

Abstract
Glycogen synthase kinase-3 (GSK3) has recently been linked to mood disorders and schizophrenia, and the neurotransmitter systems and therapeutic treatments associated with these diseases. GSK3 is a widely influential enzyme that is capable of phosphorylating, and thereby regulating, over forty known substrates. Four mechanisms regulating GSK3 (phosphorylation, protein complexes, localization, and substrate phosphorylation) combine to provide substrate-specific regulation of the actions of GSK3. Several intracellular signaling cascades converge on GSK3 to modulate its activity, and several neurotransmitter systems also regulate GSK3, including serotonergic, dopaminergic, cholinergic, and glutamatergic systems. Because of changes in these neurotransmitter systems and the actions of therapeutic drugs, GSK3 has been linked to the mood disorders, bipolar disorder and depression, and to schizophrenia. Inhibition of GSK3 may be an important therapeutic target of mood stabilizers, and regulation of GSK3 may be involved in the therapeutic effects of other drugs used in psychiatry. Dysregulated GSK3 in bipolar disorder, depression, and schizophrenia could have multiple effects that could impair neural plasticity, such as modulation of neuronal architecture, neurogenesis, gene expression, and the ability of neurons to respond to stressful, potentially lethal, conditions. In part because of these key actions of GSK3 and its associations with mood disorders and schizophrenia, much research is currently being devoted to identifying new selective inhibitors of GSK3.
Keywords: GSK3, bipolar disorder, depression, schizophrenia, Akt, serotonin, dopamine
INTRODUCTION
The association of glycogen synthase kinase-3 (GSK3) with psychiatric diseases is a relatively new concept, first having been raised only in 1996. At that time, it was discovered that GSK3 is a target of the mood stabilizer lithium [1], a primary treatment for bipolar mood disorder [2], an illness also referred to as manic-depression. During the intervening ten years, a wide variety of types of studies have contributed to the hypothesis that inhibition of GSK3 by lithium makes an important contribution to lithium’s mood stabilizing capability [3]. These recent findings not only support the proposition that inhibition of GSK3 is an important therapeutic target of mood stabilizers, but also indicate that regulation of GSK3 may be involved in the therapeutic effects of other drugs used in psychiatry. Thus, evidence has begun to accumulate suggesting that dysregulation of GSK3 may occur in depression and schizophrenia, as well as in bipolar disorder. These connections are especially supported by evidence that neurotransmitter systems that are implicated in the pathophysiology of mood disorders and schizophrenia contribute to regulating GSK3 in the brain in vivo. Equally important to substantiating the link between GSK3 and psychiatric diseases are recent studies of the functions of GSK3. These studies have provided strong mechanistic hypotheses concerning how neuronal plasticity and function could be impaired by abnormally regulated GSK3 in psychiatric diseases. These diverse findings linking GSK3 to psychiatric diseases are summarized and evaluated in this review.
REGULATION OF GSK3
The major physiological mechanism that regulates the activity of GSK3 is phosphorylation of an N-terminal serine of GSK3 (Ser9-GSK3β , the predominant brain isoform, or Ser21-GSK3α ). This serine phosphorylation inhibits the activity of GSK3 and can be catalyzed by several kinases, such as Akt (Fig. 1). Thus, many growth factors that activate receptors coupled to the sequential activation of phosphoinositide 3-kinase (PI3K) and Akt inhibit GSK3 activity by increasing the Akt-mediated phosphorylation of the regulatory serine of GSK3. As shown in Fig. (1), other prominent intracellular signaling pathways, including those that activate protein kinase A or protein kinase C, also converge on GSK3 to inhibit it via phosphorylation of the N-terminal serine. Tyrosine phosphorylation of GSK3 (Tyr216-GSK3β ; Tyr279-GSK3α ) also contributes to regulating its activity in an activating manner, but the mediating kinases remain to be clearly identified and this modification may be carried out by autophosphorylation, so its importance in regulating the activity of GSK3 in situ remains a matter of debate [4].
Fig 1. Regulation of the inhibitory serine-phosphorylation of GSK3.
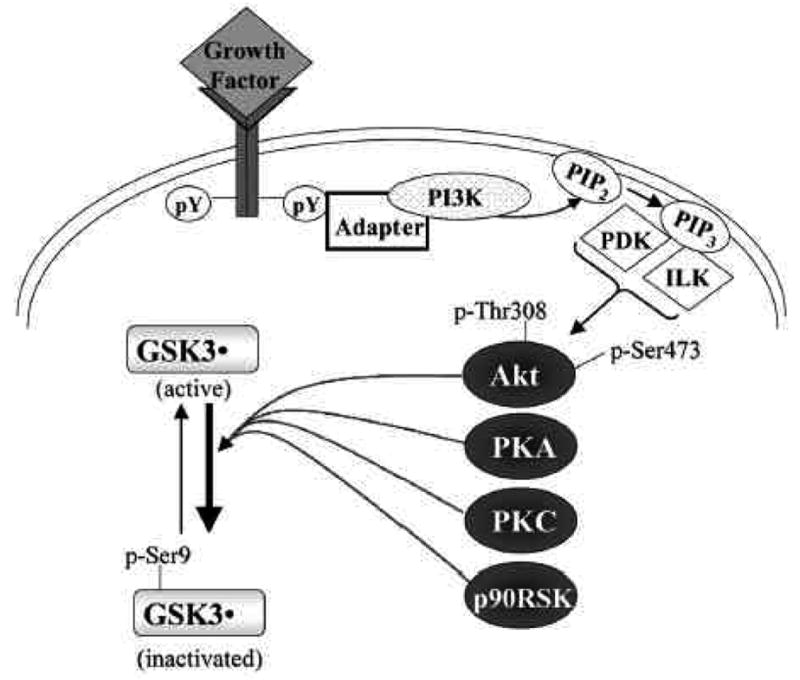
Phosphorylation of Ser-9 of GSK3β inhibits its activity. Some of the kinases reported to phosphorylate this site on GSK3β include Akt, protein kinase A (PKA; also known as cyclic AMP-dependent protein kinase), protein kinase C (PKC), and p90 ribosomal S6 kinase (p90RSK). Signaling leading from a growth factor receptor to activation of Akt is depicted. Growth factor receptor stimulation causes tyrosine phosphorylation (pY) of the receptor which interacts with various adaptor proteins to initiate a signaling cascade that results in activation of Akt via dual phosphorylation on threonine-308 and serine-473 which results in Akt-induced serine-phosphorylation and inactivation of GSK3β . ILK, integrin-linked kinase; PDK, phosphoinositide-dependent kinase; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate.
Further, substrate-selective regulation of the actions of GSK3 is also needed because GSK3 phosphorylates more than 40 substrates [5]. This large number of substrates enables GSK3 to influence many critical cellular functions, such as gene expression, cell structure, neural plasticity, and survival, so regulatory mechanisms must be invoked to selectively alter GSK3 activity to limit the substrates that it phosphorylates. GSK3-binding proteins provide one method by which cells have developed substrate-selective regulation of GSK3 (Fig. 2A). For example, in the Wnt signaling pathway axin and other proteins bind GSK3 to direct its actions to a specific substrate, β -catenin [6, 7]. Recently several additional GSK3-binding proteins have been identified and it appears that this is a common mechanism by which the action of GSK3 is directed to specific substrates [5].
Fig 2. Mechanisms contributing to substrate-selective regulation of GSK3.
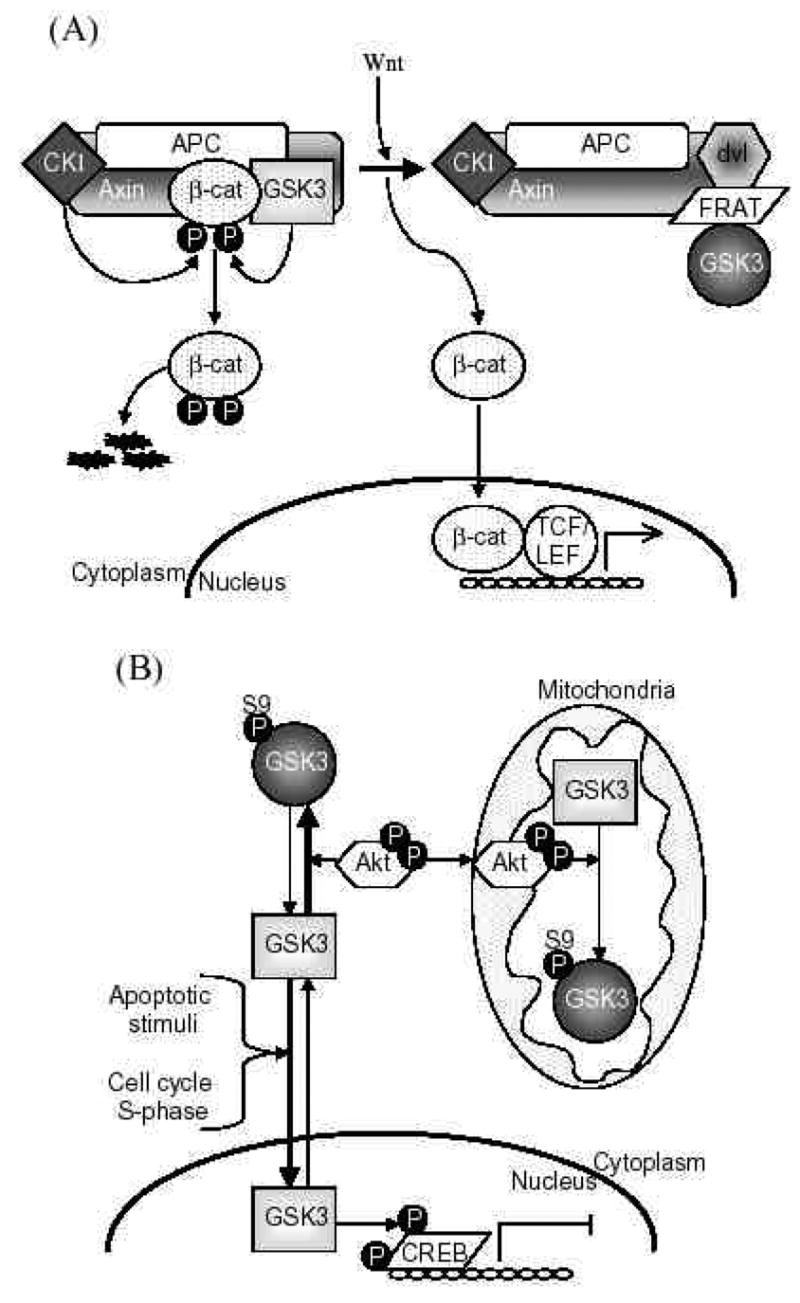
A. GSK3-binding proteins. GSK3-binding proteins regulate the action of GSK3 in the Wnt signaling pathway. Axin acts as a scaffold bringing together the substrate β -catenin with APC, the priming kinase, casein kinase 1 (CK1), and GSK3. In the absence of Wnt, phosphorylation of β -catenin by CK1 and GSK3 targets it for degradation. Wnt activation results in disheveled (dvl) and FRAT binding to GSK3, inhibiting its phosphorylation of β -catenin. This results in the stabilization and accumulation of β -catenin, its translocation to the nucleus, and facilitation of TCF/LEF-mediated transcription.
APC, adenomatous polyposis coli gene product; FRAT, frequently rearranged in advanced T-cell lymphoma; LEF, lymphoid-enhancing factor; TCF, T cell factor.
B. Subcellular distribution. GSK3 is predominantly a cytosolic protein, where it can be regulated by serine phosphorylation carried out by several kinases, such as Akt. GSK3 reversibly translocates to the nucleus where it’s level is increased in the S-phase of the cell cycle and during some types of apoptosis. Nuclear accumulation of GSK3 facilitates its actions on nuclear substrates, such as the transcription factor cyclic AMP response element-binding protein (CREB), which is inhibited following phosphorylation by GSK3. Mitochondria also contain GSK3. Although no studies have reported alterations of mitochondrial GSK3 levels, its activity can be regulated by serine phosphorylation. For example, activation of cytosolic Akt can lead to its import into mitochondria where it can serine-phosphorylate mitochondrial GSK3 to inhibit its activity.
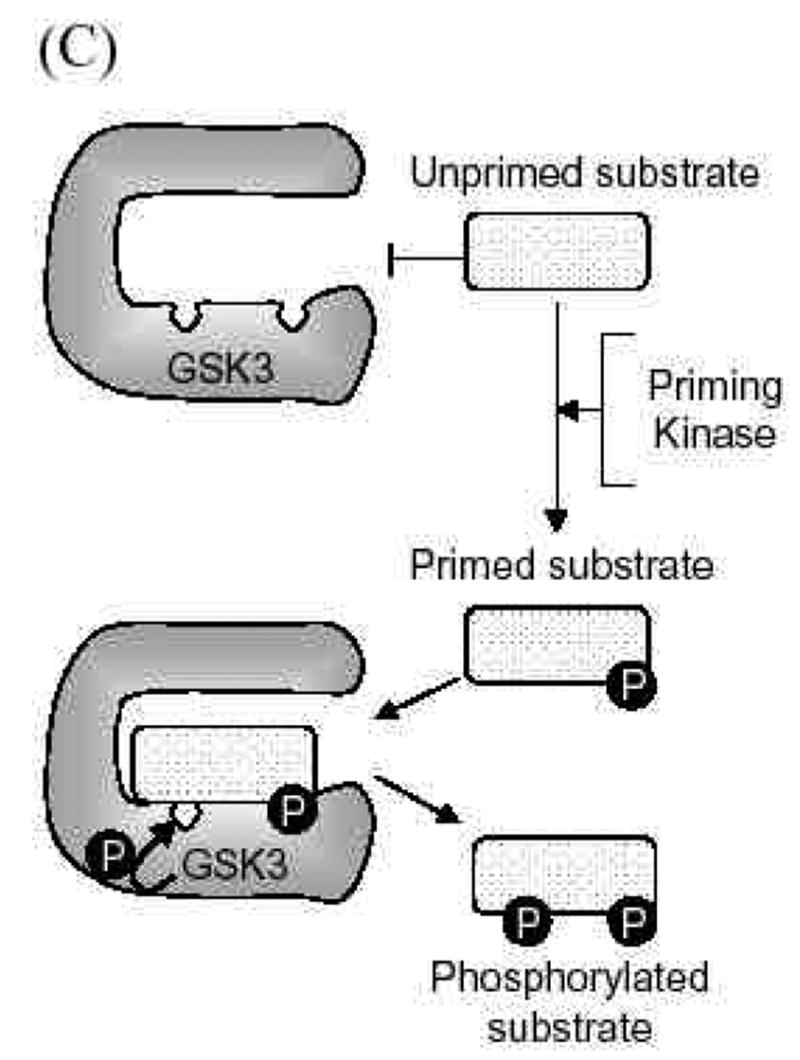
C. Priming phosphorylation of GSK3 substrates. Many substrates of GSK3 must be "primed", which means they are pre-phosphorylated at a serine/threonine four residues removed from the serine/threonine that is phosphorylated by GSK3. Thus, the consensus site for phosphorylation of primed substrates by GSK3 is S/T-X-X-X-S/T(p). This provides an important regulatory mechanism controlling the action of GSK3 because signaling pathways phosphorylating its substrates must be active before GSK3 can have any effect on such substrates. The preference of GSK3 for phosphorylating substrates that have been prephosphorylated (or "primed") 4 amino acids C-terminal to the target Ser/Thr is due to the presence of a phosphate binding pocket in GSK3. The phosphate of the primed substrate sits in this pocket and positions the phosphate acceptor site to enable efficient phosphorylation by GSK3.
Another method by which the capacity of GSK3 to interact with substrates is limited is by regulation of its subcellular localization (Fig. 2B). For example, nuclear levels of GSK3 are dynamically regulated with changes in the nuclear GSK3 level evident in several conditions, and this affects its ability to phosphorylate nuclear substrates, such as certain transcription factors [8–10]. There also appears to be subcellular compartment-selective phosphorylation of the inhibitory serine of GSK3 to locally modulate its activity, such as following activation of signals to the mitochondria which can target mitochondrial GSK3 [11]. Thus, subcellular-selective changes in the localization and phosphorylation of GSK3 can serve to regulate its actions on substrates within these compartments.
Finally, GSK3’s actions are often regulated by the phosphorylation state of its substrate, because most of GSK3’s substrates must be ‘primed’, pre-phosphorylated at a residue four-amino acids removed from the GSK3 phosphorylating site (Fig. 2C). Thus, the activity of a signaling pathway leading to the phosphorylation of the primed site of a substrate regulates the ability of GSK3 to phosphorylate the primed substrate. This, in concert with the other three regulatory mechanisms (phosphorylation, protein complexes, localization) combines to provide an integrated control to allow local and substrate-specific regulation of the actions of GSK3 [5]. In other words, GSK3 needs to be in the right phosphorylation state, in the right place, and associated with the right protein partners, at the same time that a substrate is co-localized and primed by another signaling pathway. Because of these multiple regulatory mechanisms, many proteins can be targets of phosphorylation by GSK3 in a selective manner.
The activity of GSK3 also can be regulated pharmacologically. Since GSK3 is a key component of many fundamental neuronal processes, as discussed later in this article, it was very intriguing when Klein and Melton [1] discovered that the therapeutic agent lithium is a direct inhibitor of GSK3. This raised the hypothesis that inhibition of GSK3 is important in the therapeutic actions of lithium, as discussed in the following section, and provided the first selective inhibitor of GSK3 which greatly facilitated studies of the actions of GSK3. Because of the likely importance of lithium’s inhibitory effect on GSK3 in treating bipolar disorder, and because of GSK3’s potential involvement in other prevalent diseases, including Alzheimer’s disease and diabetes, during the last few years much effort has been focused on discovering new inhibitors of GSK3, several of which have been identified [12–14]. Thus, there are now several selective GSK3 inhibitors available and there is currently a large effort directed towards finding the potential therapeutic effects of GSK3 inhibitors.
The importance of maintaining strict controls on the activity of GSK3 because of its many substrates and actions and its association with several diseases of the central nervous system [15] has raised much interest in studies of the activation state of GSK3 in vivo, as discussed in the following sections. However, two important considerations should be taken into account when conducting and evaluating studies of the in vivo activation state of GSK3. The first of these is consideration of postmortem stability of the GSK3 phosphorylation state. In mouse brain, following death there is a rapid dephosphorylation of the regulatory serine of both isoforms of GSK3, which amounts to about a 90% loss within 2 minutes of postmortem time [16]. Thus experimentally it is critically important to minimize postmortem artifacts in determining the phosphorylation state of GSK3. Furthermore, this limitation indicates that measurements of the serine phosphorylation state of GSK3 in postmortem human brain is not likely to be meaningful because of the extended postmortem times, encompassing several hours, of such tissues. It would be imperative that any such studies of postmortem human brain include more detailed verification of the identity of immunoreactive species other than relying solely on migration rates in gels. The possibility that similar rapid postmortem changes in the phosphorylation of GSK3 occurs in other tissues has not been reported. The second consideration concerning in vivo studies of the GSK3 activation state in mammalian brain is that anesthetic agents can cause large artifactual changes. Several commonly used anesthetic agents have been found to change the serine-phosphorylation state of GSK3 in mouse brain [16, 17]. For example, serine phosphorylation of GSK3 is increased approximately five-fold in mouse brain during pentobarbital-induced anesthesia. Thus, the presence of anesthetic agents and the postmortem handling of tissue are important considerations when evaluating measurements of GSK3 in brain tissue, and possibly in other tissues.
MOOD DISORDERS AND GSK3
Bipolar affective disorder, in which patients have a history of experiencing manic episodes that are often interspersed with depression, and major depression are commonly referred to as mood disorders. These are debilitating illnesses with a lifetime prevalence rate of approximately 20%, and they are life-threatening due to suicide as well as other causes [18–21]. The pathophysiological underpinnings of bipolar mood disorder and depression are unknown. Research into the causative mechanisms has been greatly hampered by the lack of adequate animal models of these diseases [22]. However, investigations of the mechanisms of action of therapeutic agents have provided leads about possible pathological mechanisms, and recent findings have revealed a number of connections linking GSK3 to the causes and, especially, to the actions of therapeutic agents used in these disorders.
For approximately the last fifty years the drug lithium has been the mainstay for the treatment of bipolar disorder, with a beneficial effect often observed in approximately 60–80% of patients and with no tolerance or sensitivity developing throughout many years of treatment [2, 23]. GSK3 was first linked to bipolar disorder in 1996 by the finding that lithium is a direct inhibitor of GSK3 [1]. They found that lithium inhibited GSK3 with an IC50 of approximately 2 mM, slightly greater than the therapeutic concentration range of lithium in serum, which is approximately 0.5–1.5 mM [1]. Soon thereafter, GSK3 was shown to be inhibited by lithium both in intact cells [24] and in mammalian brain in vivo [25]. These and subsequent examinations of many kinases confirmed that the inhibitory effect of lithium was a relatively selective action targeted to GSK3 [26].
The discovery that lithium directly inhibits GSK3 [1] introduced the concept that this action may contribute to the therapeutic effects of lithium in mood disorders and, consequently, that GSK3 may be dysregulated in mood disorders. However, because the therapeutically effective concentration of lithium in serum is lower than the IC50 of lithium for inhibition of GSK3, it seemed that the inhibition of GSK3 by lithium at therapeutic levels may be too little to contribute significantly to mood stabilization. A solution to this limitation was provided by the discovery of an in vivo amplification mechanism for lithium’s inhibition of GSK3 [27]. We found that chronic (4 weeks) in vivo treatment with a therapeutically relevant regimen of lithium administration increased by several-fold the phosphorylation of serine-9 of GSK3β in mouse brain regions [27]. Increased serine-phosphorylation of GSK3 following lithium administration indicates that the modest direct inhibitory effect of lithium on GSK3 is amplified by this phosphorylation mechanism, providing more substantial inhibition of GSK3 at a therapeutically relevant concentration of lithium than would be attained only by the direct inhibitory effect of lithium. These observations raised the exciting prospect that lithium inhibits GSK3 with amplification-mediated selectivity in the magnitude of inhibition [as reviewed in 28].
Other therapies used for mood disorders also have been linked to inhibition of the activity of GSK3. Valproic acid, originally used as an anticonvulsant and now also widely used as a mood stabilizer in bipolar disorder, was reported to directly inhibit GSK3 activity by some investigators [29–31] but not others [32]. Valproic acid treatment also increased the inhibitory serine phosphorylation of GSK3 in human neuroblastoma cells [27]. Although in vivo treatment with valproate did not increase serine phosphorylation of GSK3, valproate treatment did reduce pathophysiologically-induced dephosphorylation of both isoforms of GSK3 [17]. Thus, like lithium, valproate appears to contribute to the inhibitory control of GSK3 in mammalian brain in vivo. Increased serine phosphorylation of GSK3β in mouse brain also was induced by electroconvulsive seizure treatment of mice, another effective and widely used therapeutic intervention for bipolar disorder [33]. Thus, it is intriguing to find that three disparate mood stabilizing therapies, lithium, valproic acid, and electroconvulsive seizures, have the common action of causing inhibition of GSK3. These findings support the postulate that inhibition of GSK3 contributes to the therapeutic actions of mood stabilizers.
In summary, evidence that the actions of GSK3 are involved in the pathophysiology of bipolar mood disorder stems from the inhibitory actions on GSK3 of therapeutic interventions used in this illness. This connection should not be over-interpreted to indicate that GSK3 itself is hyperactive in bipolar disorder. Although this is one possibility, it is equally likely that upstream or downstream signals linked to GSK3 may be altered in bipolar disorder. For example, signaling activities upstream of GSK3 may be the primary deficiency, resulting in inadequate inhibitory control of GSK3 by these signaling pathways which would be bolstered by these therapies that inhibit GSK3. Alternatively, targets downstream of GSK3 may be dysfunctional, so these may be bolstered by inhibition of GSK3 by these therapies. Thus, either GSK3 itself or signaling intermediates coupled to GSK3 may be abnormal in bipolar disorder, so inhibition of GSK3 may contribute to normalizing these targets.
Dysfunctional GSK3, or molecules linked to GSK3, also may contribute to major depression. The most prevalent model for the pathological cause of depression is the monoamine hypothesis, which suggests that depression stems from inadequate serotonin (5HT) and/or norepinephrine neurotransmission. This hypothesis is supported by the findings that most antidepressants facilitate mono-aminergic neurotransmission, especially serotonergic actions, although the disease is clearly more complex [19–21]. Therefore, much research is focused on identifying intracellular signaling pathways coupled to 5HT receptors that may be involved in mood disorders and that may provide new targets for therapeutic agents. If key signaling outcomes downstream of receptors can be identified, then drugs targeting these signaling pathways may provide a therapeutic approach that is an alternative, or an add-on, to classical antidepressants. Recent evidence indicates that lithium’s inhibition of GSK3 may fulfill such a role, since GSK3 was recently found to be a downstream target in 5HT receptor-mediated signaling pathways that may not be adequately inhibited in depression (discussed below), and the GSK3 inhibitor lithium can be an effective add-on agent in antidepressant-refractory depression [34, 35].
The deficiency in serotonergic activity in depression makes especially relevant recent findings that serotonergic activity contributes to the inhibitory control of GSK3 in mammalian brain in vivo, so that serotonergic deficiency would lead to abnormally activated GSK3 [36]. In this study, serotonergic activity was increased in vivo by administration of d-fenfluramine and clorgyline to mice. D-fenfluramine induces the release of 5HT and inhibits 5HT reup-take, and clorgyline is a monoamine oxidase inhibitor that inhibits the breakdown of 5HT. Therefore, administration of d-fenfluramine and clorgyline augments 5HT levels, and this was found to increase the inhibitory serine-9 phosphorylation of GSK3β in mouse pre-frontal cortex, hippocampus, and striatum [36]. Increasing 5HT levels by inhibition of monoamine reuptake using the antidepressants fluoxetine or imipramine also increased serine-9 phosphorylation of GSK3β in mouse brain. These results demonstrate that increased serotonergic activity following the administration of anti-depressants inhibits GSK3β in brain.
Examination of 5HT receptor subtypes showed that stimulation of 5HT1A receptors in vivo caused increases in serine-9 phosphorylation of GSK3β [36]. Conversely, blockade of 5HT2 receptors by administration of a selective antagonist increased the serine-9 phosphorylation of GSK3β , indicating that 5HT2 receptors normally cause dephosphorylation of phospho-Ser9-GSK3β (Fig. 3). This balance of 5HT1A and 5HT2 receptors in regulating the phosphorylation of GSK3 is an interesting finding, since much previous evidence suggests that an imbalance between 5HT1A and 5HT2 receptors is associated with depression [37–39]. Previous studies have provided evidence that 5HT1A receptors are deficient in major depressive disorder and that 5HT2 receptors are up-regulated in depression, although this remains a subject of intense investigation [reviewed in 40]. Thus, these alterations would lead to decreases in the inhibitory serine phosphorylation of GSK3 in depression. It has been hypothesized that antidepressants exert their therapeutic effects through restoring the balance between those two receptor subtypes [39, 41], which may restore phospho-Ser9-GSK3β to normal levels. Unfortunately it is not possible to directly test this hypothesis in postmortem human brain because of the rapid postmortem dephosphorylation of GSK3 [16].
Fig 3. Schematic depiction of the regulation of GSK3 by 5HT1A and 5HT2 receptors and changes that may be associated with major depression.
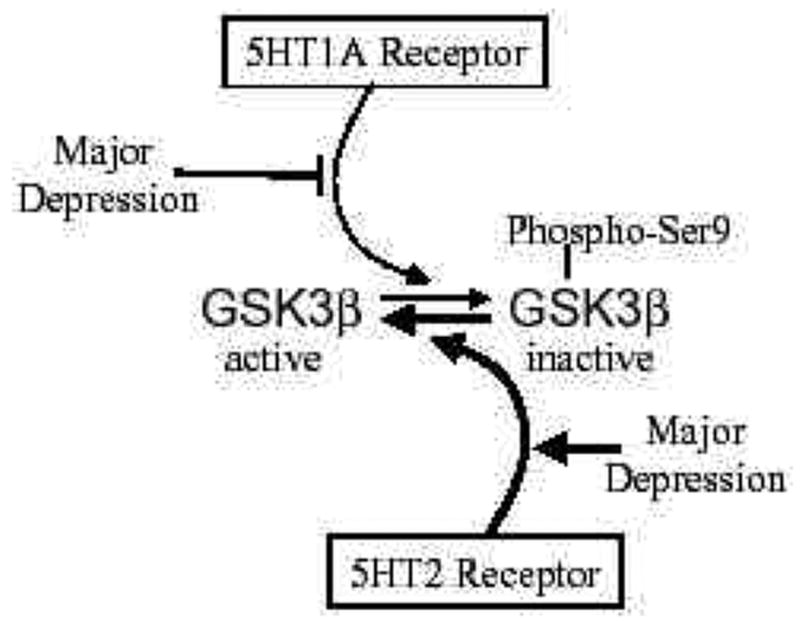
GSK3β is inhibited by phosphorylation of serine-9. This inhibitory phosphorylation is promoted by activation of 5HT1A receptors. However, decreased 5HT1A receptors occur in major depression, so there may be insufficient signaling leading to inhibition of GSK3β in depression. Conversely, activation of 5HT2 receptors cause activation of GSK3β by promoting its dephosphorylation. Increased 5HT2 receptors occur in major depression, suggesting increased activation of GSK3β . Overall, the balance between 5HT1A and 5HT2 receptors contributes to maintaining the normal activity level of GSK3β , and changes in each receptor associated with depression disrupt the 5HT receptor-mediated inhibitory signals that normally control GSK3β , suggesting that the activity of GSK3β is not adequately controlled in major depression.
The signaling pathways linking serotonergic activity to the regulation of phospho-Ser-GSK3 in brain in vivo are not known since many pathways converge on GSK3 and it is difficult to make mechanistic conclusions based on in vivo studies where many signaling pathways converge on GSK3. Most serotonergic receptors are coupled to classical second messenger pathways by one of three types of heterotrimeric G-proteins, Gq which couples receptors to the phosphoinositide second messenger system, and Gi or Gs which couple receptors to inhibition or activation, respectively, of cyclic AMP production. Gq-coupled serotonergic receptors activate protein kinase C which, in turn, is known to phosphorylate the regulatory serine of GSK3 [42, 43]. We are unaware of any reports that directly demonstrate protein kinase C-mediated serine phosphorylation of GSK3 following serotonergic receptor activation although this connection seems likely to occur. Gi-coupled receptors are known to activate the PI3K/Akt signaling pathway, so Akt activated in this manner could account for serine phosphorylation of GSK3 by increased serotonergic activity. 5HT1A receptors coupled to Gi have been shown to activate PI3K [44] and Akt [45]. Furthermore, the activity of Akt in brain samples from depressed suicide victims were below that of matched controls [45]. This indicated that depression might be associated with diminished activity of the PI3K/Akt signaling pathway which normally leads to inhibition of GSK3β , supporting the possibility that GSK3β may not be adequately inhibited in depression.
The hypothesis that hyperactive GSK3 may partially contribute to depression or behaviors associated with depression was further supported by studies of the effects of administration of GSK3 inhibitors to mice. The most intriguing aspect of these studies are results using the forced swim test, a widely used model to assess depressive behavior and the effects of antidepressant agents, measured by immobilization time which is diminished by most antidepressants. Kaidanovich-Beilin et al. [46] found that administration of a peptide inhibitor of GSK3β rapidly induced antidepressant-like behavioral effects, specifically reducing immobilization in the forced swim test. That this depression-associated behavior is modulated by GSK3 was further supported by recent studies [47, 48]. Gould et al. [48] reported that administration of an ATP competitive GSK3 inhibitor modestly reduced immobilization in the forced swim test. O’Brien et al. [47] showed more directly the relation between GSK3β and immobilization in the forced swim test. They found that large reductions in immobilization time were induced not only by inhibition of GSK3β by lithium administration, but also that lowered GSK3β levels expressed in heterozygote GSK3β +/− mice was associated with a large reduction of immobilization in the forced swim test. Taken together, these three reports demonstrate convincingly that GSK3 has a critical role in this widely used paradigm to assess depressive activity and the counteracting effects of antidepressants.
In summary, research on 5HT receptor signaling and GSK3β suggests that 5HT receptor actions, especially the balance of activities between 5HT1A and 5HT2 receptor subtypes, can regulate GSK3β in brain. Furthermore, 5HT receptor dysfunction and reduced PI3K/Akt signaling are observed in depression, each of which could contribute to GSK3β activation. Furthermore, antidepressants that enhance 5HT receptor signaling increase serine phosphorylation of GSK3. These findings consolidate the hypothesis that abnormally active GSK3 due to 5HT receptor signaling dysfunction could contribute to the pathophysiology of depression, and that a part of the therapeutic effects of increased serotonergic activity by antidepressants is mediated by GSK3 inhibition. Downstream substrates of GSK3 which may contribute to antidepressant actions, such as exemplified by the reduced immobility in the forced swim test, remain to be identified. GSK3 phosphorylates many proteins including structural proteins, transcription factors, metabolic proteins, and others. Later sections in this article describe some of these actions of GSK3 that may be linked to its actions in psychiatric diseases.
SCHIZOPHRENIA AND GSK3
Several lines of research have produced findings consistent with the hypothesis that alterations in GSK3 are connected with schizophrenia. However this association has not received the same intense scrutiny as it has in mood disorders, and contradictory findings have been reported, so the association of GSK3 with schizophrenia, while tantalizing, remains to be more thoroughly evaluated.
Schizophrenia is a prevalent and severe psychotic disorder with considerable variation among individuals in symptoms associated with thought content, perception, cognition, and affect [49, 50]. The causative factors of schizophrenia remain unknown, but dysregulated dopamine neurotransmission is likely the most widely investigated hypothesis of the pathophysiology of schizophrenia. This classical hyperdopaminergic hypothesis of schizophrenia pathology is supported by the therapeutic effects in schizophrenia of conventional antipsychotics that are dopamine D2 receptor antagonists and by the psychotogenic effects of dopamine enhancing drugs. However, although conventional antipsychotics can diminish symptoms in some patients, it is evident that the pathophysiology of schizophrenia is more complex, and diverse, than being caused only by increased dopaminergic activity in subcortical regions [49–51]. Some symptoms of schizophrenia, such as cognitive impairments, are resistant to conventional antipsychotics, and the cognitive deficits in schizophrenia are thought to arise in part from hypodopaminergic neurotransmission at dopamine D1 receptors in the pre-frontal cortex [52]. This is supported by clinical studies that show atypical antipsychotics, which increase dopamine neurotransmission at dopamine D1 receptors [53], improve cognitive symptoms in schizophrenic patients [54, 55]. Especially intriguing is the evidence showing that dopamine D1 receptor hypoactivity in the pre-frontal cortex can result in dopamine D2 receptor hyperactivity in the striatum. Imaging studies of the function of dopamine D1 receptors in the prefrontal cortex [56–58] and of dopamine D2 receptors in the striatum [51, 52] of schizophrenic patients lend further support to the view that an imbalance of cortical/subcortical dopaminergic function may be central to the pathology of schizophrenia [51]. Thus, regarding dopaminergic neurotransmission, balanced activities of dopamine D1 and D2 receptors seems to be critical, and schizophrenia appears to be associated with low dopamine D1 and/or high dopamine D2 receptor function. Beyond the dopaminergic system, many other theories of the pathology of schizophrenia have been promulgated. One of the most widely considered is the evidence of neurodevelopmental abnormalities, supporting the concept that schizophrenia represents a spectrum of diseases with multi-factorial pathologies [49]. Although studies connecting GSK3 to schizophrenia are few, they have identified links between GSK3 and these two major postulates, altered dopaminergic activity and disrupted neurodevelopment.
Abnormalities of GSK3 were first linked to schizophrenia in a series of studies reported by Agam and colleagues [59–63]. They found approximately 40% lower GSK3β mRNA levels, GSK3β protein levels, and GSK3 kinase activity in postmortem samples of frontal cortex from subjects with schizophrenia, and a 30% lower GSK3β protein level in the cerebrospinal fluid, compared with controls. However, these differences were not detected, except for the lower GSK3 mRNA level, by the same investigators in samples from a different brain collection [63]. This difference between brain collections was also encountered by another group who found differences of GSK3β protein levels in schizophrenic compared to control samples in one brain collection but not another [64, 65]. As noted earlier in this review, a lack of changes in GSK3 levels does not preclude changes in GSK3 actions because of the intracellular mechanisms that regulate its activity, such as the inhibitory effect of serine-phosphorylation, but this can not be studied in human brain samples because of the extensive loss of serine phosphorylation of GSK3 that occurs postmortem. Further studies with a greater number of samples will be necessary to draw concrete conclusions about whether or not alterations in GSK3 expression or protein levels reproducibly occur in subjects with schizophrenia.
Another approach to identifying potential links between schizophrenia and dysregulated GSK3 is to examine the modulatory influences of neurotransmitter systems that are thought to be involved in the illness, as discussed in the previous section concerning studies of the serotonergic system in mood disorders. Therefore, since there is evidence for dopaminergic dysregulation in schizophrenia, it is pertinent to consider whether dopaminergic activity has a role in regulating GSK3. This was first examined in brain in vivo by Gil et al. [66] who found that administration of a dopamine D1 receptor agonist inhibited GSK3 activity in rabbit frontal cortex and hippocampus, and this was blunted in rabbits following prenatal cocaine exposure which itself caused inhibition of GSK3β activity. Thus, this study showed for the first time that dopaminergic activity has a regulatory influence on GSK3 in brain in vivo, and that long term changes in the dopaminergic system can modulate GSK3. From this initial report, especially notable is the possibility that low dopamine D1 receptor activation that is reported to occur in schizophrenia would be associated with impaired inhibitory control of GSK3 (Fig. 4).
Fig 4. Schematic depiction of the regulation of GSK3 by dopamine D1 and dopamine D2 receptors and changes associated with schizophrenia.
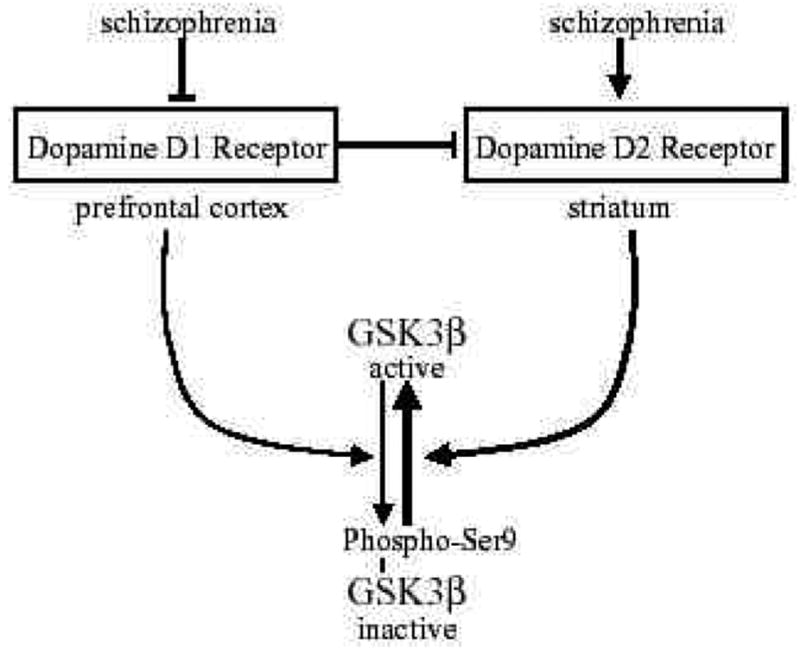
GSK3β is inhibited by phosphorylation of serine-9. This inhibitory phosphorylation is promoted by activation of dopamine D1 receptors. However, decreased dopamine D1 receptor activation occurs in schizophrenia, so there may be insufficient signaling leading to inhibition of GSK3β in schizophrenia. Conversely, activation of dopamine D2 receptors cause activation of GSK3β by promoting its dephosphorylation. Increased dopamine D2 receptor activation occurs in schizophrenia, in part by deficient inhibitory inputs from dopamine D1 receptors, suggesting increased activation of GSK3β . Overall, the balance between dopamine D1 and D2 receptors contributes to maintaining the normal activity level of GSK3β , and changes in each receptor associated with schizophrenia disrupt the control of GSK3β .
Subsequently, more extensive in vivo interactions between the dopaminergic system and GSK3 in mammalian brain were reported. In a recent definitive study, Beaulieu et al. [67] found that increased dopaminergic stimulation of dopamine D2 receptors in the striatum induced by administration of the indirect dopamine stimulant amphetamine or present in dopamine transporter knockout mice (DAT-KO), caused activation of GSK3α and GSK3β in mouse striatum. This appeared to occur because of decreased Akt activity which resulted in decreased serine-phosphorylation of GSK3. Hyperactive GSK3 was shown to contribute to the behavioral phenotype because administration of GSK3 inhibitors, including lithium, antagonized dopamine-dependent hyperactivity and stereotypy in the DAT-KO mice, and amphetamine-induced hyperactivity was lower in GSK3β +/− mice [67]. These findings clearly demonstrated that GSK3 is a downstream target of dopamine D2 receptor-mediated signaling in vivo and that GSK3 mediates some of the behavioral effects of dopamine, supporting the possibility that alterations in GSK3 activity may be relevant to schizophrenia and other dopamine-related disorders [67]. This study notably raises the possibility that GSK3 is abnormally activated in schizophrenia since dopamine D2 receptor activation is elevated, which in conjunction with impaired dopamine D1 receptor-mediated inhibition of GSK3 may synergistically contribute to hyperactivated GSK3 (Fig. 4).
More direct evidence of impaired Akt/GSK3β signaling in subjects with schizophrenia was recently reported. Emamian et al. [68] found approximately 50% decreases in the protein levels of one isoform of Akt, called Akt1, in the frontal cortex and lymphocytes of subjects with schizophrenia compared with controls. Administration to mice of haloperidol, a typical antipsychotic which is an antagonist of dopamine D2 receptors, increased the activating phosphorylation of Akt and the inhibitory serine-phosphorylation of GSK3β in brain. The decreased Akt signaling to GSK3β in schizophrenia and corrective modulation by the dopaminergic antagonist supports the potential role of dopamine receptor-coupled signaling to Akt and GSK3β in the pathogenesis of schizophrenia. This study also reported lower phospho-Ser9-GSK3β levels in samples from subjects with schizophrenia compared with controls as determined by immunoblot analysis [68]. However, the recently reported rapid postmortem serine-dephosphorylation of GSK3β [16] suggests that more thorough examination of immunoreactive bands is necessary to unequivocally identify levels of phosphoserine-GSK3 in postmortem samples. Most interestingly, Emamian et al. [68] found that a haplotype of Akt1 that was preferably transmitted to schizophrenic probands is related to a lower protein level of Akt1 and that amphetamine administration to Akt1-depleted mice showed disruption of prepulse inhibition, a representative model of impaired sensorimotor gating of schizophrenia. A recent report confirmed that Akt1 is a susceptibility gene for schizophrenia in a large population study [69] but it was not confirmed in another study [70]. This finding suggests that reduced Akt1 may contribute to schizophrenia, supporting the possibility of an association between impaired control of GSK3 and schizophrenia.
The role of GSK3β in association with the Wnt signaling pathway (Fig. 2A) is a well known factor regulating CNS development [6, 7], so altered GSK3β signaling in the brain of subjects with schizophrenia also could contribute to the neurodevelopmental abnormalities that have been linked to schizophrenia. There have been several reports of alterations in the Wnt signaling pathway, which regulates the action of GSK3, associated with schizophrenia [71–74]. Since Wnt signaling is a key component of neurodevelopment, and much evidence indicates neurodevelopmental abnormalities in schizophrenia [75–77], these are tantalizing reports, but the significance of these findings for the pathophysiology of schizophrenia remain to be investigated in greater detail.
In addition to dopaminergic activity, alterations of cholinergic and glutamatergic neurotransmission also have been linked to the pathology of schizophrenia, so it is of interest that each of these neurotransmitter systems recently was found to influence the regulation of brain GSK3 in vivo. Schizophrenia has been linked to dysregulated cholinergic neurotransmission in several studies, and especially strong is the evidence indicating association with the cognitive impairment of schizophrenia [78, 79]. Cognitive impairment is often evident in schizophrenia, and schizophrenia has been reported to be associated with reduced choline acetyltransferase, the enzyme that synthesizes acetylcholine which is a critical neuro-transmitter for cognition, and choline acetyltransferase activity was reported to be inversely correlated with cognitive impairments in schizophrenia [80–82]. There have been several reports of decreased levels of muscarinic receptors in specific brain regions of schizophrenic patients, including frontal cortex [83–85], anterior cingulate gyrus [86], hippocampus [87], Broadmann’s area 9 [88], and caudate-putamen [85, 89]. Taken together, these and other findings suggest that muscarinic receptor stimulation can be impaired in schizophrenia either at the level of acetylcholine synthesis or receptor activation. Therefore, it is of interest that a regulatory influence of cholinergic activity modulating the phosphorylation of brain GSK3 in vivo was recently identified. De Sarno et al. [90] found that cholinergic stimulation with the muscarinic receptor-selective agonist pilocarpine or the acetylcholinesterase inhibitor physostigmine rapidly increased the serine-phosphorylation of GSK3α and of GSK3β by several-fold in three mouse brain regions. This finding raised the possibility that dysfunctional cholinergic activity may cause inadequate inhibitory control of GSK3 which can be restored by stimulation of muscarinic receptors.
Much research has linked altered glutamatergic neurotransmission to schizophrenia [91]. One of the most widely used models of schizophrenia involves application of glutamatergic N-methyl D-aspartate (NMDA) receptor antagonists to animals because in healthy human subjects these agents can induce symptoms similar to those seen in schizophrenia [92]. Thus, administration of the noncompetitive NMDA receptor antagonists phencyclidine or ketamine can induce several symptoms of schizophrenia in normal control individuals, and can worsen symptoms in schizophrenic subjects [reviewed in 51]. Conversely, administration of NMDA receptor agonists examined as adjunctive treatments have been reported to improve psychotic symptoms in schizophrenia [93]. These findings support the hypothesis that activation of NMDA receptors may be impaired in schizophrenia [93].
This connection between NMDA receptor activity and schizophrenia raises the question of whether this may contribute to the regulation of GSK3, and several recent studies have provided support for this regulatory interaction. NMDA treatment of cultured hippocampal neurons caused a rapid and nearly complete dephosphorylation of phospho-Ser9-GSK3β , indicating that GSK3β is activated by NMDA receptor signaling [94]. In accordance with that conclusion, in vivo blockade of NMDA receptors by administration of the antagonist phencyclidine increased mouse brain serine-phosphorylation of GSK3 [95], a response was also observed in mouse brain following administration of memantine, an NMDA antagonist approved for use in humans [90]. A conflicting report showed that in immature rats blockade of NMDA receptors by in vivo administration of the antagonist MK-801 transiently decreased the serine-phosphorylation of GSK3 [96], a difference from the other reports that could be due to age-dependent differences in responses to NMDA receptor modulation or differences between NMDA antagonists. Thus, although still only few, the majority of studies indicate that NMDA receptor stimulation dephosphorylates GSK3 and that blockade of NMDA receptors in vivo is sufficient to cause increased levels of serine-phosphorylated GSK3.
Overall, quite a few connections have been identified between GSK3 and schizophrenia, but there are also serious contradictions in this data. Thus, some data suggests the action of GSK3 is reduced, whereas other data suggests it is increased, in association with schizophrenia. Schizophrenia-associated reductions of GSK3 are indicated by the measurements in postmortem brain samples and by the inhibitory effects of the NMDA antagonists phencyclidine and memantine. Postmortem measurements are the most direct approach to identifying disease-related links, but this strategy is also fraught with difficulties inherent in using postmortem tissue and in studying such a heterogeneous sample population. These difficulties are exemplified by the mixed results obtained in different sample sets. Countering the indications of reduced GSK3, there is substantial evidence of increased GSK3 actions in schizophrenia. This evidence comes from studies showing reduced Akt in schizophrenia and studies of neurotransmitter effects on regulating GSK3. The two studies implicating Akt deficits in schizophrenia provide strong evidence that this inhibitory regulator of GSK3 is dysfunctional, thus allowing hyperactivation of GSK3. This is corroborated by some indications in schizophrenia of low dopamine D1 receptors which inhibit GSK3, and elevated dopamine D2 receptors that activate GSK3, and that typical antipsychotics block D2 receptors, which would cause inhibition of GSK3. The altered balance of D1 and D2 receptors, along with a possible deficit in cholinergic neurotransmission, lead to the prediction that GSK3 is inadequately controlled in schizophrenia. However, lithium, a GSK3 inhibitor, has very limited therapeutic effects in schizophrenia, suggesting that if GSK3 activity is abnormal in schizophrenia it may only contribute to a subset of symptoms. Thus, although intriguing connections between schizophrenia and alterations of GSK3 have been identified, much more research is necessary to integrate the findings from studies of GSK3 in postmortem tissue, the developmental influences of GSK3, and the regulatory effects on GSK3 of neurotransmitter systems that have been shown to be involved in schizophrenia.
NEURONAL FUNCTIONS REGULATED BY GSK3 WHICH MAY UNDERLIE THE PATHOLOGY AND TREATMENT MECHANISMS IN PSYCHIATRIC DISEASES
The evidence linking GSK3 to the pathology and treatment of mood disorders and schizophrenia raises the key question of how dysregulated GSK3 might contribute to these diseases. As the understanding of the actions of GSK3 has been greatly expanded during the last few years, several effects of GSK3 have been identified as strong candidates that might link its dysregulation to these diseases. These actions are centered on neural plasticity, and this is considered below within the contexts of structural effects, neurogenesis, gene expression, and responses to stress.
Plasticity: Cell Structure and Remodelling
Perhaps the most widely accepted conceptual basis for mood disorders, and also considered in schizophrenia research, is the postulate that there is impaired neural plasticity associated with the pathophysiology of these diseases. One reason for the fairly widespread acceptance of this concept is that it can be applied to nearly every aspect of neuronal function, thus its application to mood disorders really does little to help to precisely and specifically define key functions that are dysregulated in these diseases. Therefore, for the purpose of this review, we define neural plasticity more narrowly as the ability of neurons to respond appropriately to fluctuating inputs, such as changes in neurotransmission, stress, or cellular insults. In other words, these represent adaptive changes that facilitate neuronal function in response to alterations in external inputs, and these inputs may be normal fluctuations in neurotransmission or exposure to stress. In this context, much evidence has documented the crucial role of GSK3 in neuronal plasticity, and here we consider specifically structural plasticity as an example of one of the many influences of GSK3 on neural plasticity.
GSK3 has many influences on cell biology and architecture, as recently reviewed [5]. Briefly, GSK3 phosphorylates several proteins that can bind to microtubules, protein complexes that provide structural stability to neurons but which must maintain plasticity in order to allow the dynamic changes in neuronal shape and contacts which continually occurs in neurons. These substrates of GSK3 include the microtubule-associated protein (MAP) tau, MAP1β , APC, CRMP-2, and others. By phosphorylating microtubule-associated proteins GSK3 modulates microtubule dynamics which are important in neuronal remodelling, neurite outgrowth and collapse, and axonogenesis [97–104]. These are all key processes in structural neural plasticity, emphasizing the widespread influences of GSK3 and the importance of maintaining strict control of GSK3 activity. GSK3 also phosphorylates the protein motor kinesin, thereby modulating intracellular transport of many types of cargo [105], modulates growth cone extension [106, 107], and modulates cell motility [108]. Thus, fluctuations in GSK3 activity modulate many intracellular structural dynamics of neurons. GSK3 is also an essential component of several developmentally important signaling pathways (which are also functional in adult brain), including Wnt [6, 7], Hedgehog [109, 110], Reelin [111], and Notch [112] signaling, each of which controls aspects of neuronal structure. Dysregulation of GSK3 associated with these developmental systems may be particularly relevant to schizophrenia for which there is a strong body of evidence of developmental deficiency. Thus, GSK3 has numerous effects on cell biology, architecture, and remodeling, actions that may underlie its detrimental effects in psychiatric disorders when it is not properly regulated. For example, deficient serotonergic activity, as can occur in mood disorders, can cause GSK3 in the brain to be abnormally active. This can impair neural plasticity through the actions of GSK3 on these dynamic structural targets, and bolstering serotonergic activity with antidepressants is now known to strengthen the inhibitory control of GSK3 in the brain, thereby potentially facilitating neural plasticity by modulating these structural dynamics.
Neurogenesis
Impaired neurogenesis in animal models of depression, and its correction by antidepressants, has recently brought this process into prominence as a potential crucial mechanism in depression and other psychiatric diseases. Neurogenesis in this context involves the production, survival, or integration of new neurons in the adult brain. In a simplified manner, this theory posits that depression may be causally associated with impaired neurogenesis, and that antidepressants augment one or more of the components of neurogenesis [113–115]. Thus it is intriguing that GSK3 also is a noted regulator of neurogenesis. Using a variety of experimental systems, several early studies nearly simultaneously reported increases in markers of neurogenesis following diverse treatments in animals or cells with agents that are therapeutic in mood disorders, including antidepressants [116–118], electroconvulsive shock [119, 120], and lithium [121–124]. None of these studies with lithium examined the mechanism by which neurogenesis was enhanced, but studies of GSK3 make it a target worth investigating. For example, in addition to the well known role of the Wnt signaling pathway (activation of which inhibits GSK3) on neuronal development [125], several studies reported effects of inhibitors of GSK3 on neurogenesis in embryonic stem cells [126–128]. Thus, inhibition of GSK3, which may be impaired in mood disorders, by lithium and other therapeutics may bolster neurogenesis, an action that may contribute to therapeutic outcomes. With respect to depression, especially compelling is the recent report by Santarelli et al. [118] which provided strong evidence that the behavioral effects of chronic antidepressants may be mediated by the stimulation of hippocampal neurogenesis. Since serotonergic activity is reduced in depression, it is also of interest that several studies found activation of 5HT1A receptors, which we showed [36] causes phosphorylation (inactivation) of GSK3, enhanced neurogenesis [113, 129, 130]. In summary, there is growing, but still very incomplete, data suggesting links between neurogenesis and both mood disorders and therapeutic agents, and that GSK3 may contribute to these regulatory mechanisms.
Gene Expression
Of the many substrates that are phosphorylated by GSK3, the largest category consists of transcription factors, proteins that are key regulators of gene expression, with over a dozen transcription factors known to be substrates of GSK3 [5]. These include several of the most widely studied transcription factors, such as AP-1, NFκ B, HSF-1, CREB, p53, the co-activator β -catenin, as well as others. This gives GSK3 the capacity to have a tremendous impact on gene expression and consequently to regulate many cellular functions.
Phosphorylation by GSK3 regulates these transcription factors through diverse mechanisms, examples of which include the following. The most widely studied substrate of GSK3β in this category is β -catenin, a component of the Wnt signaling pathway [6, 7]. Phosphorylation of β -catenin by GSK3 promotes its proteolysis, whereas inhibition of GSK3 allows the accumulation of β -catenin which acts in the nucleus in conjunction with transcription factors to control gene expression. Thus, GSK3 controls the level of β -catenin by facilitating its degradation. GSK3 also can regulate the nuclear localization of transcription factors, such as nuclear factor of activated T cells (NFAT). In the nucleus GSK3 phosphorylates NFAT, signaling its export to the cytosol, terminating its action as a transcription factor [131]. In addition to controlling stability and subcellular localization, GSK3 regulates the transcriptional activities of transcription factors, such as heat shock factor-1 (HSF-1) and p53. Phosphorylation by GSK3 contributes to the inactivation of HSF-1 [132], whereas inhibition of GSK3 counteracts this negative influence on HSF-1, allowing greater expression of heat shock proteins to bolster cellular plasticity and survival [133]. While nuclear GSK3 down-regulates the survival-promoting action of HSF-1, nuclear GSK3 also promotes apoptosis by contributing to the activation of p53 [10, 134, 135]. Conversely, inhibition of GSK3 greatly attenuates the actions of p53. Thus, GSK3 acts in diverse ways to either reduce or enhance transcription factor activities.
Taken together, it is evident that GSK3 uses a variety of means to regulate the activities of a broad spectrum of transcription factors, affording it the capacity to regulate the expression of numerous genes and, consequently many cell functions. Thus, phosphorylation of these substrates likely influences neuronal plasticity by regulating gene expression, and dysregulated GSK3 in psychiatric diseases can easily be envisioned as having detrimental effects on the normal regulation of these transcription factors.
CREB and BDNF
One of the most often examined transcription factor in studies of both mood disorders and schizophrenia is cyclic AMP response element binding protein (CREB). Stimulation of either serotonergic or dopaminergic receptors can activate CREB, thus alterations of CREB have been linked to the psychiatric diseases associated with each of these neurotransmitters, mood disorders and schizophrenia, respectively. CREB is activated when it is phosphorylated on Serine-133. Phospho-Ser133-CREB is a primed substrate for GSK3, which subsequently can phosphorylate Serine-129 to inactivate CREB (30, 136). Thus, inhibition of GSK3 can facilitate CREB activity (30). The expression of the key neurotrophin, brain-derived neurotrophic factor (BDNF), is regulated by CREB [137, 138]. Recently, much evidence indicates that the production and/or actions of BDNF may be deficient in depression [139, 140]. BDNF expression is reduced in animal models of stress-induced depression [141, 142], and administration of antidepressants or electro-convulsive shock can increase BDNF expression and counteract stress-induced decreases in BDNF [143–145]. Especially persuasive is the finding that centrally administered BDNF produced antide-pressant-like behavior [146, 147]. Studies of subjects with major depression also support the hypothesis that BDNF is depleted in major depression, as BDNF serum levels were reduced in major depression subjects compared to control subjects [148, 149], and postmortem measurements of brain BDNF immunoreactivity demonstrated higher levels in antidepressant-treated than non-treated subjects with major depression [150]. Thus, many studies have linked CREB-regulated BDNF expression to mood disorders and the actions of therapeutic agents, but as reviewed by Jacobsen and Mork (151) there are also contradictory findings. With CREB being regulated by GSK3, and CREB regulating BDNF, it is intriguing that links have been established between BDNF and GSK3. Treatment with lithium [151–153] or valproate [152], which are GSK3 inhibitors, increased BDNF protein levels in rat brain, although strain-dependent effects also have been reported [154]. However, it remains to be demonstrated the extent to which these actions of mood stabilizers on BDNF expression are due to inhibition of GSK3. Recent findings that antidepressants [36] and electroconvulsive shock treatment [33] can inhibit GSK3 suggest that this action may contribute to the increases in CREB, and subsequently BDNF expression, caused by these treatments. In addition to impairing the expression of BDNF, GSK3 also can impede BDNF-induced intra-cellular signaling activities [155]. Thus, in mood disorders impaired inhibitory control of GSK3 may reduce CREB activity, the expression of BDNF, and signaling induced by BDNF, to contribute to deficient BDNF actions, and the inhibitory effects of therapeutic treatments on GSK3 may contribute to facilitated CREB activity and BDNF expression and BDNF-induced activation of intracellular signaling pathways.
Stress and Cell Survival
Neural plasticity includes the capacity of cells to respond appropriately to stressful events or agents, including those that could be lethal. Experimentally, this can be measured by assessing the terminal outcome of stress-induced cell death by apoptosis. Thus, even though apoptosis may not be a causative factor in mood disorders or schizophrenia, measurements of apoptosis are a direct indicator of neural plasticity, the ability to respond and adapt to stressful insults, and can indicate the role of enzymes and therapeutic agents in these plastic responses. In this context, abundant evidence has proven that GSK3 is detrimental to neural plasticity, as one of the most replicated actions of GSK3 is that its activity impedes survival following exposure to many kinds of insults that are able to cause mitochondria-mediated apoptosis and eventual cell death.
Thus, using evidence of apoptosis as an indicator of impaired neural plasticity, GSK3 has been demonstrated to impair survival and to promote apoptosis caused by a wide variety of different conditions or agents. Among these many conditions in which GSK3 promotes apoptosis are exposure to growth factor withdrawal and inhibition of the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway [156], mitochondrial toxins [157], hypoxia/ischemia [158], glutamate excitotoxicity [159], endoplasmic reticulum stress [160], DNA damage [10], ceramide [161], oxidative stress [162], Alzheimer’s disease-related amyloid β -peptide [163], prion peptide [164], polyglutamine toxicity [165] HIV-associated conditions [166], hypertonic stress [167], and a variety of other conditions. This large and diverse number of conditions in which GSK3 impairs survival and promotes apoptosis has solidly established the conclusion that if the activity of GSK3 is not adequately controlled, then it can be severely detrimental for neural plasticity. This established role of GSK3 in impairing neural plasticity in conditions that can cause cell death can be directly extrapolated to non-lethal, but nevertheless critical, stressful conditions that may occur in psychiatric disorders. Thus, inadequately controlled GSK3 is thought to impair neural plasticity in both lethal and non-lethal conditions, giving rise to impaired responses to stressful conditions, and providing the target for lithium and other therapeutic agents to bolster neural plasticity by inhibiting GSK3.
The mechanisms whereby GSK3 impairs neural plasticity to the extent that it can facilitate apoptosis after exposure of cells to potentially lethal conditions remain to be fully clarified, but several possibilities have been raised. We summarize these mechanisms based on subcellular localization, considering nuclear, mitochondrial, and cytosolic actions.
The fact that GSK3 can promote apoptosis was clearly demonstrated by Pap and Cooper [156] who found that overexpression of GSK3 was sufficient to induce apoptosis. Concerning a mechanism for this action, they showed that this was blocked by expression of dominant-negative p53, suggesting that activation of p53 makes an important contribution to GSK3-induced apoptosis [156]. Subsequently, p53 was shown to bind GSK3 in a mutually synergistic fashion, as p53 activates GSK3 and GSK3 promotes p53-induced apoptosis [10, 134, 135]. As noted above, regulation of gene expression through modulatory effects of GSK3 on many transcription factors likely contributes to its impairment of neural plasticity and also may underlie some of its proapoptotic actions. For example, GSK3β phosphorylates CREB to inhibit its activity [15], and much evidence has shown that CREB promotes cell survival [168]. Therefore, inhibition of CREB by GSK3 would block the anti-apoptotic actions of CREB. The same situation may apply to HSF-1 which promotes cell survival by inducing the expression of heat shock proteins, an action blocked by GSK3 [133]. Interestingly the large neuroprotection provided by lithium from ischemia [169] was associated with increased activation of HSF-1 in vivo [170]. Transcription factor NFAT3 was recently identified as a key target in GSK3-promoted apoptosis in cerebellar granule neurons [171]. Thus, several of the many transcription factors that are targets of GSK3 may contribute to facilitation of apoptosis through regulation of gene expression, and notably, apoptosis can put GSK3 in the right place to regulate transcription factors because several apoptotic conditions cause a rapid accumulation of GSK3 in the nucleus [9].
GSK3 also is present in mitochondria, which is noteworthy because of the central role of mitochondria in cellular responses to stress, as well as its central role in apoptosis. Although little is known about the possibility that mitochondria also may be directly involved in the apoptotic actions of GSK3, it is notable that GSK3 in the mitochondria is activated by some apoptotic stimuli, such as DNA damage and endoplasmic reticulum stress [11]. GSK3’s apoptotic action is known to be upstream of caspase-3 [172], of caspase-9 [172], of cytochrome c release from mitochondria [134], of the permeability transition pore complex in mitochondria [173], and of activation of the proapoptotic bcl2-family member Bax, which targets to mitochondria [174, 175]. Thus, GSK3 in mitochondria, or targeting proteins that act on mitochondria such as Bax [175], may contribute to the facilitative effect of GSK3 on apoptosis.
Finally, cytosolic targets also may be important in the actions of GSK3 that impair neural plasticity and in lethal conditions can lead to cell death by apoptosis. Pap and Cooper [176] showed that inhibition of protein synthesis, which GSK3 achieves by phosphorylating and inhibiting eIF2B, is an important component of GSK3’s proapoptotic action. GSK3 also can promote apoptotic signaling pathways, such as activation of the c-Jun NH2-terminal kinase pathway [177, 178]. And as mentioned previously, actions of GSK3 that modulate microtubule dynamics and kinesin-mediated intra-cellular transport can impair neural plasticity in a manner that could promote cell death.
Overall, it is evident that following exposure to stressful conditions GSK3 can have multiple effects that could impair neural plasticity and in potentially lethal conditions could facilitate the apoptotic process. These include actions of GSK3 that both contribute to apoptosis and actions that block anti-apoptotic processes. Thus much evidence has shown that GSK3 promotes the signaling activity of mitochondria-mediated apoptosis that is induced by many insults. An important corollary to this hypothesis is that the neuro-protective properties of lithium can stem from its inhibition of GSK3, as has been demonstrated [172], allowing lithium to have the broad neuroprotective properties that have been observed. Although a single specific target of GSK3 that accounts for its promotion of apoptosis may yet be found, currently it appears that GSK3 has multiple actions that together serve to facilitate apoptosis. These include actions in the nucleus to inhibit the expression of anti-apoptotic proteins and promote p53-dependent apoptosis, in the mitochondria to impair mitochondrial function and impede energy production, and in the cytosol, such as inhibiting protein synthesis and destabilizing microtubules and other cytoskeletal structures,. Taken together, these actions make GSK3 a powerful agent for contributing to impaired neural plasticity which in extreme conditions can facilitate the apoptosis process.
CONCLUSIONS
GSK3 has a pervasive influence on neuronal function, affecting structure, remodelling, gene expression, survival, and many other aspects of cellular operations. Therefore, the activity of GSK3 is tightly controlled in a substrate-specific manner to allow modulation of select actions of GSK3 by many signaling pathways that converge upon it. As would be expected of such a multi-functional enzyme, recent studies have found that the activities of several neurotransmitter systems regulate GSK3 in the brain in a receptor subtype-selective manner. For example, 5HT1A receptor stimulation promotes inhibitory serine-phosphorylation of GSK3, whereas stimulation of 5HT2 receptors does the opposite, promoting dephosphorylation and activation of GSK3. Strong regulatory influences on GSK3 exerted by neurotransmitter systems that are involved in mood disorders and schizophrenia, especially the serotonergic and dopaminergic systems respectively, support the postulate that abnormalities in the activities of these neurotransmitters in these diseases likely are associated with abnormal control of GSK3. Taken in conjunction with the strong evidence that therapeutic agents used especially in mood disorders but also in schizophrenia regulate GSK3, this mounting evidence supports the conclusion that abnormally regulated GSK3 is associated with mood disorders and possibly schizophrenia. Thus, GSK3 and its signaling partners, both upstream and downstream of GSK3, provide potential targets for new therapeutic agents.
Acknowledgments
We gratefully acknowledge the research and discussions contributed by members of our laboratory, and the support of the National Institutes of Health (MH38752, AG021045) for research in the authors’ laboratory.
References
- 1.Klein PS, Melton DA. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jope RS. Mol Psychiat. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 3.Jope RS. Clinical Neuroscience Res. 2004;4:171–179. [Google Scholar]
- 4.Cole A, Frame S, Cohen P. Biochem J. 2004;377:249–255. doi: 10.1042/BJ20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jope RS, Johnson GVW. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Ferkey DM, Kimelman D. Dev Biol. 2000;225:471–479. doi: 10.1006/dbio.2000.9816. [DOI] [PubMed] [Google Scholar]
- 7.Yanfeng W, Saint-Jeannet JP, Klein PS. Bioessays. 2003;25:317–325. doi: 10.1002/bies.10255. [DOI] [PubMed] [Google Scholar]
- 8.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bijur GN, Jope RS. J Biol Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GVW, Jope RS. Proc Natl Acad Sci USA. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bijur GN, Jope RS. J Neurochem. 2003;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eldar-Finkelman H. Trends Mol Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- 13.Martinez A, Castro A, Dorronsoro I, Alonso M. Med Res Rev. 2002;22:373–384. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- 14.Alonso M, Martinez A. Curr Med Chem. 2004;11:755–763. doi: 10.2174/0929867043455738. [DOI] [PubMed] [Google Scholar]
- 15.Grimes CA, Jope RS. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Friedman AB, Roh MS, Jope RS. J Neurochem. 2005;92:701–704. doi: 10.1111/j.1471-4159.2004.02898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS. Biol Psychiat. 2005;57:278–286. doi: 10.1016/j.biopsych.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 18.Wong ML, Licinio J. Nat Rev Neurosci. 2001;2:343–351. doi: 10.1038/35072566. [DOI] [PubMed] [Google Scholar]
- 19.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 20.Berns GS, Nemeroff CB. Amer J Med Genet. 2003;123:76–84. doi: 10.1002/ajmg.c.20016. [DOI] [PubMed] [Google Scholar]
- 21.Belmaker RH. New Engl J Med. 2004;351:476–486. doi: 10.1056/NEJMra035354. [DOI] [PubMed] [Google Scholar]
- 22.Cryan JF, Markou A, Lucki I. Trends Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- 23.Phiel CJ, Klein PS. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- 24.Stambolic V, Ruel L, Woodgett JR. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 25.Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J. FEBS Lett. 1997;411:183–188. doi: 10.1016/s0014-5793(97)00688-1. [DOI] [PubMed] [Google Scholar]
- 26.Davies SP, Reddy H, Caivano M, Cohen P. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Sarno P, Li X, Jope RS. Neuropharmacol. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 28.Jope RS. Trends Pharmacol Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 29.Chen G, Huang LD, Jiang YM, Manji HK. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- 30.Grimes CA, Jope RS. J Neurochem. 2001;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim AJ, Shi Y, Austin RC, Werstuck GH. J Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- 32.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 33.Roh MS, Kang UG, Shin SY, Lee YH, Jung HY, Juhnn YS, Kim YS. Prog Neuropsychopharmacol Biol Psychiat. 2003;27:1–5. doi: 10.1016/s0278-5846(02)00307-x. [DOI] [PubMed] [Google Scholar]
- 34.Bauer M, Adli M, Baethge C, Berghofer A, Sasse J, Heinz A, Bschor T. Canad J Psychiat. 2003;48:440–448. doi: 10.1177/070674370304800703. [DOI] [PubMed] [Google Scholar]
- 35.Kennedy SH, Lam RW. Bipolar Disord. 2003;5(Suppl 2):36–47. doi: 10.1111/j.1399-2406.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. Neuropsychopharmacol. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borsini F. Pharmacol Res. 1994;30:1–11. doi: 10.1016/1043-6618(94)80082-0. [DOI] [PubMed] [Google Scholar]
- 38.Berendsen HH. Pharmacol Ther. 1995;66:17–37. doi: 10.1016/0163-7258(94)00075-e. [DOI] [PubMed] [Google Scholar]
- 39.Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Proc Natl Acad Sci USA. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stockmeier CA. J Psychiatr Res. 2003;37:357–373. doi: 10.1016/s0022-3956(03)00050-5. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Gray TS, D’Souza DN, Carrasco GA, Damjanoska KJ, Dudas B, Garcia F, Zainelli GM, Sullivan Hanley NR, Battaglia G, Muma NA, Van de Kar LD. J Pharmacol Exp Ther. 2004;310:59–66. doi: 10.1124/jpet.103.062224. [DOI] [PubMed] [Google Scholar]
- 42.Goode N, Hughes K, Woodgett JR, Parker PJ. J Biol Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- 43.Tsujio I, Tanaka T, Kudo T, Nishikawa T, Shinozaki K, Grundke-Iqbal I, Iqbal K, Takeda M. FEBS Lett. 2000;469:111–117. doi: 10.1016/s0014-5793(00)01234-5. [DOI] [PubMed] [Google Scholar]
- 44.Cowen DS, Sowers RS, Manning DR. J Biol Chem. 1996;271:22297–22300. doi: 10.1074/jbc.271.37.22297. [DOI] [PubMed] [Google Scholar]
- 45.Hsiung SC, Adlersberg M, Arango V, Mann JJ, Tamir H, Liu KP. J Neurochem. 2003;87:182–194. doi: 10.1046/j.1471-4159.2003.01987.x. [DOI] [PubMed] [Google Scholar]
- 46.Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Biol Psychiat. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 47.O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, Klein PS. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gould TD, Einat H, Bhat R, Manji HK. Int J Neuropsychopharmacol. 2004;7:387–390. doi: 10.1017/S1461145704004535. [DOI] [PubMed] [Google Scholar]
- 49.Mueser KT, McGurk SR. Lancet. 2004;363:2063–2072. doi: 10.1016/S0140-6736(04)16458-1. [DOI] [PubMed] [Google Scholar]
- 50.Tamminga CA, Holcomb HH. Mol Psychiat. 2005;10:27–39. doi: 10.1038/sj.mp.4001563. [DOI] [PubMed] [Google Scholar]
- 51.Laruelle M, Kegeles LS, Abi-Dargham A. Ann N Y Acad Sci. 2003;1003:138–158. doi: 10.1196/annals.1300.063. [DOI] [PubMed] [Google Scholar]
- 52.Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Psychopharmacology (Berl) 2004;174:3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- 53.Gessa GL, Devoto P, Diana M, Flore G, Melis M, Pistis M. Neuropsychopharmacol. 2000;22:642–649. doi: 10.1016/S0893-133X(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 54.Meltzer HY, McGurk SR. Schizophr Bull. 1999;25:233–255. doi: 10.1093/oxfordjournals.schbul.a033376. [DOI] [PubMed] [Google Scholar]
- 55.Purdon SE, Jones BD, Stip E, Labelle A, Addington D, David SR, Breier A, Tollefson GD. Arch Gen Psychiatry. 2000;57:249–258. doi: 10.1001/archpsyc.57.3.249. [DOI] [PubMed] [Google Scholar]
- 56.Okubo Y, Suhara T, Suzuki K, Kobayashi K, Inoue O, Terasaki O, Someya Y, Sassa T, Sudo Y, Matsushima E, Iyo M, Tateno Y, Toru M. Nature. 1997;385:634–636. doi: 10.1038/385634a0. [DOI] [PubMed] [Google Scholar]
- 57.Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, Hwang DR, Keilp J, Kochan L, Van Heertum R, Gorman JM, Laruelle M. J Neurosci. 2002;22:3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abi-Dargham A, Laruelle M. Eur Psychiat. 2005;20:15–27. doi: 10.1016/j.eurpsy.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 59.Kozlovsky N, Belmaker RH, Agam G. Amer J Psychiat. 2000;157:831–833. doi: 10.1176/appi.ajp.157.5.831. [DOI] [PubMed] [Google Scholar]
- 60.Kozlovsky N, Belmaker RH, Agam G. Schizophr Res. 2001;52:101–105. doi: 10.1016/s0920-9964(00)00174-2. [DOI] [PubMed] [Google Scholar]
- 61.Kozlovsky N, Regenold WT, Levine J, Rapoport A, Belmaker RH, Agam G. J Neural Transm. 2004;111:1093–1098. doi: 10.1007/s00702-003-0127-0. [DOI] [PubMed] [Google Scholar]
- 62.Kozlovsky N, Nadri C, Agam G. Eur Neuropsychopharmacol. 2005;15:1–11. doi: 10.1016/j.euroneuro.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 63.Nadri C, Dean B, Scarr E, Agam G. Schizophr Res. 2004;71:377–382. doi: 10.1016/j.schres.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 64.Beasley C, Cotter D, Khan N, Pollard C, Sheppard P, Varndell I, Lovestone S, Anderton B, Everall I. Neurosci Lett. 2001;302:117–120. doi: 10.1016/s0304-3940(01)01688-3. [DOI] [PubMed] [Google Scholar]
- 65.Beasley C, Cotter D, Everall I. Schizophr Res. 2002;58:63–67. doi: 10.1016/s0920-9964(01)00376-0. [DOI] [PubMed] [Google Scholar]
- 66.Gil M, Zhen X, Friedman E. Neurosci Lett. 2003;349:143–146. doi: 10.1016/s0304-3940(03)00852-8. [DOI] [PubMed] [Google Scholar]
- 67.Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Proc Natl Acad Sci USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 69.Ikeda M, Iwata N, Suzuki T, Kitajima T, Yamanouchi Y, Kinoshita Y, Inada T, Ozaki N. Biol Psychiat. 2004;56:698–700. doi: 10.1016/j.biopsych.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 70.Ohtsuki T, Inada T, Arinami T. Mol Psychiat. 2004;9:981–983. doi: 10.1038/sj.mp.4001559. [DOI] [PubMed] [Google Scholar]
- 71.Cotter D, Kerwin R, al-Sarraji S, Brion JP, Chadwich A, Lovestone S, Anderton B, Everall I. Neuroreport. 1998;9:1379–1383. doi: 10.1097/00001756-199805110-00024. [DOI] [PubMed] [Google Scholar]
- 72.Miyaoka T, Seno H, Ishino H. Schizophr Res. 1999;38:1–6. doi: 10.1016/s0920-9964(98)00179-0. [DOI] [PubMed] [Google Scholar]
- 73.Katsu T, Ujike H, Nakano T, Tanaka Y, Nomura A, Nakata K, Takaki M, Sakai A, Uchida N, Imamura T, Kuroda S. Neurosci Lett. 2003;353:53–56. doi: 10.1016/j.neulet.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Yu X, Yuan Y, Ling Y, Ruan Y, Si T, Lu T, Wu S, Gong X, Zhu Z, Yang J, Wang F, Zhang D. Am J Med Genet. 2004;129:16–19. doi: 10.1002/ajmg.b.30076. [DOI] [PubMed] [Google Scholar]
- 75.Marenco S, Weinberger DR. Dev Psychopathol. 2000;12:501–527. doi: 10.1017/s0954579400003138. [DOI] [PubMed] [Google Scholar]
- 76.Kozlovsky N, Belmaker RH, Agam G. Eur Neuropsychopharmacol. 2002;12:13–25. doi: 10.1016/s0924-977x(01)00131-6. [DOI] [PubMed] [Google Scholar]
- 77.McGrath JJ, Feron FP, Burne TH, Mackay-Sim A, Eyles DW. Ann Med. 2003;35:86–93. doi: 10.1080/07853890310010005. [DOI] [PubMed] [Google Scholar]
- 78.Tandon R. Br J Psychiatry Suppl. 1999;37:7–11. [PubMed] [Google Scholar]
- 79.Friedman JI. Psychopharmacology (Berl) 2004;174:45–53. doi: 10.1007/s00213-004-1794-x. [DOI] [PubMed] [Google Scholar]
- 80.Wu D, Hersh LB. J Neurochem. 1994;62:1653–1663. doi: 10.1046/j.1471-4159.1994.62051653.x. [DOI] [PubMed] [Google Scholar]
- 81.Powchik P, Davidson M, Haroutunian V, Gabriel SM, Purohit DP, Perl DP, Harvey PD, Davis KL. Schizophr Bull. 1998;24:325–341. doi: 10.1093/oxfordjournals.schbul.a033330. [DOI] [PubMed] [Google Scholar]
- 82.Holt DJ, Herman MM, Hyde TM, Kleinman JE, Sinton CM, German DC, Hersh LB, Graybiel AM, Saper CB. Neuroscience. 1999;94:21–31. doi: 10.1016/s0306-4522(99)00279-1. [DOI] [PubMed] [Google Scholar]
- 83.Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B. Amer J Psychiat. 2001;158:918–925. doi: 10.1176/appi.ajp.158.6.918. [DOI] [PubMed] [Google Scholar]
- 84.Mancama D, Arranz MJ, Landau S, Kerwin R. Am J Med Genet B Neuropsychiatr Genet. 2003;119:2–6. doi: 10.1002/ajmg.b.20020. [DOI] [PubMed] [Google Scholar]
- 85.Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, Egan MF, Coppola R, Weinberger DR. Am J Psychiat. 2003;160:118–127. doi: 10.1176/appi.ajp.160.1.118. [DOI] [PubMed] [Google Scholar]
- 86.Katerina Z, Andrew K, Filomena M, Xu-Feng H. Neuropsychopharmacology. 2004;29:619–625. [Google Scholar]
- 87.Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B. Biol Psychiat. 2000;48:381–388. doi: 10.1016/s0006-3223(00)00918-5. [DOI] [PubMed] [Google Scholar]
- 88.Dean B, McLeod M, Keriakous D, McKenzie J, Scarr E. Mol Psychiat. 2002;7:1083–1091. doi: 10.1038/sj.mp.4001199. [DOI] [PubMed] [Google Scholar]
- 89.Dean B, Crook JM, Opeskin K, Hill C, Keks N, Copolov DL. Mol Psychiat. 1996;1:54–58. [PubMed] [Google Scholar]
- 90.De Sarno P, Bijur GN, Zmijewska AA, Li X, Jope RS. Neurobiol Aging. 2005 doi: 10.1016/j.neurobiolaging.2005.03.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. Psychopharmacology (Berl) 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- 92.Abi-Saab WM, D’Souza DC, Moghaddam B, Krystal JH. Pharmacopsychiatry. 1998;31:104–109. doi: 10.1055/s-2007-979354. [DOI] [PubMed] [Google Scholar]
- 93.Millan MJ. Curr Drug Targets CNS Neurol Disord. 2002;1:191–213. doi: 10.2174/1568007024606258. [DOI] [PubMed] [Google Scholar]
- 94.Luo HR, Hattori H, Hossain MA, Hester L, Huang Y, Lee-Kwon W, Donowitz M, Nagata E, Snyder SH. Proc Natl Acad Sci USA. 2003;100:11712–11717. doi: 10.1073/pnas.1634990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M, McKinzie DL, Fienberg AA, Nomikos GG, Greengard P. Science. 2003;302:1412–1415. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- 96.Elyaman W, Terro F, Wong NS, Hugon J. Eur J Neurosci. 2002;15:651–660. doi: 10.1046/j.1460-9568.2002.01899.x. [DOI] [PubMed] [Google Scholar]
- 97.Hong M, Chen DC, Klein PS, Lee VM. J Biol Chem. 1997;272:25326–2532. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 98.Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC. J Cell Sci. 1998;111:1351–1361. doi: 10.1242/jcs.111.10.1351. [DOI] [PubMed] [Google Scholar]
- 99.Goold RG, Owen R, Gordon-Weeks PR. J Cell Sci. 1999;112:3373–3384. doi: 10.1242/jcs.112.19.3373. [DOI] [PubMed] [Google Scholar]
- 100.Lovestone S, Davis DR, Webster MT, Kaech S, Brion JP, Matus A, Anderton BH. Biol Psychiat. 1999;45:995–1003. doi: 10.1016/s0006-3223(98)00183-8. [DOI] [PubMed] [Google Scholar]
- 101.Hall AC, Lucas FR, Salinas PC. Cell. 2000;100:525–535. doi: 10.1016/s0092-8674(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 102.Sanchez C, Diaz-Nido J, Avila J. Prog Neurobiol. 2000;61:133–168. doi: 10.1016/s0301-0082(99)00046-5. [DOI] [PubMed] [Google Scholar]
- 103.Owen R, Gordon-Weeks PR. Mol Cell Neurosci. 2003;23:626–637. doi: 10.1016/s1044-7431(03)00095-2. [DOI] [PubMed] [Google Scholar]
- 104.Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eickholt BJ, Walsh FS, Doherty P. J Cell Biol. 2002;157:211–217. doi: 10.1083/jcb.200201098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sayas CL, Avila J, Wandosell F. Biochim Biophys Acta. 2002;1582:144–153. doi: 10.1016/s1388-1981(02)00149-x. [DOI] [PubMed] [Google Scholar]
- 108.Etienne-Manneville S, Hall A. Nature. 2003;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- 109.Price MA, Kalderon D. Cell. 2002;108:823–835. doi: 10.1016/s0092-8674(02)00664-5. [DOI] [PubMed] [Google Scholar]
- 110.Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J. Nature. 2002;416:548–552. doi: 10.1038/nature733. [DOI] [PubMed] [Google Scholar]
- 111.Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J. J Biol Chem. 2002;277:49958–49964. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- 112.Foltz DR, Santiago MC, Berechid BE, Nye JS. Curr Biol. 2002;12:1006–1011. doi: 10.1016/s0960-9822(02)00888-6. [DOI] [PubMed] [Google Scholar]
- 113.Jacobs BL, Praag H, Gage FH. Mol Psychiat. 2000;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- 114.Duman RS, Malberg J, Nakagawa S, D’Sa C. Biol Psychiat. 2000;48:732–739. doi: 10.1016/s0006-3223(00)00935-5. [DOI] [PubMed] [Google Scholar]
- 115.Kempermann G. Bipolar Disord. 2002;4:17–33. doi: 10.1034/j.1399-5618.2002.40101.x. [DOI] [PubMed] [Google Scholar]
- 116.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Manev R, Uz T, Smalheiser NR, Manev H. Eur J Pharmacol. 2001;411:67–70. doi: 10.1016/s0014-2999(00)00904-3. [DOI] [PubMed] [Google Scholar]
- 118.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 119.Scott BW, Wojtowicz JM, Burnham WM. Exp Neurol. 2000;165:231–236. doi: 10.1006/exnr.2000.7458. [DOI] [PubMed] [Google Scholar]
- 120.Madsen TM, Yeh DD, Valentine GW, Duman RS. Neuropsychopharmacology. 2005;30:27–34. doi: 10.1038/sj.npp.1300565. [DOI] [PubMed] [Google Scholar]
- 121.Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. J Neurochem. 2000;75:1729–1734. doi: 10.1046/j.1471-4159.2000.0751729.x. [DOI] [PubMed] [Google Scholar]
- 122.Hashimoto R, Senatorov V, Kanai H, Leeds P, Chuang DM. Neuroscience. 2003;117:55–61. doi: 10.1016/s0306-4522(02)00577-8. [DOI] [PubMed] [Google Scholar]
- 123.Shimomura A, Nomura R, Senda T. Neuroreport. 2003;14:1779–1782. doi: 10.1097/00001756-200310060-00004. [DOI] [PubMed] [Google Scholar]
- 124.Kim JS, Chang MY, Yu IT, Kim JH, Lee SH, Lee YS, Son H. J Neurochem. 2004;89:324–336. doi: 10.1046/j.1471-4159.2004.02329.x. [DOI] [PubMed] [Google Scholar]
- 125.Wu J, Saint-Jeannet JP, Klein PS. Trends Neurosci. 2003;26:40–45. doi: 10.1016/s0166-2236(02)00011-5. [DOI] [PubMed] [Google Scholar]
- 126.Aubert J, Dunstan H, Chambers I, Smith A. Nat Biotechnol. 2002;20:1240–1245. doi: 10.1038/nbt763. [DOI] [PubMed] [Google Scholar]
- 127.Ding S, Wu TY, Brinker A, Peters EC, Hur W, Gray NS, Schultz PG. Proc Natl Acad Sci USA. 2003;100:7632–7637. doi: 10.1073/pnas.0732087100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- 129.Gould E. Neuropsychopharmacology. 1999;21:46S–51S. doi: 10.1016/S0893-133X(99)00045-7. [DOI] [PubMed] [Google Scholar]
- 130.Brezun JM, Daszuta A. Neuroscience. 1999;89:999–1002. doi: 10.1016/s0306-4522(98)00693-9. [DOI] [PubMed] [Google Scholar]
- 131.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Science. 1997;275:1930–1933. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 132.Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK. J Biol Chem. 1996;271:30847–30857. doi: 10.1074/jbc.271.48.30847. [DOI] [PubMed] [Google Scholar]
- 133.Bijur GN, Jope RS. J Neurochem. 2000;75:2401–2408. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- 134.Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. J Biol Chem. 2003;278:48872–48879. doi: 10.1074/jbc.M305870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Ruiz-Ruiz C, Cadoret A, Capeau J, Desbois-Mouthon C. Exp Cell Res. 2004;300:354–364. doi: 10.1016/j.yexcr.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 136.Bullock BP, Habener JF. Biochemistry. 1998;37:3795–3809. doi: 10.1021/bi970982t. [DOI] [PubMed] [Google Scholar]
- 137.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 138.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 139.Duman RS. Eur Psychiat. 2002;17:306–310. doi: 10.1016/s0924-9338(02)00654-5. [DOI] [PubMed] [Google Scholar]
- 140.Hashimoto K, Shimizu E, Iyo M. Brain Res Rev. 2004;45:104–114. doi: 10.1016/j.brainresrev.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 141.Smith MA, Makino S, Kvetnansky R, Post RM. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Roceri M, Hendriks W, Racagni G, Ellenbroek BA, Riva MA. Mol Psychiat. 2002;7:609–616. doi: 10.1038/sj.mp.4001036. [DOI] [PubMed] [Google Scholar]
- 143.Nibuya M, Morinobu S, Duman RS. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nibuya M, Nestler EJ, Duman RS. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Russo-Neustadt AA, Beard RC, Huang YM, Cotman CW. Neuroscience. 2000;101:305–312. doi: 10.1016/s0306-4522(00)00349-3. [DOI] [PubMed] [Google Scholar]
- 146.Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM. Pharmacol Biochem Behav. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 147.Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry JM. Psychiat Res. 2002;109:143–148. doi: 10.1016/s0165-1781(02)00005-7. [DOI] [PubMed] [Google Scholar]
- 149.Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, Nakazato M, Watanabe H, Shinoda N, Okada S, Iyo M. Biol Psychiat. 2003;54:70–75. doi: 10.1016/s0006-3223(03)00181-1. [DOI] [PubMed] [Google Scholar]
- 150.Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Biol Psychiat. 2001;50:260–265. doi: 10.1016/s0006-3223(01)01083-6. [DOI] [PubMed] [Google Scholar]
- 151.Jacobsen JP, Mork A. Brain Res. 2004;1024:183–192. doi: 10.1016/j.brainres.2004.07.065. [DOI] [PubMed] [Google Scholar]
- 152.Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Psychopharmacology (Berl) 2001;158:100–106. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- 153.Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM. Neuropharmacol. 2002;43:1173–1179. doi: 10.1016/s0028-3908(02)00217-4. [DOI] [PubMed] [Google Scholar]
- 154.Angelucci F, Aloe L, Jimenez-Vasquez P, Mathe AA. Int J Neuropsychopharmacol. 2003;6:225–231. doi: 10.1017/S1461145703003468. [DOI] [PubMed] [Google Scholar]
- 155.Mai L, Jope RS, Li X. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- 156.Pap M, Cooper GM. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 157.King TD, Bijur GN, Jope RS. Brain Res. 2001;919:106–114. doi: 10.1016/s0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- 158.Loberg RD, Vesely E, Brosius FC., 3rd J Biol Chem. 2002;277:41667–41673. doi: 10.1074/jbc.M206405200. [DOI] [PubMed] [Google Scholar]
- 159.Hashimoto M, Sagara Y, Langford D, Everall IP, Mallory M, Everson A, Digicaylioglu M, Masliah E. J Biol Chem. 2002;277:32985–32991. doi: 10.1074/jbc.M202803200. [DOI] [PubMed] [Google Scholar]
- 160.Song L, De Sarno P, Jope RS. J Biol Chem. 2002;277:44701–44708. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- 161.Mora A, Sabio G, Risco AM, Cuenda A, Alonso JC, Soler G, Centeno F. Cell Signal. 2002;14:557–562. doi: 10.1016/s0898-6568(01)00282-0. [DOI] [PubMed] [Google Scholar]
- 162.Shin SY, Kim CG, Jho EH, Rho MS, Kim YS, Kim YH, Lee YH. Cancer Lett. 2004;212:225–231. doi: 10.1016/j.canlet.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 163.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Proc Natl Acad Sci USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Perez M, Rojo AI, Wandosell F, Diaz-Nido J, Avila J. Biochem J. 2003;372:129–136. doi: 10.1042/BJ20021596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. J Biol Chem. 2002;277:33791–33798. doi: 10.1074/jbc.M204861200. [DOI] [PubMed] [Google Scholar]
- 166.Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. J Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- 167.Rao R, Hao CM, Breyer MD. J Biol Chem. 2004;279:3949–3955. doi: 10.1074/jbc.M309325200. [DOI] [PubMed] [Google Scholar]
- 168.Lonze BE, Ginty DD. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 169.Nonaka S, Chuang DM. Neuroreport. 1998;9:2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- 170.Ren M, Senatorov VV, Chen RW, Chuang DM. Proc Natl Acad Sci USA. 2003;100:6210–6215. doi: 10.1073/pnas.0937423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirch-hausen T, Rao A, Bonni A. J Biol Chem. 2005;280:2818–2825. doi: 10.1074/jbc.M408741200. [DOI] [PubMed] [Google Scholar]
- 172.Bijur GN, De Sarno P, Jope RS. J Biol Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- 173.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Somervaille TC, Linch DC, Khwaja A. Blood. 2001;98:1374–1381. doi: 10.1182/blood.v98.5.1374. [DOI] [PubMed] [Google Scholar]
- 175.Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pap M, Cooper GM. Mol Cell Biol. 2002;22:578–586. doi: 10.1128/MCB.22.2.578-586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kim JW, Lee JE, Kim MJ, Cho EG, Cho SG, Choi EJ. J Biol Chem. 2003;278:13995–4001. doi: 10.1074/jbc.M300253200. [DOI] [PubMed] [Google Scholar]
- 178.Hongisto V, Smeds N, Brecht S, Herdegen T, Courtney MJ, Coffey ET. Mol Cell Biol. 2003;23:6027–6036. doi: 10.1128/MCB.23.17.6027-6036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]