

Abstract
Apoptotic cell death is usually accompanied by activation of a family of cysteine proteases termed caspases. Caspases mediate the selective proteolysis of multiple cellular targets often resulting in the disruption of survival pathways. Intracellular levels of the antioxidant glutathione (GSH) are an important determinant of cellular susceptibility to apoptosis. The rate-limiting step in GSH biosynthesis is mediated by glutamate-l-cysteine ligase (GCL), a heterodimeric enzyme consisting of a catalytic (GCLC) and a modifier (GCLM) subunit. In this report we demonstrate that GCLC is a direct target for caspase-mediated cleavage in multiple models of apoptotic cell death. Mutational analysis revealed that caspase-mediated cleavage of GCLC occurs at Asp499 within the sequence AVVD499G. GCLC cleavage occurs upstream of Cys553, which is thought to be important for association with GCLM. GCLC cleavage is accompanied by a rapid loss of intracellular GSH due to caspase-mediated extrusion of GSH from the cell. However, while GCLC cleavage is dependent on caspase-3, GSH extrusion occurs by a caspase-3-independent mechanism. Our identification of GCLC as a target for caspase-3-dependent cleavage during apoptotic cell death suggests that this post-translational modification may represent a novel mechanism for regulating GSH biosynthesis during apoptosis.
Apoptosis is a genetically regulated, energy-dependent form of cell death triggered by a variety of physiological and pathological stimuli. 1 Biochemically, apoptosis is characterized by the activation of a family of cysteine-dependent aspartate-directed proteases termed caspases that are responsible for the initiation and execution of apoptotic cell death. 2 Caspases are synthesized as inactive zymogens (pro-caspases) that are activated by either autocatalytic processing or cleavage by other caspases. 2 The activation of initiator caspases (eg, caspase-8 and -9) results in the cleavage and activation of downstream effector caspases (eg, caspase-3, -6, and -7), which are responsible for the selective and limited proteolysis of multiple cellular proteins involved in the morphological and biochemical changes associated with apoptosis. 2 Caspase-mediated cleavage of specific target proteins generally results in either the activation of proteins that participate in the execution of apoptosis or the inhibition of target proteins that would normally promote cell survival. In this regard, some caspase targets are components of cellular defense systems and their proteolytic inactivation plays a permissive role in facilitating the cell death process.
The tripeptide glutathione (GSH; γ-glutamylcysteinylglycine), the most abundant non-protein thiol antioxidant within the cell, is vitally important in maintenance of cellular redox status and protection against oxidative injury. 3 GSH acts as a free-radical scavenger, and through the GSH-peroxidase/GSSG-reductase system, represents a first line defense against oxidative stress. 4 GSH levels are rapidly depleted in many models of apoptosis. 5-11 Furthermore, maintaining intracellular GSH levels by the use of membrane permeable GSH analogs, supplementation with N-acetyl-cysteine, or inhibition of GSH efflux has been shown to inhibit or delay apoptosis. 7,12-18 Elevated levels of GSH have also been reported to play an important role in mediating tumor cell resistance to radiation therapy and chemotherapy. 19-21 While the molecular mechanism(s) mediating the cytoprotective effects of GSH have not been fully elucidated, depletion of intracellular GSH is closely linked to the mitochondrial dysfunction that accompanies many forms of apoptosis. 22 Alternatively, GSH can detoxify various chemotherapeutics by direct conjugation 4 and has also been shown to inhibit several putative mediators of apoptosis, including the serine protease AP24 23 and neutral sphingomyelinase. 24 These findings suggest that changes in cellular redox status associated with alterations in GSH levels have a significant effect on the apoptotic cell death process.
GSH is synthesized by two sequential reactions catalyzed by glutamate-l-cysteine ligase (GCL; also known as γ-glutamylcysteine synthetase), and glutathione synthetase. 4 GCL, a heterodimeric enzyme that catalyzes the first and rate-limiting step in GSH synthesis, consists of a catalytic subunit (GCLC, 73 kd), which contributes all of the enzymatic activity and contains all of the substrate binding sites of GCL, and a modifier subunit (GCLM, 31 kd), which modulates the affinity of GCLC for substrates and inhibitors. 4 In this regard, GCLC is highly sensitive to feedback inhibition by GSH in the absence of GCLM. 25 Heterodimerization with GCLM also lowers the apparent Km of GCLC for glutamate to within a physiological range. 25 Overexpression of the GCL subunits has been shown to elevate intracellular GSH levels and inhibit both receptor- and chemical-mediated apoptosis. 26-28 GCL expression and/or activity are also up-regulated in some cancer cell lines that exhibit chemoresistance and pharmacological inhibition of GCL leads to a reversion of the resistant phenotype. 20,21,29 While manipulation of GCL activity and GSH status can influence the cellular response to apoptotic stimuli, little is known concerning the dynamic regulation of GCL activity and GSH biosynthesis during apoptotic cell death.
In the present study we demonstrate that GCLC is a direct target for caspase-mediated cleavage during both receptor- and chemical-mediated apoptosis. While caspase-3 is required for GCLC cleavage, the caspase-dependent depletion of intracellular GSH that accompanies receptor-mediated apoptotic cell death occurs in the absence of caspase-3 and can be mechanistically dissociated from GCLC cleavage. Furthermore, cleavage of GCLC occurs upstream of the putative GCLM interaction site, suggesting that this may result in the inability of GCLC to associate with GCLM to form a functionally active GCL holoenzyme. Our identification of GCLC as a target for caspase-mediated cleavage reveals a novel post-translational modification of GCLC that may play an important role in regulating GSH biosynthesis during apoptotic cell death.
Materials and Methods
Plasmids and Reagents
Murine GCLC and GCLM were amplified by polymerase chain reaction (PCR) using pCR3.1-GCLC or pCR3.1-GCLM as templates. 30,31 GCLC(D499A) was prepared by PCR mutagenesis. PCR products were subcloned into pET28, pET21 (Clontech, Palo Alto, CA), and/or pcDNA3.1/His (Invitrogen, Carlsbad, CA) and verified by DNA sequencing. pGEX-KG-caspase-3 was provided by Dr. Dennis Templeton (Case Western Reserve University, Cleveland, OH). 32 Antisera against GCLC and GCLM have been described. 33 Caspase-3 antibodies were from Transduction Laboratories (clone 19) (Lexington, KY) or Santa Cruz Biotechnology (sc-7272) (Santa Cruz, CA). Ac-DEVD-AMC was from Alexis Biochemicals (San Diego, CA), Ac-DEVD-CHO was from Biomol (Plymouth Meeting, PA), and z-VAD-fmk was from Bachem (King of Prussia, PA). Human tumor necrosis factor-α (TNF) was from BD Pharmingen (San Diego, CA) and anti-human αFas antibody (CH-11) was from Kamiya Biochemicals (Seattle, WA). All lysis buffers were supplemented with Complete protease inhibitor mix from Roche Molecular Biochemicals (Indianapolis, IN).
Cell Lines
Jurkat cells (provided by Dr. Jonathan Graves, University of Washington, Seattle, WA) were cultured as described. 10 HeLa cells (provided by Dr. Peter Rabinovitch, University of Washington, Seattle, WA) and MCF7 cells stably expressing pcDNA3.0 vector or pcDNA3.0-caspase-3 (provided by Dr. Vince Kidd, St. Jude’s Research Hospital, Memphis, TN) were cultured as described. 34
Immunoblot Analysis and Affinity Purification of Ectopically Expressed His-GCLC
Whole cell extracts were prepared by a brief sonication on ice in TES/SB buffer (20 mmol/L Tris, pH 7.4, 1 mmol/L EDTA, 250 mmol/L sucrose, 20 mmol/L boric acid, and 1 mmol/L L-serine) (Sigma Chemical, St. Louis, MO). GCLC, GCLM, and pro-caspase-3 expression were analyzed by immunoblotting as described. 33,34 For analysis of ectopically expressed His-GCLC protein, HeLa cells were transfected by electroporation as described. 35 Cells were lysed by sonication in His lysis buffer (HLB) (20 mmol/L Tris, pH 7.9 and 500 mmol/L NaCl) and clarified extracts (500 μg) incubated with TALON metal affinity resin (Clontech, Palo Alto, CA) for 1 hour at 4°C. After extensive washing in HLB containing 40 mmol/L imidazole, affinity purified His-GCLC was analyzed by immunoblotting with anti-GCLC antibody.
Analysis of Apoptosis and Glutathione Levels
Cells were analyzed for apoptotic nuclear morphology by staining with 4′,6-diamidino-2-phenylindole (DAPI). 11 For analysis of caspase-3-like activity, cell extracts were prepared by sonication in TES/SB as described above and 50 μg of soluble protein was added to 50 μl of 2X caspase cleavage buffer (CCB) (40 mmol/L piperazine-N,N′-bis(2-ethanesylfonic acid) (PIPES), pH 7.2, 200 mmol/L NaCl, 20% sucrose, 0.2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 20 mmol/L DL-dithiothreitol (DTT) containing 40 μmol/L Ac-DEVD-AMC and incubated for 30 minutes at 37°C. Fluorescence was measured and specific activity calculated as previously described. 11 Data are presented as fold activation relative to untreated cells. For glutathione measurements, cells were harvested and lysed in TES/SB as described above and total glutathione content (GSH + GSSG) was determined by a modification of the Tietze assay. 36 In some experiments the culture media was also collected and glutathione content analyzed in a similar manner. GSH levels are reported as nmol/mg protein in the initial cell extract.
In Vitro Analysis of GCLC Cleavage
35S-labeled GCLC(WT) and GCLC(D499A) were synthesized from pET28 expression vectors with the in vitro TNT coupled transcription/translation system (Promega, Madison, WI) using T7 polymerase. BL21(DE3) bacterial cultures expressing pGEX-KG-caspase-3 were grown at 30°C and induced for 16 hours with 0.1 mmol/L isopropyl β-D-1-thiogalactopyranoxide (IPTG). Cultures were harvested, lysed by sonication in phosphate-buffered saline (PBS), and clarified supernatant used as a source of active caspase-3. 32 Cleavage assays were performed by incubating 1 μl of 35S-labeled protein with 10 μg of bacterial extract containing active caspase-3 in CCB for 1 hour at 30°C. Proteins were resolved by SDS/PAGE and analyzed by fluorography.
Results
Cleavage of GCLC and Depletion of Intracellular GSH during Receptor-Mediated Apoptosis
We have previously reported that GCLC is cleaved from a 73-kd protein to a 60-kd fragment during etoposide-induced apoptosis in acute myeloblastic leukemia cells. 29 We thus sought to determine whether GCLC was also cleaved during receptor-mediated apoptosis. Treatment of HeLa cells with TNF in the presence of cycloheximide (CHX) resulted in a time-dependent induction of apoptosis as judged by the appearance of condensed nuclei on cell staining with DAPI, with 50% of the cells displaying apoptotic morphology within 4 to 6 hours of treatment (Figure 1A ▶ , % apoptotic). Western blot analysis of GCLC revealed that apoptotic cell death was accompanied by a rapid and nearly quantitative cleavage of GCLC to a 60-kd fragment (Figure 1A ▶ , top). Cleavage was specific for GCLC as GCLM was not cleaved during apoptosis (Figure 1A ▶ , middle). GCLC cleavage correlated temporally with the processing of pro-caspase-3 (Figure 1A ▶ , bottom) and the induction of a caspase-3-like activity as measured using the fluorogenic DEVD-AMC substrate (Figure 1B ▶ , open bars). The kinetics of apoptotic cell death and GCLC cleavage also correlated with the rapid and dramatic depletion of intracellular GSH (Figure 1B ▶ , closed bars). GCLC cleavage and GSH depletion also occurred during αFas-induced apoptosis in Jurkat cells, which again correlated temporally with the processing of pro-caspase-3 and induction of a caspase-3-like DEVDase activity (Figure 2A) ▶ . GCLC cleavage was also observed during Jurkat cell apoptosis in response to multiple other apoptotic stimuli including staurosporine, cycloheximide, and ultraviolet irradiation (Figure 2B) ▶ . Furthermore, the degree of GCLC cleavage correlated with the extent of pro-caspase-3 processing (Figure 2B) ▶ . Again, there was no change in GCLM protein levels in any of these models of Jurkat cell apoptosis (Figure 2B) ▶ . In aggregate, these findings indicate that GCLC cleavage is characteristic of, and occurs during, many forms of apoptotic cell death.
Figure 1.

Cleavage of GCLC and depletion of cellular GSH during apoptotic cell death. HeLa cells were treated with human TNF (30 ng/ml) and cycloheximide (CHX, 10 μg/ml) for the time period indicated. A: GCLC, GCLM, and pro-caspase-3 were analyzed by immunoblotting. Apoptosis was quantified by assessing apoptotic nuclear morphology of DAPI-stained cells. B: Cellular glutathione levels (GSH+GSSG; closed bars) were measured by a modified Tietze assay. 36 Caspase-3-like activities (open bars) were determined using the fluorogenic DEVD-AMC substrate. Data presented are from a representative experiment performed at least three times with similar results.
Figure 2.

GCLC cleavage correlates with caspase-3 activation and GSH depletion in multiple models of Jurkat cell apoptosis. A: Jurkat cells were treated with αFas antibody (CH-11, 100 ng/ml) for the time period indicated. GCLC and pro-caspase-3 were analyzed by immunoblotting. Cellular GSH levels and caspase-3-like DEVDase activities were measured as described in Figure 1 ▶ . B: Jurkat cells were treated with staurosporine (ST, 200 nmol/L), cycloheximide (C, 20 μmol/L) or exposed to UV irradiation (200 J/m2) and harvested after 8 hours. GCLC, GCLM, and pro-caspase-3 were analyzed by immunoblotting.
Cleavage of GCLC and GSH Depletion during Receptor-Mediated Apoptosis Is Mediated by Caspase Activation
To determine whether caspase activation was necessary for GCLC cleavage and GSH depletion, HeLa cells were pretreated with the cell-permeable broad-spectrum caspase inhibitor, z-VAD-fmk, before treatment with either TNF/CHX or αFas/CHX for 6 hours to induce apoptosis. As expected, z-VAD-fmk pretreatment blocked pro-caspase-3 processing (Figure 3A ▶ , bottom) and the induction of apoptotic cell death (Figure 3A ▶ , % apoptotic) in response to both TNF and αFas. Importantly, inhibition of caspase activation abolished both TNF- and αFas-induced cleavage of GCLC (Figure 3A ▶ , top). To examine whether caspase activation was also involved in the depletion of intracellular GSH levels, identically treated cells were analyzed for GSH content. Pretreatment with z-VAD-fmk was also found to block both TNF- and αFas-induced depletion of intracellular GSH (Figure 3B) ▶ . These findings indicate that caspases mediate both GCLC cleavage and GSH depletion during receptor-mediated apoptosis.
Figure 3.

Cleavage of GCLC and GSH depletion during receptor-mediated apoptosis is mediated by caspase activation. HeLa cells were pretreated with z-VAD-fmk (50 μmol/L) for 2 hours before treatment with either TNF (30 ng/ml) or αFas antibody (100 ng/ml) in the presence of cycloheximide (CHX, 10 μg/ml) for 6 hours as indicated. A: Cellular extracts were analyzed for GCLC, GCLM, and pro-caspase-3 by immunoblotting, and apoptosis was quantitated by assessing apoptotic nuclear morphology of DAPI-stained cells. B: Glutathione levels were determined by a modified Tietze assay. Data presented are from a representative experiment performed at least three times with similar results.
GCLC Is a Direct Target for Caspase-Mediated Cleavage at AVVD499G
To assess whether GCLC was a direct target for caspase-mediated cleavage, 35S-labeled in vitro translated GCLC protein was incubated with bacterial extract expressing active recombinant caspase-3. 32 Analysis of the resulting cleavage products revealed that cleavage of GCLC in vitro produced a 60-kd fragment (Figure 4A ▶ , lane 2) identical to that observed during apoptosis in cultured cells (see Figures 1 and 2 ▶ ▶ ). This cleavage was blocked by co-treatment with DEVD-CHO, a selective inhibitor of the caspase-3 subfamily (Figure 4A ▶ , lane 3). The radiolabeling of all methionine residues during in vitro translation also permitted the detection of a small 13-kd cleavage fragment (lane 2, lower arrow). Since only the large 60-kd GCLC cleavage fragment was detected by immunoblotting with an antibody raised against the N-terminus of GCLC (amino acids 76 to 91), we examined the C-terminus for potential caspase cleavage sites. Several prospective caspase cleavage sites that could potentially generate fragments of 60 and 13 kd were identified and subjected to systematic site-directed mutagenesis. Only mutation of Asp499 to Ala within the sequence AVVD499G produced a protein resistant to caspase-3-mediated cleavage in vitro (Figure 4A ▶ , lanes 4 to 6). To verify that this was the same site cleaved during apoptosis, HeLa cells were transiently transfected with expression vectors encoding His-tagged wild-type GCLC or GCLC(D499A) mutant protein. After treatment with either TNF/CHX or αFas/CHX for 6 hours to induce apoptosis, the His-tagged proteins were affinity-purified and GCLC protein analyzed by immunoblotting. While ectopically expressed His-GCLC(WT) protein was extensively cleaved after TNF/CHX or αFas/CHX treatment (Figure 4B ▶ , lanes 4 to 6), His-GCLC(D499A) mutant protein was not cleaved in response to these apoptotic treatments (Figure 4B ▶ , lanes 7 to 9). In aggregate, these findings indicate that GCLC is a bona fide target for caspase-mediated cleavage at AVVD499G within the C-terminus of GCLC during apoptotic cell death.
Figure 4.
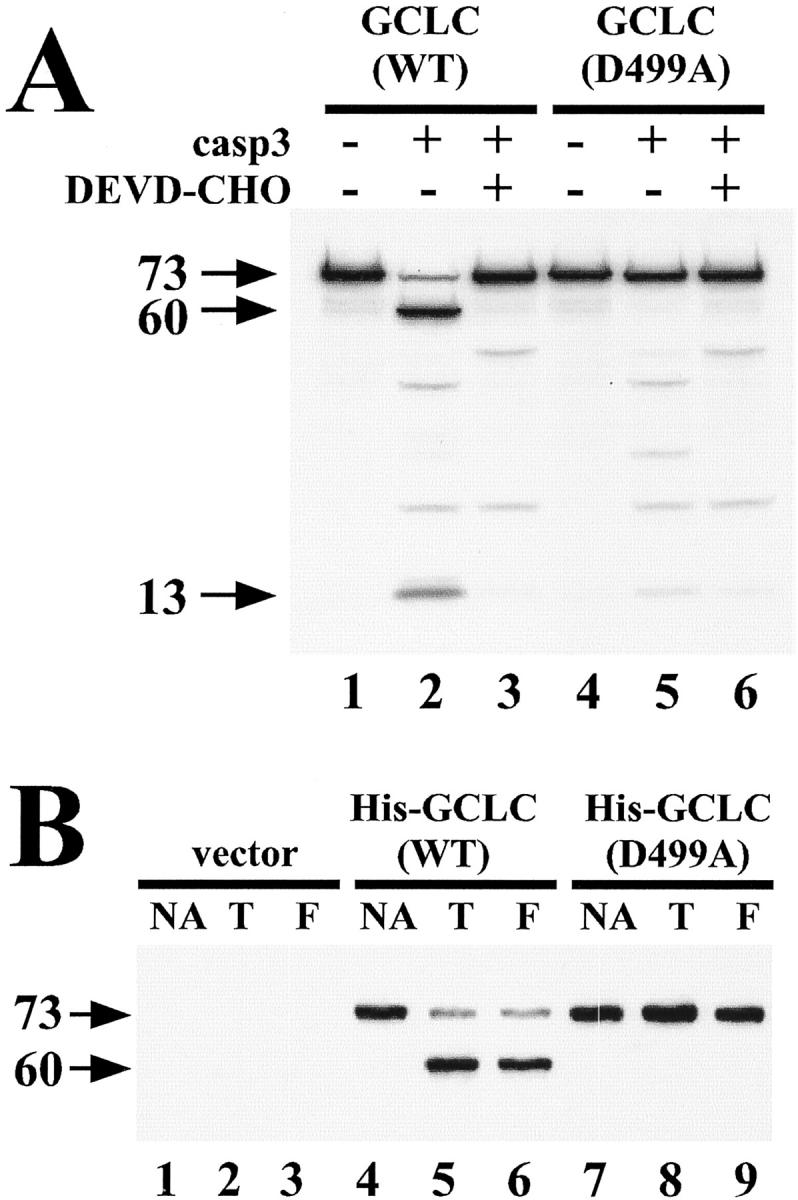
GCLC is a direct target for caspase-mediated cleavage at AVVD499G. A: 35S-labeled in vitro translated wild-type GCLC(WT) or mutant GCLC(D499A) proteins were incubated in the absence or presence of soluble extract from bacteria expressing active recombinant caspase-3 (casp3) and Ac-DEVD-CHO (10 μmol/L) for 1 hour at 30°C as indicated. Radiolabeled proteins were resolved by SDS-PAGE and visualized by fluorography. Arrows denote full-length 73-kd GCLC protein and the 60-kd and 13-kd cleavage fragments. B: HeLa cells were transiently transfected with pcDNA3.1-His vector, pcDNA3.1-His-GCLC(WT) or pcDNA3.1-His-GCLC(D499A) and 24 hours post-transfection were treated for 6 hours with either TNF (30 ng/ml) (T) or αFas antibody (100 ng/ml) (F) in the presence of cycloheximide (10 μg/ml). His-tagged GCLC proteins were recovered by metal affinity chromatography and analyzed by immunoblotting with anti-GCLC antisera. Data presented are from a representative experiment performed at least three times with similar results.
Caspase-3 Is Required for GCLC Cleavage during TNF-Induced Apoptosis
While caspase-3 was capable of directly mediating GCLC cleavage in vitro, it was not clear whether caspase-3 activation was necessary or sufficient for mediating GCLC cleavage during apoptosis. To further explore the role of caspase-3 in mediating GCLC cleavage, GCLC cleavage was analyzed during TNF-induced apoptosis in both the caspase-3-deficient MCF7 breast carcinoma cell line and an MCF7 cell line stably transfected with pro-caspase-3 (MCF7/casp3). 34,37 TNF/CHX treatment of MCF7/casp3 cells led to a time-dependent processing of pro-caspase-3 (Figure 5A ▶ , right bottom), which correlated with the appearance of a caspase-3-like DEVDase activity (Figure 5A ▶ , right). In contrast, vector transfected MCF7 cells (MCF7/neo) expressed no pro-caspase-3 (Figure 5A ▶ , left bottom) and exhibited no DEVDase activity in response to TNF/CHX treatment (Figure 5A ▶ , left). Importantly, cleavage of GCLC to a 60-kd fragment occurred only during TNF-induced apoptosis in the MCF7/casp3 cell line, while no cleavage could be detected in the MCF7/neo cell line, even though cell death was evident (Figure 5A ▶ , top). Again, no cleavage or alteration in GCLM expression was observed in either cell line (Figure 5A ▶ , middle). These findings are consistent with a model in which caspase-3, or a caspase downstream of caspase-3, mediates GCLC cleavage during apoptotic cell death.
Figure 5.
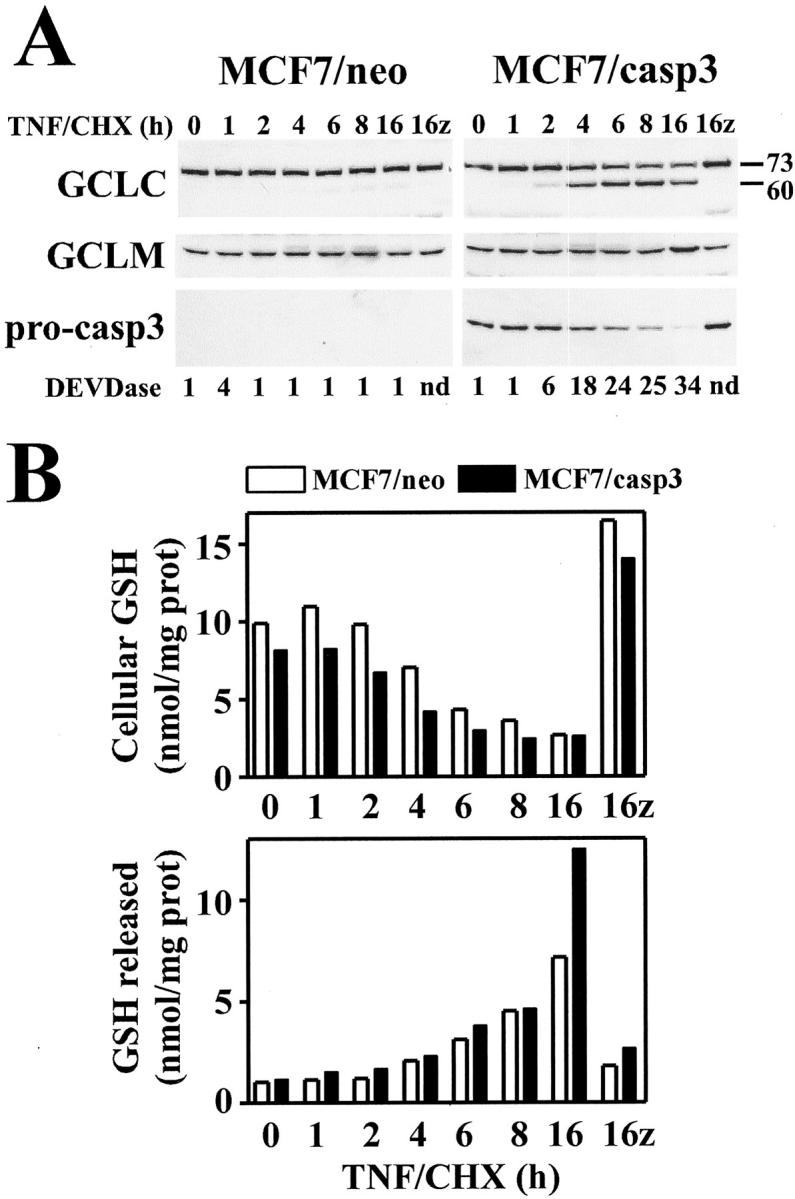
Caspase-3 is required for GCLC cleavage, but not GSH depletion, during TNF-induced apoptosis. A: MCF7 cells stably expressing either pcDNA3.0 vector (MCF7/neo) or pcDNA3.0-caspase-3 (MCF7/casp3) were treated with TNF (30 ng/ml) and cycloheximide (CHX, 10 μg/ml) for the time periods indicated. Some cultures were pretreated for 2 hours with z-VAD-fmk (50 μmol/L) before treatment with TNF/CHX for 16 hours (16+z) as indicated. Cell extracts were analyzed for GCLC, GCLM, and pro-caspase-3 by immunoblotting. Caspase-3-like DEVDase activity was determined as described for Figure 1 ▶ . B: MCF7/neo (open bars) or MCF7/casp3 (closed bars) cells were treated exactly as described above. Cells were harvested and both the cell pellet (top) and media (bottom) were analyzed for GSH content. Data presented are from a representative experiment performed at least three times with similar results.
Distinct Caspase-Dependent Mechanisms Mediate GCLC Cleavage and GSH Efflux during TNF-Induced Apoptosis in MCF7 Cells
While the temporal correlation between GCLC cleavage and GSH depletion suggested a causal relationship between these cellular responses during receptor-mediated apoptosis, several reports indicate that GSH depletion occurs by the extrusion of reduced GSH from the cell during apoptosis. 6,10,12 To determine whether efflux of GSH might account for the rapid depletion of GSH during TNF-induced apoptosis in MCF7 cells, intracellular GSH and GSH released into the culture media was measured in cells treated with TNF/CHX. There was a time-dependent recovery of GSH in the media of both MCF7/neo and MCF7/casp3 cells treated with TNF/CHX that correlated with the loss of intracellular GSH during apoptosis (Figure 5B) ▶ . Furthermore, pretreatment with the pan-caspase inhibitor z-VAD-fmk (50 μmol/L) blocked the TNF-induced depletion of intracellular GSH as well as its appearance in the media in both MCF7 cell lines irrespective of caspase-3 expression and activation (Figure 5B) ▶ . Similar results were obtained at z-VAD-fmk concentrations as low at 1 to 5 μmol/L, yet no inhibitory effect was observed using the caspase-1-specific inhibitor z-YVAD-fmk even at concentrations up to 50 μmol/L (data not shown). Thus, in contrast to GCLC cleavage, which requires caspase-3, GSH extrusion occurs by a caspase-dependent mechanism that does not involve the activation of caspase-3.
Caspase-3-Dependent GCLC Cleavage Correlates with Enhanced TNF-Induced GSH Depletion
The results described in Figure 5 ▶ indicate that TNF-induced depletion of GSH can occur independent of GCLC cleavage. Thus, it is unclear whether GCLC cleavage results in the functional inactivation of GCL and a reduction in GSH biosynthesis during apoptosis. However, the high doses of TNF used in these studies may result in unregulated GSH extrusion that masks a more subtle effect of GCLC cleavage on de novo GSH biosynthesis and replenishment of GSH stores. To determine whether GCLC cleavage altered the kinetics of GSH depletion we examined the efficacy of TNF-induced GSH depletion in the MCF7/neo and MCF7/casp3 cell lines. TNF induced a dose-dependent depletion of cellular GSH in both cell lines (Figure 6A) ▶ . However, there was an approximate threefold difference in the efficacy of TNF-induced GSH depletion in MCF7/casp3 cells (EC50 = 0.54 ± 0.15 ng/ml) when compared to MCF7/neo cells (EC50 = 1.54 ± 0.42 ng/ml). MCF7/casp3 cells were also ∼ sixfold more sensitive to TNF-induced cell death than MCF7/neo cells (EC50 = 1.1 ± 0.1 vs. 6.5 ± 0.1 ng/ml, respectively). Thus, GSH depletion and cell death were enhanced under conditions that resulted in GCLC cleavage. Furthermore, we were able to demonstrate a time-dependent difference in GSH depletion between these two cell lines using a submaximal dose of TNF (Figure 6B) ▶ . Treatment of both cell lines with 1 ng/ml TNF resulted in the time-dependent depletion of GSH (Figure 6B) ▶ . However, GSH levels were reduced to 50% of control within 4 to 6 hours and approached 75% by 10 hours of TNF treatment in MCF7/casp3 cells. In contrast, GSH levels were never reduced by more than 50% of control in MCF7/neo cells and this level was only observed after 10 hours of TNF treatment.
Figure 6.

Caspase-3 activation enhances TNF-induced GSH depletion. A: MCF7/neo (open symbols) or MCF7/casp3 (closed symbols) cells were treated for 6 hours with the indicated concentration of TNF in the presence of 10 μg/ml cycloheximide (A) or for the time period indicated with 1 ng/ml TNF in the presence of cycloheximide (B). Cells were harvested and cellular GSH content analyzed as previously described. The data presented are from a representative experiment repeated either five times (A) or two times (B).
Discussion
Pharmacological manipulation of GSH homeostasis is known to modulate cellular sensitivity to apoptotic cell death. 4,38 For example, supplementation with GSH precursors or overexpression of the GCL subunits promotes cellular resistance to receptor- and chemical-mediated apoptosis, 7,12-18,26-28 and depletion of intracellular GSH often results in increased sensitivity to various apoptotic stimuli. 4 While such findings highlight the crucial role of GSH homeostasis in directing either cell death or survival, it is unknown whether GSH biosynthesis per se is dysregulated during apoptosis. In this study, we demonstrate that the catalytic subunit of the rate-limiting enzyme in GSH biosynthesis, GCLC, undergoes caspase-mediated cleavage during many forms of apoptotic cell death in multiple cell types. Furthermore, we establish that GCLC is a direct target for caspase-mediated cleavage in vitro and demonstrate that GCLC cleavage is dependent on caspase-3 activation during apoptotic cell death.
GCLC cleavage is accompanied by a rapid depletion of intracellular GSH, which occurs in multiple models of apoptosis and may play a role in potentiating apoptotic cell death. 5-11 The detection of GSH in the media of apoptotic MCF7 cells suggests that GSH extrusion is responsible for the rapid loss of intracellular GSH during TNF-induced apoptosis, which is consistent with previous reports in other models of apoptosis. 6,10,12 GSH efflux is inhibited by the pan-caspase inhibitor z-VAD-fmk (Figure 5B ▶ and 10 ) as well as bromosulfophthalein (data not shown and 10 ), an inhibitor of GSH transport, 39 indicating the involvement of a caspase-activated transporter-mediated efflux of GSH and not leakage due to cell lysis. A particularly salient feature of these studies is our finding that GCLC cleavage requires caspase-3 activation, whereas GSH efflux occurs by a caspase-dependent mechanism that does not involve caspase-3. Thus, distinct caspase-dependent mechanisms mediate GSH extrusion and GCLC cleavage during apoptotic cell death. We exploited this differential regulation of GCLC cleavage and GSH efflux in MCF7 cells to demonstrate that GCLC cleavage correlates with an enhanced sensitivity to TNF-induced GSH depletion. As GSH efflux occurs independent of caspase-3 activation (Figure 5B) ▶ , we postulate that the more rapid and complete depletion of GSH during TNF-induced apoptosis in MCF7/casp3 cells is the result of reduced repletion of intracellular GSH pools due to decreased GSH biosynthesis. However, we cannot rule out the possibility that altered utilization of GSH may also contribute to this response.
While our studies in MCF7 cells provide indirect evidence that GCLC cleavage inhibits GSH biosynthesis, previous studies suggest that cleavage of GCLC does not abolish GCL enzymatic activity per se. 29 Thus, cleavage of GCLC may exert subtle effects on GCL activity not detected under the assay conditions used in that study. Specifically, GCL activity in our previous studies was determined using saturating substrate conditions. 29 While all of the enzymatic activity of GCL resides in GCLC, GCLC must dimerize with GCLM to be functionally active under physiological concentrations of glutamate and GSH. 4 Interestingly, the GCLC cleavage site (Asp499) was mapped N-terminal to a cysteine residue (Cys553) reported to be important for the functional interaction between GCLC and GCLM. 40 Thus, cleavage of GCLC at Asp499 could potentially abolish GCL holoenzyme formation by removal of this GCLM interaction site. As the kinetics of GCLC enzymatic activity are highly dependent on heterodimer formation with GCLM, 4 this would essentially result in the inactivation of GCL under physiological conditions. Alternatively, it is possible that GCLC cleavage induces a conformational change in either GCLC or GCL that alters GCL kinetics without influencing the physical interaction between GCLC and GCLM. GCLC cleavage may also complement additional GCLC post-translational modifications that occur during apoptotic cell death. In this regard, GCL activity is modulated by intracellular oxidation 41 and would likely be stimulated in response to increased ROS production following apoptosis-associated mitochondrial dysfunction. GCLC activity is also under the negative feedback regulation of GSH. 4 While physiological concentrations of GSH favor feedback inhibition of GCL, oxidizing conditions associated with low GSH promote intermolecular disulfide bond formation resulting in a conformational change in GCL increasing the affinity for glutamate. 42 As GSH levels are rapidly depleted during apoptotic cell death, GCL activity should increase due to loss of this negative regulatory effect. Caspase-mediated cleavage of GCLC may prevent the conformational change that mediates these stimulatory effects on GCL activity inhibiting the resynthesis of GSH that would normally occur under conditions of GSH depletion. Additional enzymatic analyses are clearly warranted to determine whether GCLC cleavage alters the kinetic parameters of the GCL holoenzyme under conditions that faithfully reproduce those observed during apoptosis in vivo.
In summary, our identification of GCLC as a target for caspase-mediated cleavage during apoptosis reveals a novel post-translational modification that may play a role in regulating GSH biosynthesis and GSH homeostasis during apoptotic cell death. Interestingly, the NRF2 transcription factor, which plays an important role in the inducible expression of GCLC, 43-45 has also been identified as a caspase target. 46 Thus, at least three distinct caspase-mediated mechanisms potentially contribute to the regulation of intracellular GSH status during apoptotic cell death; GSH extrusion, proteolytic processing of GCLC, and inhibition of GCLC transcription. While the exact role of GSH in regulating apoptotic cell death is unclear, these multiple levels of regulation highlight the importance of GCL and GSH in controlling key regulatory events during cell death.
Acknowledgments
We thank Drs. Vince Kidd, Dennis Templeton, Jonathan Graves, and Peter Rabinovitch for their generous gifts of cell lines and reagents, and members of the Fausto lab for their many helpful comments.
Footnotes
Address reprint requests to Christopher C. Franklin, University of Washington, Department of Pathology, Box 357705, Seattle, WA 98195-7470. E-mail: cfrankli@u.washington.edu.
Supported by National Institutes of Health grants CA75316 (to CCF), ES07032 (to CMK), CA23226 and CA74131 (to NF), and ES04696, AG01751, and ES07033 (to TJK).
Christopher C. Franklin and Cecile M. Krejsa contributed equally to this work.
Current address of Cecile M. Krejsa is Cerep Inc., Redmond, WA.
References
- 1.Thompson CB: Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267:1456-1462 [DOI] [PubMed] [Google Scholar]
- 2.Earnshaw WC, Martins LM, Kaufmann SH: Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem 1999, 68:383-424 [DOI] [PubMed] [Google Scholar]
- 3.Meister A, Anderson ME: Glutathione. Annu Rev Biochem 1983, 52:711-760 [DOI] [PubMed] [Google Scholar]
- 4.Griffith OW, Mulcahy RT: The enzymes of glutathione synthesis: γ-glutamylcysteine synthetase. Adv Enzymol Relat Areas Mol Biol 1999, 73:209-267 [DOI] [PubMed] [Google Scholar]
- 5.Backway KL, McCulloch EA, Chow S, Hedley DW: Relationships between the mitochondrial permeability transition and oxidative stress during ara-C toxicity. Cancer Res 1997, 57:2446-2451 [PubMed] [Google Scholar]
- 6.Ghibelli L, Coppola S, Rotilio G, Lafavia E, Maresca V, Ciriolo MR: Non-oxidative loss of glutathione in apoptosis via GSH extrusion. Biochem Biophys Res Commun 1995, 216:313-320 [DOI] [PubMed] [Google Scholar]
- 7.Fernandez A, Kiefer J, Fosdick L, McConkey DJ: Oxygen radical production and thiol depletion are required for Ca(2+)-mediated endogenous endonuclease activation in apoptotic thymocytes. J Immunol 1995, 155:5133-5139 [PubMed] [Google Scholar]
- 8.Macho A, Hirsch T, Marzo I, Marchetti P, Dallaporta B, Susin SA, Zamzami N, Kroemer G: Glutathione depletion is an early and calcium elevation is a late event of thymocyte apoptosis. J Immunol 1997, 158:4612-4619 [PubMed] [Google Scholar]
- 9.Slater AF, Stefan C, Nobel I, van den Dobbelsteen DJ, Orrenius S: Signalling mechanisms and oxidative stress in apoptosis. Toxicol Lett 1995, 82–83:149-153 [DOI] [PubMed] [Google Scholar]
- 10.van den Dobbelsteen DJ, Nobel CSI, Schlegel J, Cotgreave IA, Orrenius S, Slater AFG: Rapid and specific efflux of reduced glutathione during apoptosis induced by anti-Fas/APO-1 antibody. J Biol Chem 1996, 271:15420-15427 [DOI] [PubMed] [Google Scholar]
- 11.Pierce RH, Campbell JS, Stephenson AB, Franklin CC, Chaisson M, Poot M, Kavanagh TJ, Rabinovitch PS, Fausto N: Disruption of redox homeostasis in tumor necrosis factor-induced apoptosis in a murine hepatocyte cell line. Am J Pathol 2000, 157:221-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghibelli L, Fanelli C, Rotilio G, Lafavia E, Coppola S, Colussi C, Civitareale P, Ciriolo MR: Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J 1998, 12:479-486 [DOI] [PubMed] [Google Scholar]
- 13.Chiba T, Takahashi S, Sato N, Ishii S, Kikuchi K: Fas-mediated apoptosis is modulated by intracellular glutathione in human T cells. Eur J Immunol 1996, 26:1164-1169 [DOI] [PubMed] [Google Scholar]
- 14.Deas O, Dumont C, Mollereau B, Metivier D, Pasquier C, Bernard-Pomier G, Hirsch F, Charpentier B, Senik A: Thiol-mediated inhibition of FAS and CD2 apoptotic signaling in activated human peripheral T cells. Int Immunol 1997, 9:117-125 [DOI] [PubMed] [Google Scholar]
- 15.Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA: Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-alpha-induced cell death. J Biol Chem 1998, 273:11313-11320 [DOI] [PubMed] [Google Scholar]
- 16.Um HD, Orenstein JM, Wahl SM: Fas mediates apoptosis in human monocytes by a reactive oxygen intermediate dependent pathway. J Immunol 1996, 156:3469-3477 [PubMed] [Google Scholar]
- 17.Zhao Z, Francis CE, Welch G, Loscalzo J, Ravid K: Reduced glutathione prevents nitric oxide-induced apoptosis in vascular smooth muscle cells. Biochim Biophys Acta 1997, 1359:143-152 [DOI] [PubMed] [Google Scholar]
- 18.Singh I, Pahan K, Khan M, Singh AK: Cytokine-mediated induction of ceramide production is redox-sensitive: implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem 1998, 273:20354-20362 [DOI] [PubMed] [Google Scholar]
- 19.Schroder CP, Godwin AK, O’Dwyer PJ, Tew KD, Hamilton TC, Ozols RF: Glutathione and drug resistance. Cancer Invest 1996, 14:158-168 [DOI] [PubMed] [Google Scholar]
- 20.Komiya S, Gebhardt MC, Mangham DC, Inoue A: Role of glutathione in cisplatin resistance in osteosarcoma cell lines. J Orthop Res 1998, 16:15-22 [DOI] [PubMed] [Google Scholar]
- 21.Godwin AK, Meister A, O’Dwyer PJ, Huang CS, Hamilton TC, Anderson ME: High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA 1992, 89:3070-3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kroemer G, Dallaporta B, Resche-Rigon M: The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 1998, 60:619-642 [DOI] [PubMed] [Google Scholar]
- 23.Wright SC, Wang H, Wei QS, Kinder DH, Larrick JW: Bcl-2-mediated resistance to apoptosis is associated with glutathione-induced inhibition of AP24 activation of nuclear DNA fragmentation. Cancer Res 1998, 58:5570-5576 [PubMed] [Google Scholar]
- 24.Liu B, Hannun YA: Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione. J Biol Chem 1997, 272:16281-16287 [DOI] [PubMed] [Google Scholar]
- 25.Huang CS, Chang LS, Anderson ME, Meister A: Catalytic and regulatory properties of the heavy subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem 1993, 268:19675-19680 [PubMed] [Google Scholar]
- 26.Mulcahy RT, Bailey HH, Gipp JJ: Transfection of complementary DNAs for the heavy and light subunits of human γ-glutamylcysteine synthetase results in an elevation of intracellular glutathione and resistance to melphalan. Cancer Res 1996, 1995:55:4771-4775 [PubMed] [Google Scholar]
- 27.Manna SK, Kuo MT, Aggarwal BB: Overexpression of γ-glutamylcysteine synthetase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappa B and activator protein-1. Oncogene 1999, 18:4371-4382 [DOI] [PubMed] [Google Scholar]
- 28.Tipnis SR, Blake DG, Shepherd AG, McLellan LI: Overexpression of the regulatory subunit of γ-glutamylcysteine synthetase in HeLa cells increases γ-glutamylcysteine synthetase activity and confers drug resistance. Biochem J 1999, 337:559-566 [PMC free article] [PubMed] [Google Scholar]
- 29.Siitonen T, Alaruikka P, Mantymaa P, Savolainen ER, Kavanagh TJ, Krejsa CM, Franklin CC, Kinnula V, Koistinen P: Protection of acute myeloblastic leukemia cells against apoptotic cell death by high glutathione and γ-glutamylcysteine synthetase levels during etoposide-induced oxidative stress. Ann Oncol 1999, 10:1361-1367 [DOI] [PubMed] [Google Scholar]
- 30.Reid LL, Botta D, Lu Y, Gallagher EP, Kavanagh TJ: Molecular cloning and sequencing of the cDNA encoding the catalytic subunit of mouse glutamate-cysteine ligase. Biochim Biophys Acta 1997, 1352:233-237 [DOI] [PubMed] [Google Scholar]
- 31.Reid LL, Botta D, Shao J, Hudson FN, Kavanagh TJ: Molecular cloning and sequencing of the cDNA encoding mouse glutamate-cysteine ligase regulatory subunit. Biochim Biophys Acta 1997, 1353:107-110 [DOI] [PubMed] [Google Scholar]
- 32.Deak JC, Cross JV, Lewis M, Qian Y, Parrott LA, Distelhorst CW, Templeton DJ: Fas-induced proteolytic activation and intracellular redistribution of the stress-signaling kinase MEKK1. Proc Natl Acad Sci USA 1998, 95:5595-5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson SA, White CC, Krejsa CM, Diaz D, Woods JS, Eaton DL, Kavanagh TJ: Induction of glutamate-cysteine ligase (γ-glutamylcysteine synthetase) in the brains of adult female mice subchronically exposed to methylmercury. Toxicol Lett 1999, 110:1-9 [DOI] [PubMed] [Google Scholar]
- 34.Tang D, Kidd VJ: Cleavage of DFF-45/ICAD by multiple caspases is essential for its function during apoptosis. J Biol Chem 1998, 273:28549-28552 [DOI] [PubMed] [Google Scholar]
- 35.Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, Chernoff J, Clark EA, Krebs EG: Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J 1998, 17:2224-2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker MA, Cerniglia GJ, Zaman A: Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem 1990, 190:360-365 [DOI] [PubMed] [Google Scholar]
- 37.Janicke RU, Sprengart ML, Wati MR, Porter AG: Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 1998, 273:9357-9360 [DOI] [PubMed] [Google Scholar]
- 38.Griffith OW: Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med 1999, 27:922-935 [DOI] [PubMed] [Google Scholar]
- 39.Ballatori N, Dutczak WJ: Identification and characterization of high and low affinity transport systems for reduced glutathione in liver cell canalicular membranes. J Biol Chem 1994, 269:19731-19737 [PubMed] [Google Scholar]
- 40.Tu Z, Anders MW: Identification of an important cysteine residue in human glutamate-cysteine ligase catalytic subunit by site-directed mutagenesis. Biochem J 1998, 336:675-680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ochi T: Hydrogen peroxide increases the activity of γ-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Arch Toxicol 1995, 70:96-103 [DOI] [PubMed] [Google Scholar]
- 42.Soltaninassab SR, Sekhar KR, Meredith MJ, Freeman ML: Multi-faceted regulation of γ-glutamylcysteine synthetase. J Cell Physiol 2000, 182:163-170 [DOI] [PubMed] [Google Scholar]
- 43.Jeyapaul J, Jaiswal AK: Nrf2 and c-Jun regulation of antioxidant response element (ARE)- mediated expression and induction of γ-glutamylcysteine synthetase heavy subunit gene. Biochem Pharmacol 2000, 59:1433-1439 [DOI] [PubMed] [Google Scholar]
- 44.Moinova HR, Mulcahy RT: Up-regulation of the human γ-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem Biophys Res Commun 1999, 261:661-668 [DOI] [PubMed] [Google Scholar]
- 45.Wild AC, Mulcahy RT: Regulation of γ-glutamylcysteine synthetase subunit gene expression: insights into transcriptional control of antioxidant defenses. Free Radic Res 2000, 32:281-301 [DOI] [PubMed] [Google Scholar]
- 46.Ohtsubo T, Kamada S, Mikami T, Murakami H, Tsujimoto Y: Identification of NRF2, a member of the NF-E2 family of transcription factors, as a substrate for caspase-3(-like) proteases. Cell Death Differ 1999, 6:865-872 [DOI] [PubMed] [Google Scholar]