances the development of DEN-induced hepatic tumors. 11,12 Indeed, by 5 months, liver tumors were grossly evident in the TGF-α transgenic mice, and 90% of the mice examined at 5 to 6 months had liver tumors. Histological examination revealed that the tumors were well-differentiated hepatocellular carcinomas of a solid pattern. In contrast, livers of TGF-α/Bcl-2 double, Bcl-2 single transgenic, and WT mice through 6 months of age had no tumors by gross or histological examination. Thus, co-expression of Bcl-2 greatly inhibited the tumor-promoting effects of TGF-α. By 8 to 10 months, liver tumors were detected in all of the TGF-α single transgenic and Bcl-2 transgenic, 88% of the WT, and 75% of the TGF-α/Bcl-2 double transgenic mice. At 13 months, 100% of the WT mice and 67% of the Bcl-2 mice had developed liver tumors. Analysis of tumorigenesis in TGF-α single transgenics was terminated at 8 to 10 months as animals died or had very large tumors. Tumor histology did not differ among the four genotypes. In normal livers of non-transgenic mice, endogenous Bcl-2 is expressed in biliary epithelium but not in hepatocytes. Expression of the Bcl-2 transgene, as assessed by immunohistochemistry in the livers of Bcl-2 single transgenics and TGF-α/Bcl-2 double transgenic mice, was uniform throughout the tissue. In TGF-α single transgenic mice, Bcl-2 expression was detected in single cells or cells in small clusters without preferential localization (data not shown).
Figure 2.
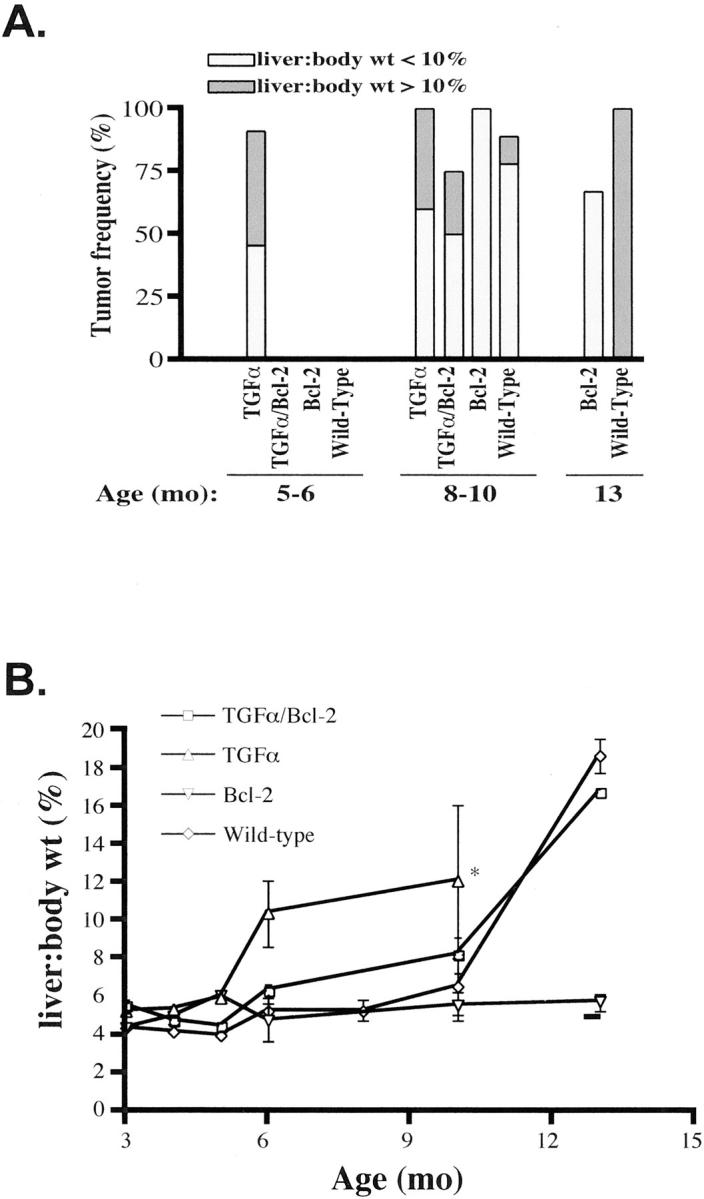
Liver tumor frequency and tumor burden in DEN-injected mice of 4 different genotypes. Mice were analyzed at 5–6, 8–10, and 13 months. A: TGF-α transgenic mice developed liver tumors as early as 5 months of age, whereas tumors were not detected in TGF-α/Bcl-2 double, Bcl-2 single transgenic, and WT mice until 8–10 months of age. Liver tumor burden is represented by the overall liver-to-body weight ratio. The percentage of mice with liver tumors, in which the liver-to-body weight ratio is less than 10%, is shown by the light gray bars; the percentage of mice with liver tumors, in which the liver-to-body weight ratio is greater than 10%, is shown by the dark gray bars. B: Increased liver-to-body weight ratios, measured by dividing the total weight of the liver by the total body weight and expressed as a percentage, are indicative of liver tumor burden. Although liver tumors developed in Bcl-2 single transgenic mice at 8–10 months, there was no significant increase in liver-to-body weight ratios. *All TGF-α transgenic mice either died or were killed at 10 months because of the presence of large liver tumors.
Although tumors developed in the majority of mice at 8 to 10 months, regardless of genotype, the overall tumor mass varied greatly among the different genotypes. Because it was difficult to separate individual tumors in the liver tissue, we estimated the tumor burden by determining liver-to-body weight ratios (Figure 2A) ▶ . Approximately 50% of TGF-α single transgenics that developed tumors at 5 to 6 and 8 to 10 months had a liver-to-body weight ratio exceeding 10% (normal value is approximately 5%; see Figure 2B ▶ ; also note that all TGF-α transgenics either died or were killed at 10 months). In contrast, none of the Bcl-2 single transgenic mice that developed tumors at 8 to 10 and 13 months had liver-to-body weight ratios higher than 10%, indicating that these mice had a much smaller tumor burden than that of TGF-α or WT mice. Despite the presence of tumors in livers of Bcl-2 transgenic mice at 8 to 10 months of age, tumor mass was much lower than that of all other groups (Figure 2B) ▶ . The protective effect of Bcl-2 expression is also illustrated by comparing liver-to-body weight ratios between TGF-α single transgenics and TGF-α/Bcl-2 double transgenic mice (Figure 2B) ▶ . Tumor growth was clearly delayed in the double transgenic animals, despite continuous expression of the TGF-α transgene (data not shown).
Development of Proliferative Hepatic Foci after DEN Injection
Tumor development in DEN-injected mice is preceded by the formation of abnormal foci in liver tissue. We examined liver sections of the 4 genotype groups at 3 to 4 months and 5 to 6 months of age to determine the number of foci present/cm 2 of liver tissue, the percentage of tissue involved by the foci and the proliferative activity of the cells of the foci measured by PCNA labeling (Table 1) ▶ . At 3 to 4 months, the number of foci/cm 2 as well as the percentage of tissue involved by the foci was significantly higher in WT and TGF-α transgenic mice than in Bcl-2 and TGF-α/Bcl-2. At 5 to 6 months, the number of foci/cm2 was ninefold higher in WT and TGF-α transgenics compared to Bcl-2 or TGF-α/Bcl-2 transgenics. The very large tissue involvement in 5- to 6-month-old TGF-α transgenic mice reflects the development of tumors in these animals. Even before the development of neoplastic lesions at 5 to 6 months, the amount of tissue involvement by proliferative foci was much larger in TGF-α transgenics than in all other groups. (See data for percent tissue involved for 4-month animals in Table 1 ▶ ). PCNA labeling showed a similar trend, that is, it was higher in WT and TGF-α transgenics compared to Bcl-2 and TGF-α/Bcl-2 transgenics with TGF-α transgenics having the highest levels. (Table 1) ▶ . The labeling data entirely agreed with measurements of proliferative indices in abnormal liver foci done in a small subset of animals which received BrdU by osmotic pump for 3 days (Figure 3) ▶ .
Table 1.
Development of Proliferative Foci in DEN-Injected Mice
| Genotype | Age (mo) | No. of mice | No. of foci | No. of foci/cm2 | % Tissue involved | % PCNA labeled* |
|---|---|---|---|---|---|---|
| WT | 4 | 8 | 9 | 1.06 ± 0.53 | 0.06 ± 0.04 | 4.6 ± 1.4 (9) |
| Bcl-2 | 4 | 9 | 2 | 0.097 ± 0.07 | 0.008 ± 0.006 | —† |
| TGF-α | 4 | 7 | 8 | 1.24 ± 0.48 | 0.60 ± 0.38 | 10.6 ± 3.6 (6) |
| TGF-α/Bcl-2 | 4 | 10 | 1 | 0.08 | 0.01 | —† |
| WT | 5–6 | 13 | 198 | 10.5 ± 3.1 | 3.28 ± 1.2 | 11.6 ± 1.0 (31) |
| Bcl-2 | 5–6 | 6 | 15 | 1.29 ± 0.65 | 0.27 ± 0.13 | 12.4 ± 2.1 (14) |
| TGF-α | 5–6 | 11 | 158 | 10.1 ± 1.91 | 41.04 ± 10.0 | 33.1 ± 2.9 (29) |
| TGF-α/Bcl-2 | 5–6 | 9 | 15 | 1.09 ± 0.29 | 0.33 ± 0.13 | 10.9 ± 2.0 (10) |
Average ± SEM are presented. For 4-month data: foci/cm2 in Bcl-2 or Bcl-2/TGF-α transgenics are significantly different from WT or TGF-α transgenics (P values 0.01 to 0.04); % tissue involved in TGF-α transgenics is significantly different from Bcl-2, Bcl-2/TGF-α transgenics and WT (P values 0.01 to 0.02). The 5- to 6-month data show the same differences with higher levels of significance. For PCNA counts, TGF-α transgenics (5- to 6-month data) differ significantly from all other genotypes (P < 0.001).
*% of PCNA-labeled cells per focus and number of foci examined in parentheses.
†% only 1 or 2 foci developed in these animals, not sufficient for analysis.
Figure 3.
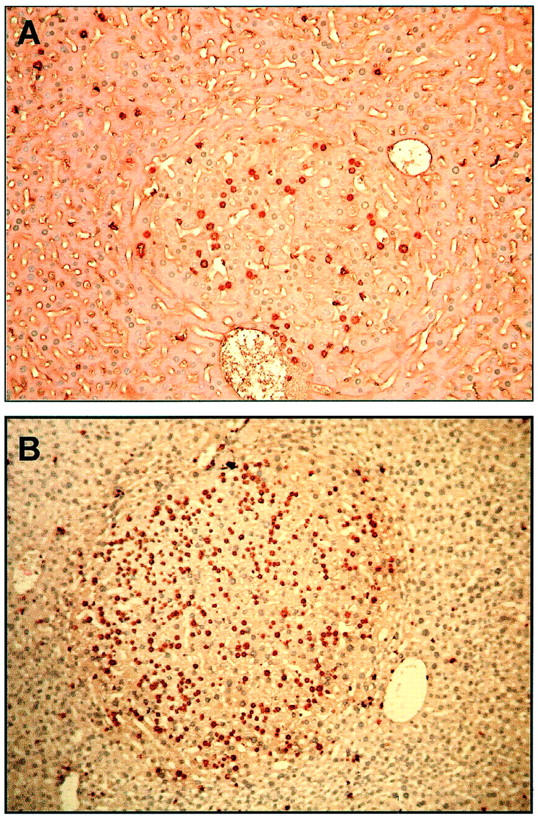
Representative proliferative foci from DEN-injected mice. TGF-α/Bcl-2 double (A), and TGF-α single (B) transgenic mice, both at 5 months. Proliferative hepatocytes were detected by BrdU incorporation and immunohistochemistry. Note the smaller focus size and fewer number of BrdU-positive hepatocytes in the TGF-α/Bcl-2 double transgenic mouse. Magnification, ×100.
Discussion
Our previous work 8 showed that Bcl-2 expression inhibits hepatocarcinogenesis induced by TGF-α. de La Coste et al 10 reported a similar inhibitory effect of Bcl-2 in c-myc transgenic mice. Our goal in the present work was to determine whether Bcl-2 inhibits chemical hepatocarcinogenesis and if it would slow the progress of proliferative foci in early carcinogenesis. We found the following: 1) Bcl-2 expression inhibits and delays hepatocarcinogenesis induced by DEN, a chemical carcinogen, and antagonizes the enhancing effect of TGF-α in TGF-α/Bcl-2 double transgenic mice. 2) Inhibition of tumorigenesis by Bcl-2 is preceded by a delay in development and cell proliferation of altered hepatocyte foci. The effect of Bcl-2 on the proliferative activity of altered foci is consistent with our data demonstrating that expression of the protein interferes with cell cycle progression in regenerating liver hepatocytes. 9 Similarly, the antagonistic effect of Bcl-2 on TGF-α enhancement of DEN carcinogenesis suggests that Bcl-2 acts as an anti-proliferative agent in the promotion of tumorigenesis. Nevertheless, we cannot completely exclude the possibility that Bcl-2 may also act at the initiation stage by preventing DEN genotoxic damage or facilitating DNA repair. A major difficulty in distinguishing promotion from initiation effects is that available markers for the identification of initiated cells in mouse liver are not completely reliable. Analysis of the expression of some potential markers (glutamine synthetase and glutamate cysteine ligase) showed that they were detectable in only a fraction of proliferative foci (data not shown). Moreover, expression of these markers in the livers of mice of the 4 genotypes did not reveal clear differences or a consistent pattern. Markers for initiated cells with a higher degree of specificity and reproducibility, which might become available in the future, may permit a more precise analysis of this issue. Nevertheless, the data presented clearly demonstrate that Bcl-2 expression inhibits DEN-induced liver tumorigenesis by inhibiting proliferation and delaying the progression of hyperplastic foci at the early stages of carcinogenesis.
An interesting finding of our work with TGF-α-induced carcinogenesis was that Bcl-2 expression not only inhibited and delayed hepatocarcinogenesis but also changed the morphological features of tumors, which eventually developed in TGF-α/Bcl-2 double transgenic mice. 8 These animals did not develop typical hepatocellular carcinomas but instead had lesions containing large hepatocytes in single rows lined by endothelial cells and separated by large vascular spaces. In contrast to these findings, tumors that developed in DEN-injected TGF-α/Bcl-2 mice were typical hepatocellular carcinomas, which did not differ in morphology from carcinomas of TGF-α single transgenics. We do not have an explanation for the morphological differences between liver tumors that develop in growth factor- and DEN-induced carcinogenesis in TGF-α/Bcl-2 double transgenic mice. However, the proliferative foci that developed in TGF-α/Bcl-2 transgenics in DEN injected animals as well as in non-injected TGF-α/Bcl-2 animals studied previously, 8 were almost always basophilic. This type of focus was rarely present in TGF-α transgenics.
We do not know whether the anti-proliferative effects of Bcl-2 are directly linked to its anti-apoptotic activity and if other anti-apoptotic genes of the Bcl-2 family also interfere with cell cycle progression. The Bcl-2 effects on tumorigenesis are not confined to the liver because its expression also inhibits the early stages of mammary carcinogenesis. 13,14 Moreover, cell cycle inhibition by Bcl-2 has been described in different types of cells in culture. 15-17 The N-terminal region of Bcl-2 adjacent to tyrosine 28 is a binding site for Bcl-2 cell cycle inhibition. Mutants have been generated which retain anti-apoptotic activity but have lost the ability to interfere with the cell cycle. 14,15,18 It would be of interest to determine whether these Bcl-2 mutants would inhibit hepatocarcinogenesis when expressed in transgenic mice.
The general implication of our findings is that attempts to block the early stages of liver tumorigenesis through the inhibition of Bcl-2 expression may result in the opposite effect, that is, enhancement of tumor formation caused by unrestrained cell proliferation. Interestingly high Bcl-2 expression in human endometrial carcinomas correlate with a low growth fraction in the tumors, 19 and in some cancers high Bcl-2 expression is associated with a better prognosis. 20-22
Acknowledgments
We thank Linda Johnstone for slide preparation (supported by NIEHS grant ES0 1247) and John T. Brooling for technical assistance.
Footnotes
Address reprint requests to Nelson Fausto, M.D., Department of Pathology, University of Washington, K-078 Health Sciences Building, Box 357705, Seattle, WA 98195-7705. E-mail: nfausto@u.washington.edu.
Supported by NIH grant CA74131 from the National Cancer Institute.
R.H.P. and M.E.V. contributed equally to this work.
Dr. Pierce’s current address: Department of Pathology and Laboratory Medicine, University of Rochester, Rochester, NY.
Dr. Vail’s current address: Science Education Partnership Program, Fred Hutchinson Cancer Research Center, Seattle, WA.
References
- 1.Korsmeyer SJ: Bcl-2: an antidote to programmed cell death. Cancer Surv 1992, 15:105-118 [PubMed] [Google Scholar]
- 2.Vaux DL, Cory S, Adams JM: Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335:440-442 [DOI] [PubMed] [Google Scholar]
- 3.Bursch W, Lauer B, Timmermann-Trosiener I, Barthel G, Schuppler J, Schulte-Hermann R: Controlled death (apoptosis) of normal and putative preneoplastic cells in rat liver following withdrawal of tumor promoters. Carcinogenesis 1984, 5:453-458 [DOI] [PubMed] [Google Scholar]
- 4.Grasl-Kraupp B, Ruttkay-Nedecky B, Mullauer L, Taper H, Huber W, Bursch W, Schulte-Hermann R: Inherent increase of apoptosis in liver tumors: implications for carcinogenesis and tumor regression. Hepatology 1997, 25:906-912 [DOI] [PubMed] [Google Scholar]
- 5.Grasl-Kraupp B, Luebeck G, Wagner A, Low-Baselli A, de Gunst M, Waldhor T, Moolgavkar S, Schulte-Hermann R: Quantitative analysis of tumor initiation in rat liver: role of cell replication and cell death (apoptosis). Carcinogenesis 2000, 21:1411-1421 [PubMed] [Google Scholar]
- 6.Schulte-Hermann R, Bursch W, Grasl-Kraupp B, Mullauer L, Ruttkay-Nedecky B: Apoptosis and multistage carcinogenesis in rat liver. Mutat Res 1995, 333:81-87 [DOI] [PubMed] [Google Scholar]
- 7.Snibson KJ, Bhathal PS, Hardy CL, Brandon MR, Adams TE: High, persistent hepatocellular proliferation and apoptosis precede hepatocarcinogenesis in growth hormone transgenic mice. Liver 1999, 19:242-252 [DOI] [PubMed] [Google Scholar]
- 8.Vail ME, Pierce RH, Fausto N: Bcl-2 delays and alters hepatic carcinogenesis induced by transforming growth factor α. Cancer Res 2001, 61:594-601 [PubMed] [Google Scholar]
- 9.Vail ME, Chaisson ML, Thompson J, Fausto N: Bcl-2 expression delays hepatocyte cell cycle progression during liver regeneration. Oncogene 2002, 21:1548-1555 [DOI] [PubMed] [Google Scholar]
- 10.de La Coste A, Mignon A, Fabre M, Gilbert E, Porteu A, Van Dyke T, Kahn A, Perret C: Paradoxical inhibition of c-myc-induced carcinogenesis by Bcl-2 in transgenic mice. Cancer Res 1999, 59:5017-5022 [PubMed] [Google Scholar]
- 11.Tamano S, Merlino GT, Ward JM: Rapid development of hepatic tumors in transforming growth factor α transgenic mice associated with increased cell proliferation in precancerous hepatocellular lesions initiated by N-nitrosodiethylamine and promoted by phenobarbital. Carcinogenesis 1994, 15:1791-1798 [DOI] [PubMed] [Google Scholar]
- 12.Takagi H, Sharp R, Takayama H, Anver MR, Ward JM, Merlino G: Collaboration between growth factors and diverse chemical carcinogens in hepatocarcinogenesis of transforming growth factor α transgenic mice. Cancer Res 1993, 53:4329-4336 [PubMed] [Google Scholar]
- 13.Murphy KL, Kittrell FS, Gay JP, Jager R, Medina D, Rosen JM: Bcl-2 expression delays mammary tumor development in dimethylbenz(a)anthracene-treated transgenic mice. Oncogene 1999, 18:6597-6604 [DOI] [PubMed] [Google Scholar]
- 14.Furth PA, Bar-Peled U, Li M, Lewis A, Laucirica R, Jager R, Weiher H, Russell RG: Loss of anti-mitotic effects of Bcl-2 with retention of anti-apoptotic activity during tumor progression in a mouse model. Oncogene 1999, 18:6589-6596 [DOI] [PubMed] [Google Scholar]
- 15.Vairo G, Soos TJ, Upton TM, Zalvide J, DeCaprio JA, Ewen ME, Koff A, Adams JM: Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol Cell Biol 2000, 20:4745-4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazel S, Burtrum D, Petrie HT: Regulation of cell division cycle progression by bcl-2 expression: a potential mechanism for inhibition of programmed cell death. J Exp Med 1996, 183:2219-2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Reilly LA, Huang DC, Strasser A: The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J 1996, 15:6979-6990 [PMC free article] [PubMed] [Google Scholar]
- 18.Huang DC, O’Reilly LA, Strasser A, Cory S: The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J 1997, 16:4628-4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuwashima Y, Kobayashi Y, Kurosumi M, Tanuma J, Shiromizu K, Kishi K: Inverse correlation between bcl-2 expression and cell growth fraction in human endometrial adenocarcinoma tissue. Anticancer Res 1997, 17:3773-3776 [PubMed] [Google Scholar]
- 20.Silvestrini R, Benini E, Veneroni S, Daidone MG, Tomasic G, Squicciarini P, Salvadori B: p53 and bcl-2 expression correlates with clinical outcome in a series of node-positive breast cancer patients. J Clin Oncol 1996, 14:1604-1610 [DOI] [PubMed] [Google Scholar]
- 21.Sinicrope FA, Hart J, Michelassi F, Lee JJ: Prognostic value of bcl-2 oncoprotein expression in stage II colon carcinoma. Clin Cancer Res 1995, 1:1103-1110 [PubMed] [Google Scholar]
- 22.Winter JN, Andersen J, Reed JC, Krajewski S, Variakojis D, Bauer KD, Fisher RI, Gordon LI, Oken MM, Jiang S, Jeffries D, Domer P: BCL-2 expression correlates with lower proliferative activity in the intermediate- and high-grade non-Hodgkin’s lymphomas: an Eastern Cooperative Oncology Group and Southwest Oncology Group cooperative laboratory study. Blood 1998, 91:1391-1398 [PubMed] [Google Scholar]