

Abstract
Pancreatic intraductal neoplasia (PanIN) is thought to be the precursor to infiltrating pancreatic ductal adenocarcinoma. We have previously shown that the preproenkephalin (ppENK) and p16 genes are aberrantly methylated in pancreatic adenocarcinoma. In this study we define the methylation status of the ppENK and p16 genes in various grades of PanINs. One hundred seventy-four samples (28 nonneoplastic pancreatic epithelia, 7 reactive epithelia, 29 PanIN-1A, 48 PanIN-1B, 27 PanIN-2, 14 PanIN-3, 15 invasive ductal adenocarcinomas, and 6 miscellaneous pancreatic neoplasms) were microdissected from 29 formalin-fixed paraffin-embedded surgically resected pancreata, and were analyzed by methylation-specific polymerase chain reaction. Fourteen of 15 (93.3%) invasive pancreatic ductal adenocarcinomas showed methylation of the ppENK gene and 4 of 15 (26.7%) showed methylation of the p16 gene. Nonneoplastic pancreatic epithelia did not harbor methylation of either gene. The prevalence of methylation of the ppENK gene increased significantly with increasing PanIN grade. A similar nonsignificant trend was noted for p16 methylation. Aberrant methylation of the ppENK gene was found in 7.7% of PanIN-1A, 7.3% of PanIN-1B, 22.7% of PanIN-2, and 46.2% of PanIN-3. Aberrant methylation of the p16 gene was found in 12% of PanIN-1A, 2.6% of PanIN-1B, 4.5% of PanIN-2, and 21.4% of PanIN-3. All but one of the PanINs from the 14 pancreata without pancreatic carcinoma was unmethylated with respect to either the p16 or ppENK gene. Our results suggest that methylation-related inactivation of the ppENK and p16 genes is an intermediate or late event during pancreatic carcinogenesis. Because aberrant methylation of ppENK or p16 was more often detected in similar grade PanINs from patients with pancreatic carcinoma than in those with other pancreatic diseases, it may be a useful indicator of the potential malignancy of epithelial cells of the pancreas.
Intraductal lesions are commonly found in the pancreatic ducts adjacent to invasive ductal adenocarcinoma, and have been regarded as precursor lesions of pancreatic ductal adenocarcinoma. 1-3 Recently, pancreatic ductal lesions, previously described as hyperplasias or dysplasias, have been reclassified into four groups of pancreatic intraepithelial neoplasias (PanINs): PanIN-1A, -1B, -2, and -3. 2,3 Molecular analysis of PanIN lesions could help refine the PanIN classification and provide insights into molecular events important to the pathogenesis of early pancreatic ductal adenocarcinoma.
The K-ras oncogene is frequently mutated in pancreatic carcinomas as are a number of tumor suppressor genes such as p53, p16, DPC4/SMAD4, 4-8 and less often BRCA2, TGFβR1, TGFβR2, BRCA2, ALK4, STK11, and MKK4. 9-12 Some PanINs harbor the same genetic changes seen in invasive ductal adenocarcinomas, albeit at lower frequency. 4-9 Mutations of the K-ras and p16 genes are occasionally found in low-grade PanINs but are observed more frequently in high-grade PanINs. 4-6 SMAD4 inactivation is observed at the PanIN-3 stage. 7 Similarly, p53 gene mutations and inactivation of the BRCA2 gene seem to occur only in high-grade PanINs. 8,9 These studies support the multiple step model of pancreatic carcinogenesis. 13
Aberrant methylation of CpG islands of several tumor-suppressor genes such as p16 is associated with the transcriptional silencing in pancreatic 14 and other carcinomas. 15-18 The p16 gene is reported to be inactivated in 15% of pancreatic carcinomas by hypermethylation of the CpG island. 3 Loss of p16 protein expression occurs more frequently with increasing PanIN grade, 6 but the role of aberrant methylation of the p16 gene in PanIN progression is unknown. We recently demonstrated, using methylated CpG island amplification (MCA) coupled with representational difference analysis (RDA), that the ppENK gene is also aberrantly methylated in pancreatic carcinomas (>90%). 19
The ppENK gene encodes met-enkephalin, which is a tonically active inhibitory factor that interacts with the opioid growth factor receptor. Zagon and colleagues 20 reported that met-enkephalin inhibited the growth of several human tumors including pancreatic cancer. Comb and colleagues 21 reported that methylation of the CpG island of ppENK inhibited its expression by directly interfering with the binding of a positively acting transcription factor. Therefore, methylation of the ppENK gene during pancreatic carcinogenesis may promote cell growth.
To understand the role of methylation of the ppENK and p16 genes in early pancreatic carcinogenesis, we analyzed the CpG islands of these genes in various grades of PanINs using the methylation-specific polymerase chain reaction (MSP).
Materials and Methods
Twenty-nine pancreata surgically resected at the Johns Hopkins Hospital were selected for the presence of PanIN lesions. Fifteen resections were performed for pancreatic ductal adenocarcinoma and six for nonductal adenocarcinoma, including two ampullary adenocarcinomas, one ampullary adenoma, one common bile duct carcinoma, one mucinous cystadenoma, and one endocrine tumor. Eight resections were performed for chronic pancreatitis. The mean age of the patients with pancreatic ductal adenocarcinoma was 62.8 years (nine men and six women), with peri-ampullary adenocarcinoma the mean age was 67.2 years (four men and two women), and with chronic pancreatitis the mean age was 60.3 years (four men and four women). The 15 ductal adenocarcinomas included 4 poorly, 8 moderately-to-poorly, and 3 moderately differentiated adenocarcinomas. The mean tumor size of the ductal adenocarcinoma was 3.2 cm (range, 1.5 to 10.0 cm).
PanINs were classified into PanIN-1A, PanIN-1B, PanIN-2, and PanIN-3 by two authors (NF and RHH) as has been previously described. 2 Briefly, the criteria for classifying these lesions can be summarized as follows: PanIN-1A are flat epithelial lesions and PanIN-1B are papillary or micropapillary lesions composed of tall columnar cells with basally located nuclei with minimal atypia (Figure 1, B and C) ▶ . PanIN-2 show mild-to-moderate architectural and cytological atypia (Figure 1D) ▶ . PanIN-3 usually show significant architectural and cytological atypia such as cribriforming, nuclear pleomorphism, and prominent nucleoli (Figure 1E) ▶ . Cancerization of ducts is recognized as the extension of infiltrating carcinoma into pancreatic ducts and ductules (Figure 1F) ▶ . Reactive changes may mimic PanIN. The presence of significant inflammatory cell infiltrates, particularly when there are numerous polymorphonuclear leukocytes and when these involve the epithelium, should raise the possibility of reactive changes. In addition, ducts with reactive changes typically lack the architectural changes that can be seen in PanINs. Finally, despite the presence of nuclear enlargement and nucleolar prominence, the nuclei in reactive epithelium often have smooth contours and finely dispersed chromatin (Figure 1G) ▶ .
Figure 1.
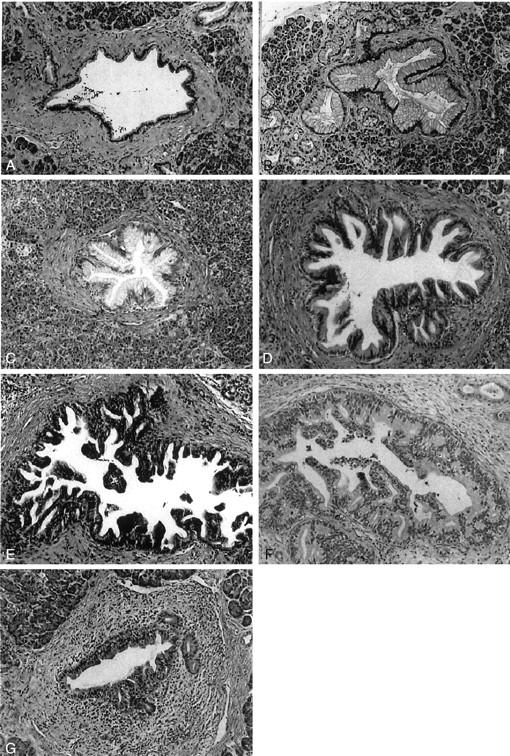
Pancreatic intraepithelial lesions. A: Normal pancreatic duct. The epithelium is composed of cuboidal to low columnar cells. B: PanIN-1A. The epithelial lesions are composed of tall columnar cells with abundant mucin in cytoplasm. The nuclei are small and located in the basal portion of the cells. C: PanIN-1B. It shows a papillary structure. The epithelial cells are similar to those in PanIN-1A. D: PanIN-2. The mucinous epithelial lesion shows mild to moderate irregularity of the papillary structure. Mild nuclear pseudostratification and enlargement (elongation) are seen. E: PanIN-3. Irregularity of the papillary structure is obvious. Budding-off of small clusters of epithelial cells into the lumen is seen. The epithelial cells show increased nuclear-to-cytoplasmic ratio, nuclear enlargement, and irregularity. F: Cancerization of ducts. This lesion is similar to PanIN-3. In surrounding stromal tissue, however, there is an infiltrating adenocarcinoma composed of cells similar to those in the intraductal lesion. G: Reactive change. There is inflammatory cell infiltration around the ductal lesion. Although mild to moderate nuclear enlargement is seen, other nuclear atypia such as hyperchromasia and irregularity of shape are not identified. H&E stain; original magnifications, ×100.
One hundred seventy-four samples were selected and microdissected from formalin-fixed and paraffin-embedded tissue blocks prepared from the 29 pancreata. These included 28 nonneoplastic pancreatic epithelia, 7 reactive epithelia, 118 PanINs, 15 invasive ductal adenocarcinomas, and 6 miscellaneous pancreatic neoplasms. Eight-μm serial sections of formalin-fixed and paraffin-embedded tissue were deparaffinized by incubating the slides in xylene for longer than 1 hour and dehydrating in ethanol; they were then stained with hematoxylin and eosin. Microdissection was performed using a small blade and needle. Pancreatic tissue surrounding the lesion of interest was first removed by blade and needle (Figure 2) ▶ ; then after placing drops of 1× TK buffer (200 μg/ml of proteinase K and 0.5% Tween 20) over the microdissected lesion the epithelial cells were collected by scratching the slide and aspirating the tissue. Whenever possible larger diameter PanINs were chosen for microdissection; these were usually 0.5-mm to 3-mm wide and ranged in cellularity from 500 to 10,000 cells (average, ∼1500 cells). The cells were collected from two or three serial sections. It was estimated that <5 to 20% of the cells collected were surrounding nonductal cells. Microdissected tissues were transferred to a tube containing 50 μl of 1× TK buffer, and incubated at 56°C overnight. The tubes were placed in a 100°C block for 10 minutes for inactivation of the proteinase K.
Figure 2.
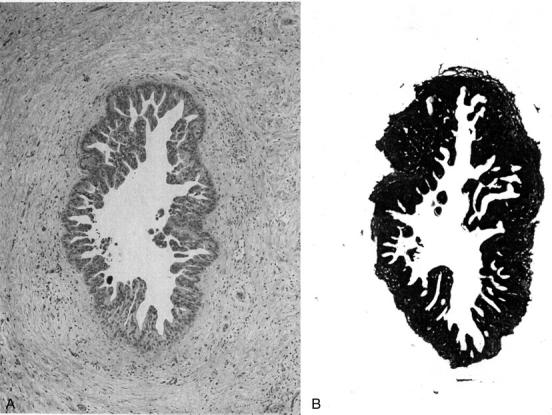
An example of microdissection. A: An intraductal epithelial lesion present in the histological section (H&E stain). B: Surrounding tissue was scratched and removed by the blade. The PanIN is easily collected by scratching the slide and aspirating the tissue after drops of TK buffer have been added.
Bisulfite Modification and MSP
The bisulfite treatment was performed on each lesion dissected (a 50-μl sample containing 500 to 10,000 cells) by incubating the DNA at 50°C for 16 hours as previously described. 14,22 The modified DNA was purified using the Wizard DNA Clean-up System (Promega, Madison, WI). After that, polymerase chain reaction (PCR) was performed without ethanol precipitation to decrease the loss of DNA.
MSP was performed on 1 μl of bisulfite-modified DNA using primers specific for unmethylated ppENK (sense: 5′-TTGTGTGGGGAGTTATTGAGT-3′; antisense: 5′-CACCTTCACAAAAAAAATCAATC-3′) or p16 (sense: 5′-GGGTGGATTGTGTGTGTTTG-3′; antisense: 5′-CCATAACCAACCAATCAACCA-3′) or methylated ppENK (5′-TGTGGGGAGTTATCGAGC-3′ and 5′-GCCTTCGCGAAAAAAATCG-3′) or p16 (sense: 5′-GGCGGATCGCGTGCGTTC-3′; antisense: 5′-CGTAACCAAATCAACCG-3′). PCR conditions were as follows: 95°C for 3 minutes; 45 cycles of 95°C for 20 seconds, the specific annealing temperature (ppENK, 62°C; p16, 65°C) for 30 seconds, and 72°C for 30 seconds; and a final extension of 3 minutes at 72°C.
Six μl of each PCR product were loaded onto 3% agarose gels stained with ethidium bromide, and visualized under UV light. All PCR reactions were performed with positive controls for both unmethylated and methylated alleles and a negative control (no DNA loaded).
To determine an approximate lower limit of detection of the MSP assays for ppENK and p16, two pancreatic adenocarcinomas known to harbor methylation of either p16 or ppENK, were microdissected from paraffin-embedded slides in the same manner as the PanIN dissections, and the number of cells dissected were counted. One sample contained ∼4000 cells and the other had 8000 cells. After doing bisulfite modification, we performed ppENK-MSP analysis using the range of DNA concentrations of the modified DNA. We found that ppENK amplification was possible with an initial 50 μl of DNA sample containing ∼200 to 400 dissected cells. More uniform amplification was obtained when >400 cells were bisulfite modified and PCR amplified.
MSP assays were repeated at least twice to determine the reproducibility of the assay. In the cases showing an unstable result (ie, methylated at the first time and unmethylated at the second), we repeated the microdissection of the corresponding PanIN using different serial sections and repeated MSP using larger amounts of modified DNA (∼6.5 μl of 50-μl modified DNA).
Statistical Analysis
Statistical analysis was performed using the StatView 5.0 statistical software package (SAS Institute Inc., Cary, NC). We used chi-square analysis to compare methylation prevalence in different grades and groups. The age of the patients was compared using a t-test.
Results
A summary of the demographics of the patients included in this study is shown in Table 1 ▶ . PanIN methylation data were divided into three groups according to the patient populations from which the samples were derived: PanINs from patients with pancreatic ductal adenocarcinoma, PanINs from patients with other neoplasms, and PanINs from patients with chronic pancreatitis. DNA was successfully amplified in 158 of the 174 samples (90.8%) using methylation-specific primers for the ppENK gene and in 154 of 174 samples (88.5%) using MSP primers for the p16 gene. Sixteen lesions (3 PanIN-1A, 7 PanIN-1B, 5 PanIN-2, and 1 PanIN-3) did not amplify for ppENK and 20 lesions (4 PanIN-1A, 10 PanIN-1B, 5 PanIN-2, and 1 reactive lesion) did not amplify for P16. Eight lesions were randomly chosen for repeat microdissection using different serial sections and MSP was repeated using larger amounts of modified DNA (>1000 cells), but DNA from these PanINs still did not amplify. For 11 PanINs neither ppENK nor p16 MSP could be amplified.
Table 1.
Case Characteristics
| Case no. | Age | Sex | Pathology | Tumor size, cm | ||||
|---|---|---|---|---|---|---|---|---|
| Patients with pancreatic ductal adenocarcinoma | ||||||||
| 1 | 66 | F | Adenocarcinoma, mod-por | 2 | ||||
| 2 | 50 | F | Adenocarcinoma, mod-por | 2 | ||||
| 3 | 61 | M | Adenocarcinoma, por | 1.5 | ||||
| 4 | 66 | M | Adenocarcinoma, mod-por | 3.5 | ||||
| 5 | 79 | M | Adenocarcinoma, mod-por | 1.5 | ||||
| 6 | 79 | M | Adenocarcinoma, mod-por | 10 | ||||
| 7 | 66 | M | Adenocarcinoma, mod | 3.5 | ||||
| 8 | 51 | F | Adenocarcinoma, mod-por | 3.5 | ||||
| 9 | 57 | M | Adenocarcinoma, mod-por | 3 | ||||
| 10 | 65 | F | Adenocarcinoma, por | 4 | ||||
| 11 | 62 | F | Adenocarcinoma, mod-por | 2.5 | ||||
| 12 | 70 | M | Adenocarcinoma, por | 3.5 | ||||
| 13 | 65 | M | Adenocarcinoma, por | 2 | ||||
| 14 | 58 | M | Adenocarcinoma, mod | 3 | ||||
| 15 | 53 | F | Adenocarcinoma, mod | 2.5 | ||||
| Patients with other neoplasms | ||||||||
| 16 | 82 | M | Adenocarcinoma, mod, common bile duct | 1.2 | ||||
| 17 | 57 | M | Adenocarcinoma, mod-por, papilla Vater | 2.5 | ||||
| 18 | 68 | F | Adenocarcinoma, mod, papilla Vater | 4 | ||||
| 19 | 69 | M | Tubulovillous denoma, papilla Vater | 4 | ||||
| 20 | 61 | F | Mucinous cystadenoma, pancreas | 2.5 | ||||
| 21 | 66 | M | Endocrine tumor, pancreas | 4.5 | ||||
| Patients with chronic pancreatitis | ||||||||
| 22 | 60 | M | Chronic pancreatitis | |||||
| 23 | 50 | M | Chronic pancreatitis | |||||
| 24 | 63 | F | Chronic pancreatitis | |||||
| 25 | 69 | F | Chronic pancreatitis | |||||
| 26 | 69 | F | Chronic pancreatitis | |||||
| 27 | 53 | M | Chronic pancreatitis | |||||
| 28 | 53 | F | Chronic pancreatitis | |||||
| 29 | 65 | M | Chronic pancreatitis | |||||
Mod, moderately differentiated; por, poorly differentiated.
Eight of 102 lesions that amplified for ppENK and 7 of the 99 lesions that amplified for p16 showed unstable results. These were redissected and bisulfite modified. None of the 28 normal pancreatic epithelia showed methylation of p16 or ppENK. All methylated PanIN lesions also contained unmethylated bands, although density of the bands was variable (∼50%). The presence of unmethylated templates in otherwise methylated PanINs probably reflects the inclusion of surrounding nonductal cells such as stromal cells and lymphocytes during the dissection or collection of cells, or partial methylation within each lesion. A summary of the methylation profiles of the PanINs from each patient group is shown in Figure 3 ▶ . An example of MSP analysis is shown in Figure 4 ▶ .
Figure 3.
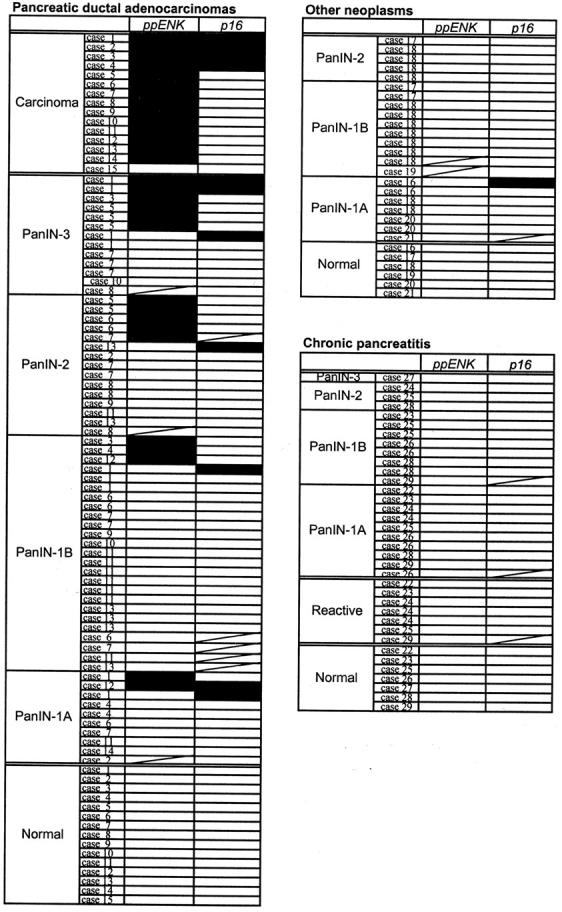
Methylation profiles of the ppENK and p16 genes in PanINs. The PanINs that were unable to be amplified were not included in this figure. Filled boxes, methylated results; open boxes, unmethylated results.
Figure 4.

The results of MSP of the ppENK and p16 genes’ CpG islands. A visible PCR product in lane U indicates the presence of unmethylated gene promoters; the presence of product in lane M indicates the presence of promoter methylation. ppENK (case 6): lanes 1 to 3, cancerization of duct; lane 4, PanIN-1B; lane 5, PanIN-1A; lane 6, carcinoma; lane 7, PanIN-1B; lane 8, normal pancreatic duct. p16 (cases 12 and 14): lane 1, PanIN-1B (case 12); lane 2, PanIN-1A (case 12); lane 3, normal pancreatic duct (case 12); lane 4, carcinoma (case 12); lane 5, carcinoma (case14); lane 6, PanIN-1A (case 14); lane 7, normal pancreatic tissue (frozen tissue); lane 8, Colo357 (cell line).
Methylation of Invasive Pancreatic Ductal Adenocarcinoma
The CpG island of the ppENK gene was frequently methylated (14 of 15, 93.3%) in invasive ductal adenocarcinomas whereas methylation of the p16 gene promoter was found in 4 of 15 cases (26.7%). These results obtained from formalin-fixed paraffin-embedded samples are similar to our previous results obtained from fresh frozen sections of invasive ductal adenocarcinoma. 11 All four of the invasive carcinomas showing p16 CpG island methylation also showed ppENK gene methylation.
Methylation of PanINs
Among the 102 PanINs from which DNA was amplified, the prevalence of ppENK CpG island methylation increased with histological grade of PanIN as follows: PanIN-1A (2 of 26, 7.7%), PanIN-1B (3 of 41, 7.3%), PanIN-2 (5 of 22, 22.7%), and PanIN-3 (6 of 13, 46.2%) (Figures 3 and 5) ▶ ▶ . ppENK methylation was present in high-grade PanINs (PanIN-3) more often than it was in low-grade PanINs (PanIN-1 and PanIN-2) (P = 0.005). Eight of 99 PanINs showed methylation of p16 CpG islands. These included 3 of 25 (12%) PanIN-1A, 1 of 38 (2.6%) PanIN-1B, 1 of 22 (4.5%) PanIN-2, and 3 of 14 (21.4%) of PanIN-3. As was true for ppENK, p16 methylation was seen more frequently in high-grade PanINs than it was in low-grade PanINs, but this difference was not statistically significant (P = 0.146).
Figure 5.

Incidence of ppENK (filled squares) and p16 (open squares) methylation in PanINs from all cases in relation to PanIN grades. The incidence of methylated CpG islands of the ppENK gene in high-grade PanINs (PanIN-3) is significantly higher than in low-grade PanINs (PanINs-1 and -2) (*, P = 0.005), but that of p16 shows no statistical difference (**, P = 0.146).
Methylation of PanINs from Patients with Ductal Adenocarcinoma
Seventy-one PanINs (10 PanIN-1A, 30 PanIN-1B, 18 PanIN-2, and 13 PanIN-3) were analyzed from the pancreata resected for pancreatic ductal adenocarcinoma and DNA was amplified from 60 of 71 PanINs. The prevalence of ppENK CpG island methylation in the 60 PanINs tended to increase with histological grade as follows: PanIN-1A (2 of 9, 22.2%), PanIN-1B (3 of 25, 12.0%), PanIN-2 (5 of 14, 35.7%), and PanIN-3 (6 of 12, 50%) (Figure 3) ▶ . Fifty-eight PanINs microdissected from pancreata with an invasive ductal adenocarcinoma had DNA amplified for p16 and seven of the lesions showed methylation of p16 CpG islands as follows: PanIN-1A (2 of 10, 20%), PanIN-1B (1 of 21, 4.8%), PanIN-2 (1 of 14, 7.1%) and PanIN-3 (3 of 13, 23.1%). In this group of PanINs ppENK methylation was more frequently seen in high-grade PanINs than in low-grade PanINs (P < 0.001). There was no significant difference in the frequency of p16 methylation (P = 0.368).
Methylation of PanINs from Patients with Other Peri-Ampullary Neoplasms
Seventeen PanINs were obtained from two pancreata resected for ampullary adenocarcinoma, two PanINs were from pancreata resected for common bile duct carcinoma, another two PanINs were from a pancreas with a mucinous cystadenoma, one was from a pancreas with an ampullary adenoma and one was from a pancreas with an endocrine tumor of the pancreas (7 PanIN-1A, 10 PanIN-1B, and 6 PanIN-2). None of these PanINs showed CpG island methylation of ppENK, and only one PanIN-1A lesion showed methylation of p16 (from a patient with common bile duct adenocarcinoma).
Methylation of ppENK was found in one of one invasive common bile duct adenocarcinomas, one of two invasive ampullary adenocarcinomas, and one of one islet cell tumor. The invasive common bile duct adenocarcinoma and islet cell tumor also showed p16 CpG island hypermethylation.
Methylation of PanINs from Patients with Chronic Pancreatitis
Twenty-four PanINs were analyzed from the pancreata resected for chronic pancreatitis (12 PanIN-1A, 8 PanIN-1B, 3 PanIN-2, and 1 PanIN-3). None of the intraepithelial lesions including PanINs and reactive epithelia isolated from these pancreata with chronic pancreatitis showed methylation of the CpG island of either ppENK or p16. There was less ppENK methylation in PanINs microdissected from patients with chronic pancreatitis than there was from PanINs microdissected from pancreata with pancreatic adenocarcinoma (P = 0.017); however, no significant difference was seen in that of p16 methylation (P = 0.240). When we limited our analysis to PanIN-1, we found no difference in the prevalence of methylation of ppENK or p16 in patients with chronic pancreatitis compared to pancreatic cancer. The lack of methylation of PanINs in the setting of pancreatitis was not too surprising because they were all of an early PanIN grade, but we considered the possibility that the aberrant methylation of pancreatic cancer-associated PanINs arose from contamination. Of 20 aberrantly methylated PanINs from patients with cancer, 4 PanINs (1 PanIN-1A, 2 PanIN-1B, 1 PanIN-2) with methylation were obtained from slides that did not harbor any cancer on the slide used for microdissection. In another two PanINs that harbored p16 methylation, the patient’s primary cancer did not harbor p16 methylation. This is not surprising because the PanINs and the cancers are distinct neoplasms. In the remaining 14 PanINs that harbored methylation (2 PanIN-1A, 1 PanIN-1B, 5 PanIN-2, and 6 PanIN-3), invasive adenocarcinoma was present on the same sections. To check for the possibility of contamination, we microdissected noncancerous tissue surrounding the invasive carcinoma from the eight sections that contained the 14 PanINs using same method as for the PanIN dissections. All eight samples harbored unmethylated templates by MSP.
Correlation of Methylation of PanINs with Patient Age
The mean age of the seven patients who had PanINs showing ppENK CpG island methylation was 69.6 years, whereas the mean age of the 22 patients without ppENK methylation was 61.2 years (P = 0.019). The mean age of the eight patients with pancreatic ductal adenocarcinoma showing no ppENK hypermethylation in their PanINs was 57.6 years. Similarly, the mean age of 4 patients who had PanINs showing p16 CpG island methylation was also older (70.8 years) than that of the other 25 patients (62.0 years). However, this difference did not show statistical significance (P = 0.053).
Discussion
In this study we demonstrated that the prevalence of methylation of CpG islands of the ppENK genes increases with PanIN grade. A similar trend of increasing methylation with increasing PanIN grade was noted for the p16 gene but did not reach statistical significance. Our data suggest that inactivation of the ppENK and p16 gene through methylation is usually an intermediate or late event during pancreatic neoplastic development. Because we were amplifying archival DNA and the level of detection of our assay was more than one template, we cannot rule out the possibility that the initial methylation of these genes could begin as an early event in the natural history of PanINs. Methylation of a few templates in such an early PanIN might occur if there is partial methylation of the CpG island that is not sufficient for silencing, or if methylation only occurred in the CpG island of one of the two alleles in the PanIN. Methylation of a few templates would probably not be associated with a selective advantage until the methylated gene was completely switched off by methylation, at which stage gene silencing and selection would occur and the presence of aberrant methylation would be more readily identifiable. Our results are consistent with several studies that have recently demonstrated that aberrant methylation of genes silenced by methylation in the cancer can be traced to precursor neoplasms. 15-18,23,24
We also found that the mean age of patients who had PanINs showing ppENK or p16 gene methylation was significantly older (∼8 years) than those patients without such methylation. These results were based on a small number of patients but raise the possibility that either the ppENK and p16 genes undergo methylation as a function of age, or alternatively that PanINs in older patients are more likely to undergo aberrant methylation of these genes. We and other investigators have found aberrant methylation more often in cancers from older patients. 19,25 The differences in the age of patients with methylated versus nonmethylated PanINs could not be accounted for by differences in PanIN grade, but it is possible that PanIN grade is a poor indicator of the age of a PanIN. Issa and colleagues 26 and Nakagawa and colleagues 27 have reported that multiple genes undergo age-related methylation in normal tissues and ulcerative colitis including hMLH1 and E-cadherin. We have not observed CpG island methylation of the ppENK and p16 gene in normal pancreatic tissues, but it is possible that such aberrant methylation quickly results in clonal expansion and is thus rarely seen in normal epithelium.
Chronic pancreatitis is associated with an increased risk of pancreatic adenocarcinoma but the mechanism by which this risk is mediated is unknown. 28,29 Although aberrant methylation of the p16 gene has been observed in Barrett’s lesions of the esophagus, 30 there is no direct evidence that inflammation induces changes in DNA methylation. Chronic inflammation within the setting of ulcerative colitis is associated with chromosomal instability, and an increase in age-related methylation. 26,31,32 We compared the methylation of PanINs from patients with chronic pancreatitis to determine whether chronic pancreatitis evolves into pancreatic cancer through progressive methylation of PanINs. Surprisingly, we found that none of the PanINs isolated from the patients with chronic pancreatitis harbored methylation of either the ppENK or p16 gene. However this may reflect the fact that most of the PanINs in the chronic pancreatitis group were PanIN-1, and the difference in the prevalence of methylation in the PanIN-1 from patients with chronic pancreatitis and pancreatic adenocarcinomas was not statistically significant. Only one other report has analyzed p16 CpG island hypermethylation in PanINs microdissected from cases of chronic pancreatitis. 33 In this study, 2 of 10 PanIN-1A lesions showed hypermethylation of the p16 CpG islands. Further studies of methylation status of the PanINs associated with chronic pancreatitis will help determine whether the relative lack of methylation observed in those lesions is a general phenomenon or particular to the p16 and ppENK genes. Comparison of the methylation status of invasive pancreatic cancers that develop after long-standing chronic pancreatitis to the methylation status of usual invasive pancreatic cancers may also shed light on this question.
Our data also suggest that the presence of aberrant methylation of the ppENK and p16 genes may be a useful indicator in biopsy, fluid, or cytology samples of the presence of high-grade PanINs or pancreatic adenocarcinoma. Given the high rate of methylation of ppENK (93.3%) and to a lesser extent, p16 (26.7%), in pancreatic adenocarcinoma, detection of aberrant methylation of these genes by MSP in the pancreatic juice of patients at high risk of developing pancreatic cancer or suspected of the disease may be worthwhile.
In conclusion, our results demonstrate that methylation of the ppENK and p16 genes increases with PanIN grade suggesting that methylation of these genes is not an initiating event in pancreatic carcinogenesis. Because ppENK methylation is rare in low-grade PanINs and very commonly observed in invasive pancreatic adenocarcinoma, it may be a useful indicator of the potential malignancy of epithelial cells of the pancreas.
Footnotes
Address reprint requests to Michael Goggins, M.D., Department of Pathology, Medicine, and Oncology, The Johns Hopkins Medical Institutions, 632 Ross Building, 720 Rutland Ave., Baltimore, MD 21205-2196. E-mail: mgoggins@jhmi.edu.
Supported by a Specialized Program in Research Excellence (SPORE) grant in Gastrointestinal Cancer (CA62924) from the National Cancer Institute (CA91968), the Lustgarten Foundation for Pancreatic Cancer Research, the Michael Rolfe Foundation, and the Uehara Memorial Foundation.
References
- 1.Kozuka S, Sassa R, Taki T, Masamoto K, Nagasawa S, Saga S, Hasegawa K, Takeuchi M: Relation of pancreatic duct hyperplasia to carcinoma. Cancer 1979, 43:1418-1428 [DOI] [PubMed] [Google Scholar]
- 2.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Klöppel G, Longnecker DS, Lüttges J, Offerhaus GJ: Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 2001, 25:579-586 [DOI] [PubMed] [Google Scholar]
- 3.Klöppel G, Hruban RH, Longnecker DS, Adler G, Kern SE, Partanen TJ: Ductal adenocarcinoma of the pancreas. Hamilton SR Aaltonen LA eds. Pathology and Genetics, Tumors of the Digestive System, World Health Organization Classification of Tumors. 2000, :pp 221-230 IARCP Press, Lyon [Google Scholar]
- 4.Moskaluk CA, Hruban RH, Kern SE: p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res 1997, 57:2140-2143 [PubMed] [Google Scholar]
- 5.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R, Kishi K, Omata M: Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology 1996, 110:227-231 [DOI] [PubMed] [Google Scholar]
- 6.Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, Yeo CJ, Kern SE, Hruban RH: Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998, 58:4740-4744 [PubMed] [Google Scholar]
- 7.Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ, Kern SE, Hruban RH: Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res 2000, 60:2002-2006 [PubMed] [Google Scholar]
- 8.DiGiuseppe JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allison DC, Cameron JL, Offerhaus GJA: Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994, 101:684-688 [DOI] [PubMed] [Google Scholar]
- 9.Goggins M, Hruban RH, Kern SE: The late temporal pattern of BRCA2 inactivation in pancreatic intraductal neoplasia: evidence and implications. Am J Pathol 2000, 156:1767-1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su GH, Bansal R, Murphy KM, Montgomery E, Yeo CJ, Hruban RH, Kern SE: ACVR1B (ALK4, activin receptor type 1B) gene mutations in pancreatic carcinoma. Proc Natl Acad Sci USA 2001, 98:3254-3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su GH, Hruban RH, Bansal RK, Bova GS, Tang DJ, Shekher MC, Westerman AM, Entius MM, Goggins M, Yeo CJ, Kern SE: Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999, 154:1835-1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su GH, Hilgers W, Shekher MC, Tang DJ, Yeo CJ, Hruban RH, Kern SE: Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res 1998, 58:2339-2342 [PubMed] [Google Scholar]
- 13.Hruban RH, Wilentz RE, Kern SE: Genetic progression in the pancreatic ducts. Am J Pathol 2000, 156:1821-1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, Goggins M: Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res 2000, 60:1835-1839 [PubMed] [Google Scholar]
- 15.Esteller M, Catasus L, Matias-Guiu X, Mutter GL, Prat J, Baylin SB, Herman JG: hMLH1 promoter hypermethylation is an early event in human endometrial tumorigenesis. Am J Pathol 1999, 155:1767-1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Virmani AK, Muller C, Rathi A, Zoechbauer-Mueller S, Mathis M, Gazdar AF: Aberrant methylation during cervical carcinogenesis. Clin Cancer Res 2001, 7:584-589 [PubMed] [Google Scholar]
- 17.Kang GH, Shim YH, Jung HY, Kim WH, Ro JY, Rhyu MG: CpG island methylation in premalignant stages of gastric carcinoma. Cancer Res 2001, 61:2847-2851 [PubMed] [Google Scholar]
- 18.Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J, Sukumar S: Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene 2001, 20:3348-3353 [DOI] [PubMed] [Google Scholar]
- 19.Ueki T, Toyota M, Skinner H, Walter KM, Yeo CJ, Issa J-PJ, Hruban RH, Goggins M: Identification and characterization of differentially methylated CpG island in pancreatic carcinoma. Cancer Res 2001, 61:8540-8546 [PubMed] [Google Scholar]
- 20.Zagon IS, Roesener CD, Verderame MF, Ohlsson-Wilhelm BM, Levin RJ, McLaughlin PJ: Opioid growth factor regulates the cell cycle of human neoplasias. Int J Oncol 2000, 17:1053-1061 [DOI] [PubMed] [Google Scholar]
- 21.Comb M, Goodman HM: CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res 1990, 18:3975-3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB: Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong DJ, Paulson, Prevo LJ, Galipeau PC, Longton G, Blount PL, Reid BJ: p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett’s metaplastic epithelium. Cancer Res 2001, 61:8284-8289 [PubMed] [Google Scholar]
- 24.Nuovo GJ, Plaia TW, Belinsky SA, Baylin SB, Herman JG: In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci USA 1999, 96:12754-12759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soria JC, Rodriguez M, Liu DD, Lee JJ, Hong WK, Mao L: Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res 2002, 62:351-355 [PubMed] [Google Scholar]
- 26.Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA: Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res 2001, 61:3573-3577 [PubMed] [Google Scholar]
- 27.Nakagawa H, Nuovo GJ, Zervos EE, Martin EW, Jr, Salovaara R, Aaltonen LA, de la Chapelle A: Age-related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res 2001, 61:6991-6995 [PubMed] [Google Scholar]
- 28.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L: Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med 1993, 328:1433-1437 [DOI] [PubMed] [Google Scholar]
- 29.Talamini G, Falconi M, Bassi C, Sartori N, Salvia R, Caldiron E, Frulloni L, Di Francesco V, Vaona B, Bovo P, Vantini I, Pederzoli P, Cavallini G: Incidence of cancer in the course of chronic pancreatitis. Am J Gastroenterol 1999, 94:1253-1260 [DOI] [PubMed] [Google Scholar]
- 30.Klump B, Hsieh CJ, Holzmann K, Gregor M, Porschen R: Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology 1998, 115:1381-1386 [DOI] [PubMed] [Google Scholar]
- 31.Rabinovitch PS, Dziadon S, Brentnall TA, Emond MJ, Crispin DA, Haggitt RC, Bronner MP: Pancolonic chromosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res 1999, 59:5148-5153 [PubMed] [Google Scholar]
- 32.Ishitsuka T, Kashiwagi H, Konishi F: Microsatellite instability in inflamed and neoplastic epithelium in ulcerative colitis. J Clin Pathol 2001, 54:526-532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerdes B, Ramaswamy A, Kersting M, Ernst M, Lang S, Schuermann M, Wild A, Bartsch DK: p16(INK4a) alterations in chronic pancreatitis-indicator for high-risk lesions for pancreatic cancer. Surgery 2001, 129:490-497 [DOI] [PubMed] [Google Scholar]