

Abstract
In 1998, an outbreak of acute encephalitis with high mortality rates among pig handlers in Malaysia led to the discovery of a novel paramyxovirus named Nipah virus. A multidisciplinary investigation that included epidemiology, microbiology, molecular biology, and pathology was pivotal in the discovery of this new human infection. Clinical and autopsy findings were derived from a series of 32 fatal human cases of Nipah virus infection. Diagnosis was established in all cases by a combination of immunohistochemistry (IHC) and serology. Routine histological stains, IHC, and electron microscopy were used to examine autopsy tissues. The main histopathological findings included a systemic vasculitis with extensive thrombosis and parenchymal necrosis, particularly in the central nervous system. Endothelial cell damage, necrosis, and syncytial giant cell formation were seen in affected vessels. Characteristic viral inclusions were seen by light and electron microscopy. IHC analysis showed widespread presence of Nipah virus antigens in endothelial and smooth muscle cells of blood vessels. Abundant viral antigens were also seen in various parenchymal cells, particularly in neurons. Infection of endothelial cells and neurons as well as vasculitis and thrombosis seem to be critical to the pathogenesis of this new human disease.
An outbreak of a previously unrecognized paramyxovirus infection that caused an encephalitic syndrome mainly among pig handlers occurred in Malaysia and Singapore in late 1998 and early 1999. 1,2 In Malaysia, the outbreak seemed to have started around pig farms in Ipoh, in the state of Perak, and was spread by the movement of sick pigs to a second epicenter ∼160 miles south in the state of Negri Sembilan. 1 Later, the infection spread to workers in an abattoir in Singapore, where pigs originating from Negri Sembilan were held and slaughtered. 3,4 There were 105 deaths among 265 reported cases of encephalitis, a mortality rate of nearly 40%. 5 Most patients presented with a severe acute encephalitic syndrome, but some also had significant pulmonary manifestations. 3,6,7 A syncytium-forming virus was isolated from the cerebrospinal fluid (CSF) of several patients. Electron microscopy (EM) examination revealed an enveloped virus with filamentous nucleocapsids, which when negatively stained showed a herringbone structure, characteristic of the family Paramyxoviridae. 8 Reactivity of infected culture cells and tissues from fatal cases with anti-Hendra antibodies by immunofluorescence and immunohistochemistry (IHC) as well as detection of anti-Hendra IgM antibodies in the serum and CSF suggested the possibility of Hendra or Hendra-like virus infection. 1 Preliminary autopsy findings showed that the central nervous system (CNS) seemed to be a major target. 2,3,8 Viral genomic sequencing provided evidence that this previously unknown virus is related to, but distinct from, Hendra virus. 8,9 The virus was subsequently named Nipah virus after Kampung Sungai Nipah (Nipah River Village), where the first viral isolates were obtained. 7,10
This study is based on the clinical and autopsy findings for 32 patients who died of Nipah virus infection. We examined the relative usefulness of various laboratory tests, including IHC, serology, and virus isolation, for the diagnosis of this emerging infectious disease. We also describe the pathological findings, including EM and tissue immunolocalization of viral antigens, and discuss the pathogenesis of Nipah virus infection based on these findings.
Materials and Methods
Demographic and Clinical Data
A series of 32 fatal cases of Nipah infection, which comprised all of the cases autopsied from late 1998 to mid-1999, was drawn from five hospitals in Malaysia. Fifteen cases were from the Seremban hospital (Table 1 ▶ , cases 1 to 15); three from the Kuala Lumpur hospital (cases 16 to 18); nine from the Ipoh hospital (cases 19 to 27); four from the University of Malaya Medical Center, Kuala Lumpur (cases 28 to 31); and one from the Kelang hospital (case 32). Medical records from the various hospitals were systematically reviewed, and demographic, clinical, and other data were extracted.
Table 1.
Thirty-Two Fatal Cases of Nipah Virus Infection: Available Clinical and Laboratory Data
| Case no. | Age (year/sex) | Prodrome (no. days) | Total duration of illness (no. days) | Serology | Virus isolation | IHC | |||
|---|---|---|---|---|---|---|---|---|---|
| CSF | Serum | ||||||||
| IgM | IgG | IgM | IgG | ||||||
| 1 | 41/M | 3 | 10 | nd | nd | + | − | + | + |
| 2 | 52/M | 6 | 8 | − | − | + | − | + | + |
| 3 | 24/M | 5 | 7 | − | − | + | − | + | + |
| 4 | 65/M | 2 | 6 | − | − | + | − | nd | + |
| 5 | 22/M | 4 | 8 | nd | nd | + | − | + | + |
| 6 | 27/M | 4 | 10 | nd | nd | + | − | nd | + |
| 7 | 54/M | 1 | 2 | + | − | + | − | nd | + |
| 8 | 30/M | 4 | 6 | + | − | + | − | nd | + |
| 9 | 42/M | 5 | 6 | nd | nd | + | − | + | + |
| 10 | 46/M | 1 | 6 | + | − | + | + | nd | + |
| 11 | 20/M | 4 | 11 | + | − | + | + | nd | + |
| 12 | 71/M | 3 | 7 | + | − | + | + | nd | + |
| 13 | 49/M | 2 | 7 | + | − | + | − | nd | + |
| 14 | 44/M | 5 | 14 | + | − | nd | nd | nd | − |
| 15 | 13/F | 2 | 5 | nd | nd | nd | nd | nd | + |
| 16 | 36/M | 3 | 6 | − | − | nd | nd | nd | + |
| 17 | 50/F | 3 | 3 | − | − | + | − | nd | + |
| 18 | 49/F | 3 | 7 | nd | nd | + | − | nd | + |
| 19 | 54/M | 4 | 31 | nd | nd | + | + | nd | − |
| 20 | 39/M | 3 | 17 | nd | nd | + | + | nd | − |
| 21 | 46/M | 2 | 3 | nd | nd | + | − | nd | + |
| 22 | 37/M | 4 | 7 | nd | nd | + | − | nd | + |
| 23 | 53/M | 1 | 3 | nd | nd | + | − | nd | + |
| 24 | 51/M | 7 | 9 | + | + | + | − | nd | + |
| 25 | 29/M | 3 | 8 | nd | nd | + | − | nd | + |
| 26 | 55/M | 2 | 25 | + | + | + | − | nd | + |
| 27 | 75/M | 2 | 4 | nd | nd | − | − | nd | + |
| 28 | 51/M | 3 | 7 | + | − | + | − | + | + |
| 29 | 52/M | 7 | 11 | + | + | + | − | + | + |
| 30 | 34/M | 3 | 8 | + | − | − | − | nd | + |
| 31 | 31/M | 2 | 34 | + | + | − | − | − | − |
| 32 | 26/M | * | * | nd | nd | − | + | nd | + |
Positive, +; negative, −; nd, not done.
Prodrome, onset of fever to admission. Total duration of illness, onset of fever to death.
*Patient with relapse encephalitis who had onset of symptoms a few months before last hospital admission. Duration of illness for admission was 11 days.
Autopsy Tissues
Of the 32 autopsies, 29 were full autopsies and 3 were limited to the brain. Tissues were fixed in 10% buffered formalin, paraffin-embedded, and stained with hematoxylin and eosin (H&E).
Antibodies, Cell and Tissue Controls
The antibody initially used for Nipah viral antigen detection was anti-Hendra hyperimmune mouse ascitic fluid [Centers for Disease Control and Prevention (CDC), Atlanta, GA]. Subsequently, hyperimmune mouse ascitic fluid against Nipah virus was generated at the CDC and reactivity was compared with that of the mouse anti-Hendra antibody. The specificity and sensitivity of these antibodies in the IHC analyses were tested by using Vero E6 cells (Vero clone CRL 1586; American Type Culture Collection, Rockville, MD) that were uninfected or infected with Hendra or Nipah virus. Antibody specificities were further confirmed by testing specimens from patients with Japanese encephalitis (JE), measles, eastern equine encephalitis, enterovirus71, or influenza, and tissue culture cells infected with western equine encephalitis virus, measles virus, or La Crosse encephalitis viruses.
Negative antibody controls for each slide in the IHC analysis included replacing primary antibody with normal mouse ascitic fluid or with the primary antibody absorbed with Nipah virus antigens. Because there was a high initial suspicion of JE, specimens from all patients were also stained for JE viral antigens by using a cross-reactive anti-flavivirus antibody.
IHC
Tissue blocks were chosen for IHC analysis after slides of the H&E-stained specimens were reviewed. The IHC was based on a method described previously for hantavirus. 11 Briefly, 4-μm sections were deparaffinized and rehydrated through graded alcohol and distilled water. They were predigested by 0.1 mg/ml of proteinase K (Boehringer-Mannheim Corp., Indianapolis, IN) in 0.6 mol/L of Tris (pH 7.5)/0.1 CaCl2 for 15 minutes at room temperature and blocked with normal swine serum. Primary antibodies were applied for 1 hour at room temperature. Optimal conditions for primary antibody and digestion conditions were previously determined by titration experiments. For anti-Hendra and anti-Nipah antibodies, the dilutions were 1:4000 and 1:2000, respectively. This step was followed by sequential application of biotinylated link antibody, alkaline phosphatase-conjugated streptavidin, and napthol fast red according to the manufacturer’s protocol (LSAB2 Universal Alkaline Phosphatase Kit; DAKO Corporation, Carpinteria, CA). Sections were then counterstained in Meyer’s hematoxylin (Fisher Scientific, Pittsburgh, PA) and mounted with an aqueous mounting medium (Faramount; DAKO Corporation). Specimens from all cases were tested with anti-Hendra antibody and selected cases with anti-Nipah antibody.
EM
Formalin-fixed brain tissues were postfixed with 1% osmium tetroxide in 0.1 mol/L of phosphate buffer, en bloc stained with 4% aqueous uranyl acetate, dehydrated through a graded series of alcohols and propylene oxide, and embedded in a mixture of epon substitute and Araldite. 12 Ultrathin sections were stained with 4% uranyl acetate and Reynolds’ lead citrate.
Nipah Antibody Detection Assays
IgM and IgG antibodies to Nipah virus in patients were detected by enzyme-linked immunosorbent assay using cross-reactive inactivated Hendra virus antigens. Both tests followed methods previously described for Ebola virus. 13 In brief, the IgM assay was performed in a Mu-capture format and used Hendra antigens obtained from virus-infected γ-irradiated Vero E6 cells and anti-Hendra hyperimmune mouse ascitic fluid antibody. The IgG assay used a detergent-extracted Hendra antigen obtained from infected Vero E6 cells and inactivated by γ irradiation; antigen was adsorbed directly onto microtiter plates. Control IgG and IgM assays were also performed with antigens from mock-infected Vero E6 cells. The IgM-capture assay used goat anti-human Mu to capture IgM (Biosource, Camarilla, CA) and a horseradish peroxidase-conjugated goat anti-mouse IgG (Biosource). The IgG assay used a horseradish peroxidase-conjugated mouse anti-human γ-chain-specific antibody (Accurate Chemical, Westbury, NY) to detect bound IgG.
Sera were tested in a fourfold dilution series from 1:100 to 1:6400, and CSF was similarly tested in a fourfold series from 1:20 to 1:1280. A sample of negative donors was used to validate the cutoff values for the assays. Samples were considered positive for the IgM assay if the sum of the adjusted optical densities from all of the dilutions (infected antigen less the mock-infected antigen) was >0.45 through the entire dilution series and the titer was ≥1:400 (1:16 for CSF). Samples were likewise considered positive in the IgG assay if the sum for the adjusted optical densities from all of the dilutions (infected antigen less the mock-infected antigen) was >0.90 through the entire dilution series and the titer was ≥1:400 (≥1:80 for CSF).
Virus Isolation and Identification
Virus isolation and identification were attempted with specimens from eight patients by use of a previously described method. 14 In brief, 100 μl of CSF was innoculated on previously seeded wells with 105 Vero cells (CCL-81; American Type Culture Collection); and the cells were transferred to 1 ml of Eagle’s minimal essential growth medium containing 10% fetal calf serum (Flowlab, Sydney, Australia). After incubation at 37°C, positive identification of virus was made by immunofluorescence using anti-Hendra hyperimmune mouse ascitic fluid and goat anti-mouse IgG fluorescein-isothiocyanate conjugate (Sigma, St. Louis, MO) secondary antibody.
Results
Demographic and Clinical Features
The age of patients in this study ranged from 13 to 75 years (mean, 43 years; median, 44 years) and the male-to-female ratio was 29:3. The prodrome, defined as the time from fever onset to the day of hospital admission, averaged 3.3 days (range, 1 to 7 days) (Table 1) ▶ . The duration of illness, defined as the time from fever onset to death, averaged 9.5 days (range, 2 to 34 days). Four patients survived >14 days before death. Case 32 had a similar clinical illness a few months earlier, improved, but subsequently relapsed and died.
Clinical symptoms and signs are summarized in Table 2 ▶ . All patients had fever. More than 70% of patients complained of drowsiness, headache, and disorientation or confusion. The most frequent clinical sign among patients was reduced consciousness. Case 31 was recovering in the hospital ward when he developed massive fatal intracerebral hemorrhage.
Table 2.
Frequency of Clinical Symptoms and Signs in 32 Fatal Cases of Nipah Virus Infection
| % | |
|---|---|
| Symptoms | |
| Fever | 100 |
| Drowsiness | 88 |
| Headache | 82 |
| Disorientation/confusion | 76 |
| Giddiness | 61 |
| Myalgia | 54 |
| Cough/Respiratory symptoms | 40 |
| Convulsion | 28 |
| Vomiting | 19 |
| Signs | |
| Reduced consciousness | 89 |
| Segmental myoclonus | 50 |
| Hyporeflexia/areflexia | 50 |
| Seizure | 40 |
| Cranial nerve palsy | 29 |
| Pyramidal signs | 21 |
| Nystagamus/cerebellar signs | 17 |
| Meningism | 10 |
| Dysphasia | 5 |
Pathological Features
The macroscopic features were nonspecific. In the CNS, lesions were generally difficult to identify; however, in a few cases, small, discrete, occasionally hemorrhagic, necrotic lesions were found. Only 2 of 10 brains examined showed unequivocal herniation. Case 29 had cerebellar tonsil herniation, and case 31 had uncal herniation and showed a large intracerebral clot in the frontal lobe with intraventricular extension and Duret hemorrhages in the midbrain and pons. Histopathological changes were seen in the blood vessels and parenchyma of multiple organs and are presented accordingly.
Blood Vessels
The distribution of histopathological lesions and immunostaining is shown in Table 3 ▶ . Extensive involvement of blood vessels in the CNS, lung, heart, and kidney was observed in Nipah virus infection. However, blood vessels in the CNS were the most severely involved. Typically, small arteries, arterioles, capillaries, and venules showed evidence of vasculitis. Vasculitis was not found in medium-sized vessels (eg, renal artery and vein, anterior, and middle cerebral arteries) or large arteries (eg, aorta and pulmonary trunk). No vasculitis was found in the relapse encephalitis case (case 32; Table 1 ▶ ).
Table 3.
Fatal Nipah Virus Infection: Frequency of Necrosis, Vasculitis, and Immunostaining of Viral Antigens in Major Organs
| Pathologic findings | Brain no. (%) | Lung no. (%) | Heart no. (%) | Kidney no. (%) | Spleen no. (%) |
|---|---|---|---|---|---|
| Necrosis* | 28/30 | 17/29 | 1/29 | 10/29 | 10/24 |
| (93) | (59) | (3) | (34) | (42) | |
| Vasculitis | 24/30 | 18/29 | 9/29 | 7/29 | 0/24 |
| (80) | (62) | (31) | (24) | (0) | |
| Viral antigens | 27/32 | 7/29 | 4/24 | 6/25 | 1/21 |
| (84) | (24) | (17) | (24) | (5) |
*Necrosis was in the form of parenchymal necrotic plaques in brain, fibrinoid alveolar necrosis in lung, and fibrinoid glomerular necrosis in kidney. In the spleen, acute necrotic inflammation was found in the area of the periarteriolar lymphoid sheath.
Note: The percentage of cases involved was calculated by using as the numerator the number of cases with one or more findings. The denominator is the total number of cases for which tissues were available for study.
Vasculitis was characterized by various degrees of segmental endothelial destruction, mural necrosis, and karyorrhexis (Figure 1; A to D) ▶ . Mural necrosis often appeared fibrinoid. Inflammatory cell infiltration of vascular walls by neutrophils and mononuclear cells was usually focal and either partial or transmural (Figure 1, A and B) ▶ . Thrombosis was found in both inflamed and noninflamed vessels (Figure 1C) ▶ . Necrosis and hemorrhage adjacent to vasculitic or thrombotic vessels were frequently seen (Figure 1E) ▶ .
Figure 1.
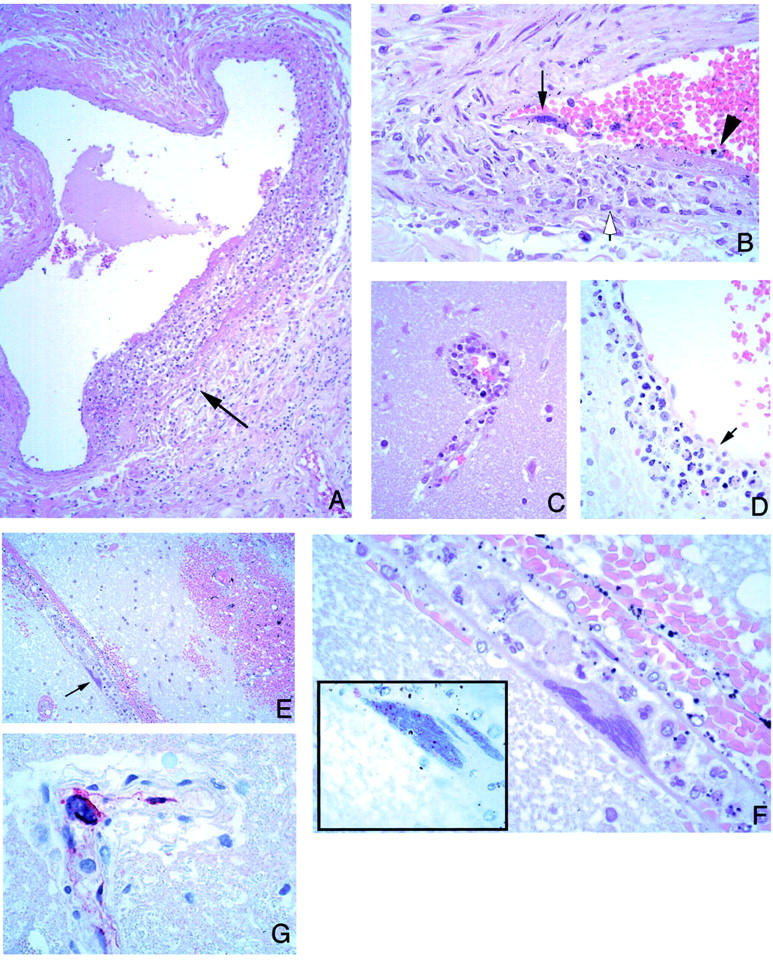
Vascular pathology and viral immunolocalization in Nipah virus infection. A: Vasculitis in a lung artery. Note focal and transmural mixed inflammatory infiltrate (arrow). B: High-power magnification of another pulmonary vessel showing endothelial syncytium (arrow) and ulceration (arrowhead). Note focal transmural inflammation (open arrow). C: Vasculitis in a small cerebral vessel. D: A cerebral venule showing endothelial ulceration (arrow) associated with inflammatory cellular debris. E: Cerebral hemorrhage adjacent to a vessel with a multinucleated giant cell (arrow). F: Higher-power magnification showing endothelial origin of syncytium seen in E. Inset: Intranuclear immunostaining of viral antigens in same cell. G: Positive immunostaining is also seen in the cytoplasm of an endothelial syncytial cell protruding in lumen of a cerebral vessel. H&E stain: A-F and inset in F; immunoalkaline phosphatase with napthol fast red substrate and hematoxylin counterstain, G. Original magnifications: ×25 (A); ×100 (B-D); ×50 (E); ×250 (F, inset); ×158 (F, G).
Syncytial or multinucleated giant endothelial cells were seen in blood vessels of various organs (Figure 1; B, E, F, and G) ▶ . In the CNS, they were found in 27% of the cases (Table 4) ▶ , mostly in patients whose duration of illness ranged from 6 to 15 days (Figure 2) ▶ . The syncytia typically consisted of several overlapping or sharply molded nuclei with moderate to abundant cytoplasm (Figure 1F) ▶ . Occasionally, syncytia protruded prominently into the vascular lumen (Figure 1, F and G) ▶ and were accompanied by vasculitis (Figure 1B) ▶ .
Table 4.
Microscopic Features in the Central Nervous System in 30 Fatal Cases of Nipah Virus Infection*
| Histopathologic lesion | Frequency, % |
|---|---|
| Necrotic plaque | 93 |
| Perivascular cuffing | 90 |
| Thrombosis | 87 |
| Vasculitis | 80 |
| Parenchymal inflammation | 67 |
| Viral inclusions | 63 |
| Meningitis | 57 |
| Syncytia | 27 |
*Brain tissues from two cases (cases 14 and 15) had severe freezing artifact that precluded adequate histopathologic examination.
Figure 2.

Temporal distribution of microscopic findings and viral antigen in the CNS in fatal Nipah virus infection. Cases were grouped into four subgroups by duration of illness: 0 to 5 days (n = 6), 6 to 10 days (n = 18), 11 to 15 days (n = 3), and 16 to 35 days (n = 4). The percentage of a particular histopathological lesion found in each subgroup was calculated by using as the numerator the number of CNS sections with at least one positive lesion found therein. The denominator is the total number of CNS sections in that subgroup. Because the duration of illness for case 32 (Table 1) ▶ could not be determined with accuracy, it was excluded.
CNS
In the CNS, the main pathological findings were vasculitis, thrombosis, parenchymal necrosis, and presence of viral inclusions (Table 4 ▶ and Figures 1, 3, 4, and 5 ▶ ▶ ▶ ). Vascular involvement of gray and white matter was seen throughout the CNS. The spinal cord was examined in eight cases and showed similar pathological lesions in three cases as observed elsewhere in CNS. Pathological lesions similar to those seen elsewhere in CNS were seen in spinal cords of three of eight cases examined. The olfactory bulb was examined in nine cases and did not show any significant histopathology. Common histopathological lesions and their relative frequency in the CNS are summarized in Table 4 ▶ .
Figure 3.
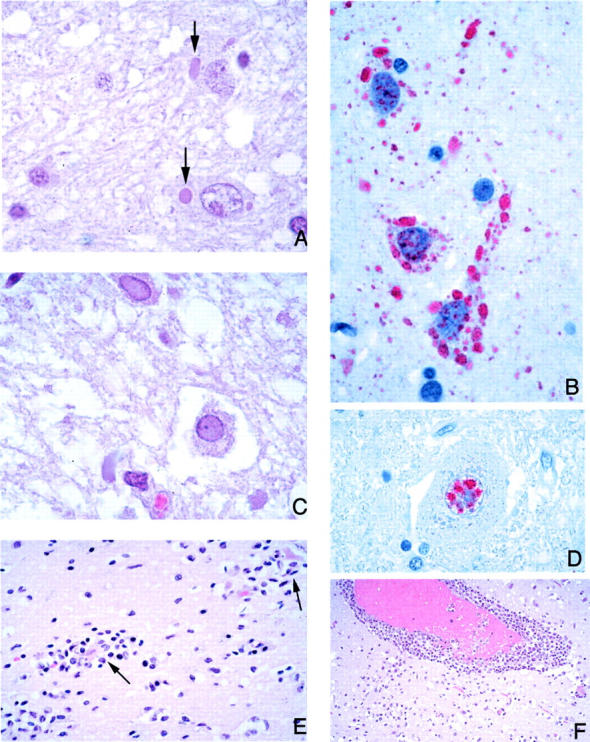
CNS pathology and viral immunolocalization in Nipah virus infection. A: Typical eosinophilic viral inclusions in cytoplasm of several neurons (arrows). B: The viral nature of these inclusions is evidenced by immunostaining of Nipah viral antigens. C: Less-common intranuclear neuronal inclusions occupying most of the nucleus and pushing the chromatin to the periphery. D: These inclusions are also immunostained for viral antigens. E and F: As in other viral encephalitides, there is significant parenchymal inflammation (E), including neuronophagia (arrows), and perivascular cuffing (F). H&E stain, A, C, E, F; immunoalkaline phosphatase with napthol fast red substrate and hematoxylin counterstain, B and D. Original magnifications: ×250 (A, B, D); ×158 (C); ×100 (E); ×50 (F).
Figure 4.
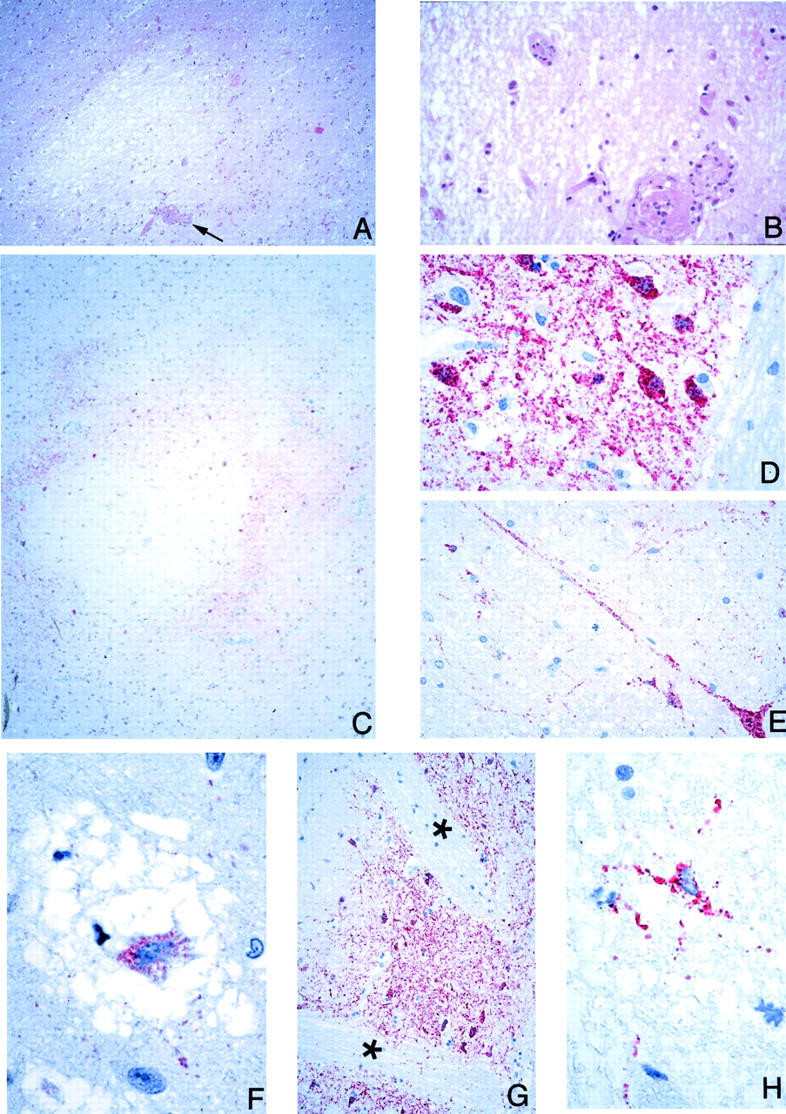
CNS pathology and viral immunolocalization in Nipah virus infection. A: Well-circumscribed, necrotic plaque associated with a vasculitic and thrombotic arteriole (arrow). B: Higher-power magnification of vessel seen in A showing complete arteriolar obstruction. C: Concentric immunostaining pattern of viral antigens around a necrotic plaque. D and E: High-power magnification showing Nipah virus antigens in neurons and neuronal processes. F: Neuronal body in a microcystic area with positive immunostaining. G: Cells in the white matter are only rarely immunostained. The pencil bundles of Wilson (asterisks) in the putamen are immunonegative even though adjacent neuronal areas are positive. H: Rare glial cell in the white matter showing immunostaining. H&E stain, A and B; immunoalkaline phosphatase with napthol fast red substrate and hematoxylin counterstain, C-H. Original magnifications: ×25 (A); ×100 (B, E); ×12.5 (C); ×158 (D); ×250 (F, H); ×50 (G).
Plaques with various degrees of necrosis were found in both the gray and white matter (Figure 4A) ▶ . These necrotic plaques were round or oval with diameters that ranged from ∼0.2 mm to ≥5 mm. Vasculitis, thrombosis, and various degrees of parenchymal edema and inflammation were frequently found in the vicinity of these plaques (Figure 4, A and B) ▶ . The inflammatory cellular infiltrate consisted of neutrophils, macrophages, lymphocytes, and reactive microglia. Areas of microcystic degeneration were seen, most commonly, in the vicinity of necrotic plaques (Figure 4F) ▶ . Microcystic change with no adjacent plaques was also occasionally seen. In white matter, damaged axons occasionally formed axonal spheroids similar to those seen in diffuse axonal injury. No large geographic infarctions of the type associated with occlusion of medium-sized or larger arteries were seen. Elsewhere in the parenchyma, focal neuronophagia, microglial nodule formation, and perivascular cuffing (Figure 3, E and F) ▶ were seen. Overall, parenchymal inflammation was present in 67% of cases (Table 4) ▶ .
Viral inclusions were found in the cytoplasm and nuclei of neurons, although the latter were generally harder to find. Most inclusions were found near vasculitic vessels or necrotic plaques. Cytoplasmic inclusions were usually small, discrete, eosinophilic, and sometimes multiple (Figure 3A) ▶ . Nuclear inclusions were less commonly found and occupied most of the nucleus except for a thin rim of chromatin at the periphery (Figure 3C) ▶ . Although inclusions were found in 63% of cases (Table 4) ▶ , in many instances they were found in only a few neurons after an extensive search.
The CNS pathology in the relapse encephalitis case (case 32; Table 1 ▶ ) was somewhat different from other cases (Figure 5) ▶ . Viral inclusions were much more extensive and prominent, occupying either the entire neuronal cytoplasm or having a more peripheral distribution; inclusions were also abundant in the neuropil. The parenchymal lesions were larger and more confluent, occasionally hemorrhagic, and associated with severe neuronal loss, gliosis, and abundant macrophages. No vasculitis or typical necrotic plaques were seen and perivascular cuffing was not a prominent feature.
Figure 5.
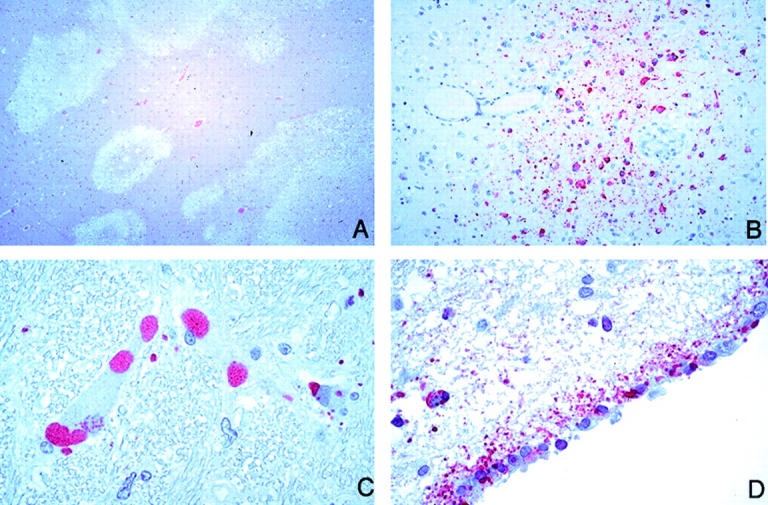
CNS pathology and viral immunolocalization in relapse Nipah virus encephalitis (case 32). A: Relapse encephalitis differs from typical acute cases in that the lesions are confluent and geographical in distribution and accompanied with severe neuronal loss and gliosis. B: Numerous Nipah virus antigen-positive macrophages are seen. C: Viral inclusions as seen by IHC; inclusions are usually larger and more abundant than in typical cases. D: Ependymal immunostaining, generally rare in acute Nipah encephalitis, is more prominent in this case. H&E stain, A; immunoalkaline phosphatase with napthol fast red substrate and hematoxylin counterstain, B-D. Original magnifications: ×12.5 (A); ×50 (B); ×158 (C, D).
Non-CNS Organs
In the lung, vasculitis was seen in 62% of cases (Table 3 ▶ ; Figure 1, A and B ▶ ) and fibrinoid necrosis was found in 59% of cases. Fibrinoid necrosis often involved several adjacent alveoli and was frequently associated with small vessel vasculitis (Table 3 ▶ , Figure 6A ▶ ). Multinucleated giant cells with intranuclear inclusions were occasionally noted in alveolar spaces adjacent to necrotic areas (Figure 6; B, C, and D) ▶ . Alveolar hemorrhage, pulmonary edema, and aspiration pneumonia were often encountered. Histopathological changes of bronchiolar epithelium were uncommon except in one case in which a large bronchus showed severe transmural inflammation and ulceration (case 22; Table 1 ▶ ).
Figure 6.
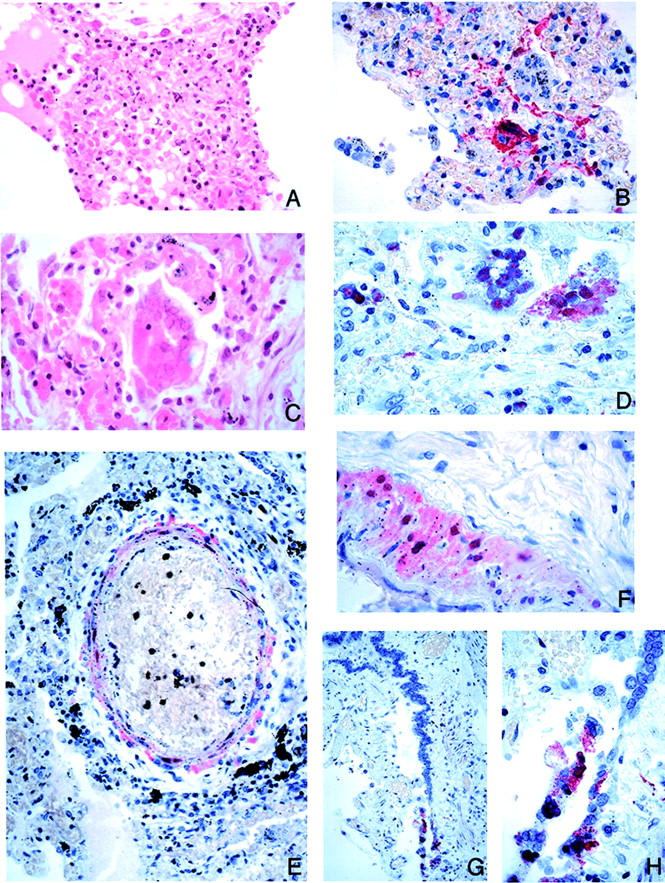
Pulmonary pathology and viral immunolocalization in Nipah virus infection. A: Focal alveolar fibrinoid necrosis and inflammation. B: These necrotic lesions show prominent immunostaining of viral antigens. C: Multinucleated giant cell with nuclear inclusions in the alveolar space. D: Viral nature of giant cells as evidenced by immunostaining for Nipah viral antigens. E and F: Nipah viral antigens in tunica media of small arteries. G and H: Rare instance of immunostaining of bronchiolar epithelium. H&E stain, A and C; immunoalkaline phosphatase with napthol fast red substrate and hematoxylin counterstain, B, D, E, G, H. Original magnifications: ×100 (A); ×50 (B, E, G); ×158 (C, D, F, H).
The spleen showed white pulp depletion and acute necrotizing inflammation in the periarteriolar sheaths (Figure 7; A, B, and C) ▶ . In addition, large prominent multinucleated giant cells with intranuclear inclusions were seen in the parenchyma of one case (case 22; Table 1 ▶ and Figure 7D ▶ ). No vasculitis of large blood vessels was seen. Lymph nodes showed large reactive mononuclear cells and occasional necrosis and hemophagocytosis. Rarely, multinucleated giant cells were encountered in cells lining the subcapsular sinusoids of lymph nodes (Figure 7, E and F) ▶ . In the kidney, focal glomerular fibrinoid necrosis was seen in 34% of cases (Table 3) ▶ . In some cases, the glomeruli were totally destroyed by inflammation. Vasculitis, thrombosis, and interstitial inflammation were occasionally seen (Figure 8A) ▶ . Syncytial formation involving the periphery of the glomerulus and tubular epithelium was rarely seen (Figure 8, C and D) ▶ . No syncytia were seen in transitional epithelium of the renal calyx and pelvis. In the heart, vasculitis was noted in 31% of cases (Table 3 ▶ , Figure 8E ▶ ). A large myocardial infarction associated with vasculitis was found in a patient comatose for >2 weeks (case 20; Table 1 ▶ ). In another patient who survived more than a month (case 31; Table 1 ▶ ), focal myocardial fibrosis associated with vasculitis was noted. In other organs, vasculitis was occasionally seen in small arteries in the mesentery, adrenal gland (Figure 8H) ▶ , and pancreas. No significant pathology was seen in the liver, skeletal muscle, and other tissues examined.
Figure 7.
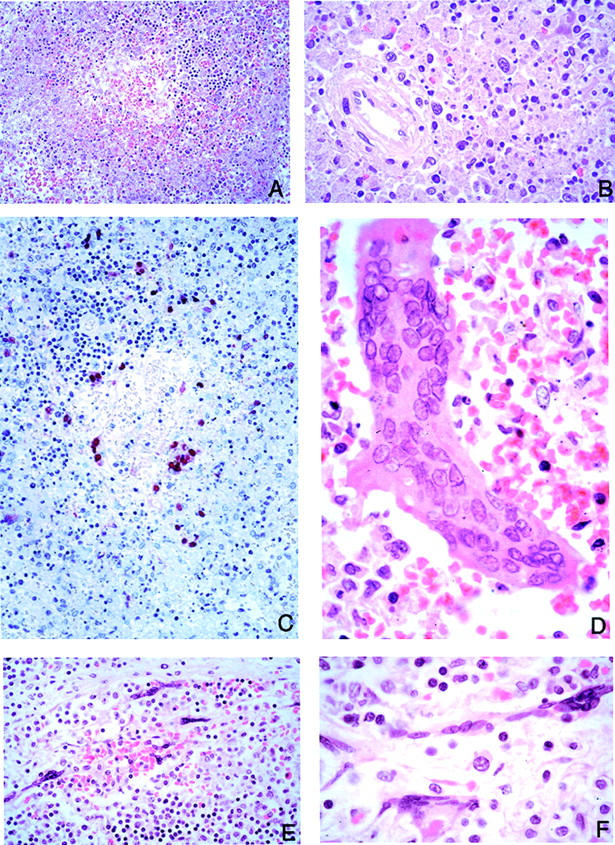
Lymphoid tissue pathology and viral immunolocalization in Nipah virus infection. A: Prominent lymphoid necrosis and depletion in the spleen. B: Splenic necrosis is particularly evident in the periarteriolar sheath. C: Same areas showing viral immunostaining. D: Multinucleated giant cells with nuclear inclusions as seen in the spleen parenchyma. E and F: Low- and high-power magnification of lymph node showing multinucleated giant cells in the subcapsular sinus. H&E stain, A, B, D-F; immunoalkaline phosphatase with napthol fast red substrate and hematoxylin counterstain, C. Original magnifications: ×50 (A, C); ×158 (B, D, F); ×100 (E).
Figure 8.
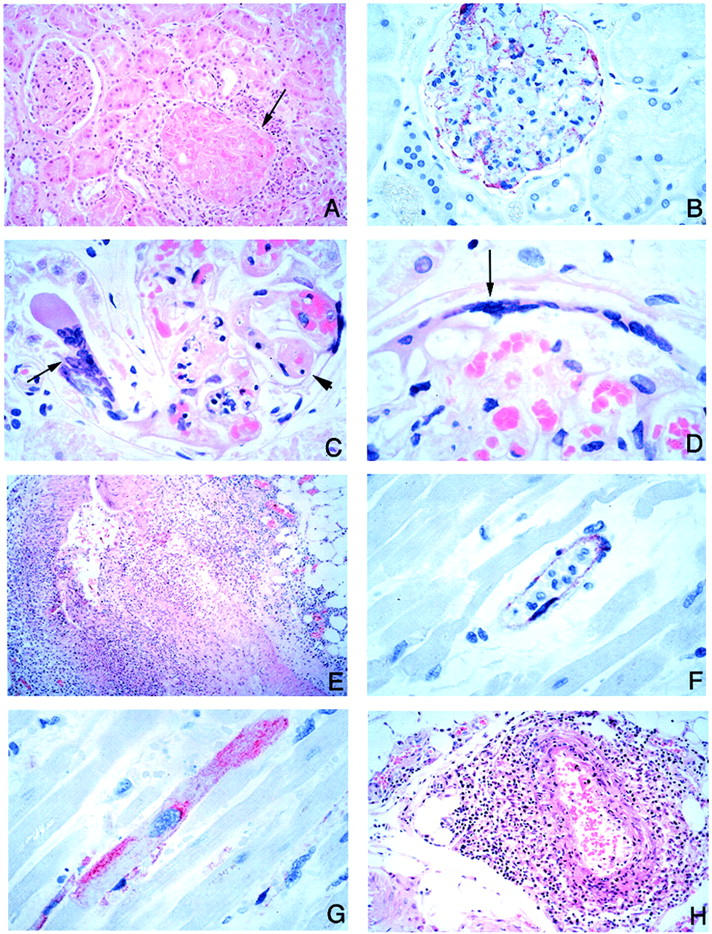
Pathology and viral immunolocalization in kidney, heart, and adrenal gland in Nipah virus infection. A: Fibrinoid necrosis of glomerulus and surrounding inflammation (arrow). B: Prominent endothelial immunostaining in glomerular capillaries. C: Affected glomerular capillaries are often thrombosed (arrowhead). Very rarely, syncytia may be seen rising from tubular epithelium (arrow). D: Multinucleated syncytial cell seen at the edge of a glomerulus (arrow). E: Severe myocardial arteritis. F: Positive immunostaining in the endothelium of a small blood vessel in the myocardium. G: Rare cardiac myocyte showing immunostaining of viral antigens. H: Periadrenal arteritis. H&E stain, A, C-E, H; immunoalkaline phosphatase with napthol fast red substrate and hematoxylin counterstain, B, F, G. Original magnifications: ×50 (A, H); ×100 (B); ×158 (C, D, F, G); ×25 (E).
EM
EM examination of CNS specimens showed inclusions typical of viruses of the family Paramyxoviridae consisting of smooth, filamentous nucleocapsids associated with dense amorphous material (Figure 9, A and B) ▶ . These inclusions were generally difficult to locate by ultrastructural examination and were mostly seen in neuronal bodies and dendritic processes (Figure 9, A and B) ▶ and occasionally within endothelial cells (Figure 9, C and D) ▶ . In addition, unusual cytoplasmic inclusions composed of aggregates of smooth curvilinear membranes were found in neurons (Figure 9E) ▶ . No mature viral particles were found in specimens examined.
Figure 9.
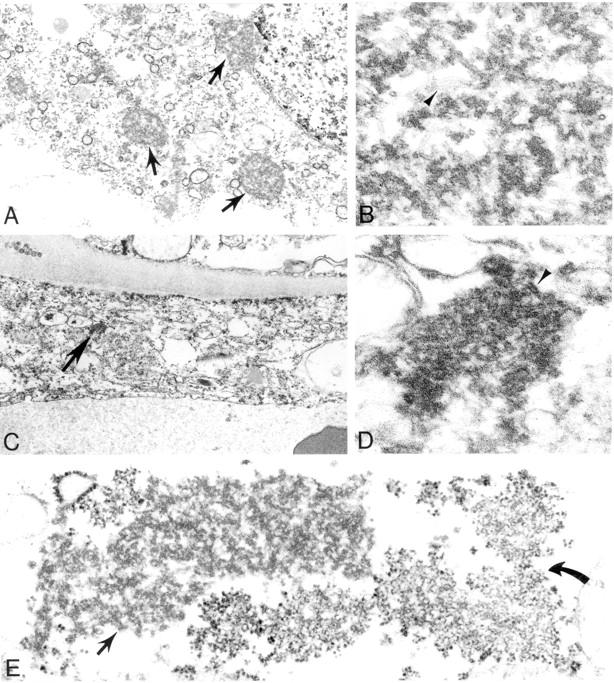
Ultrastructural appearance of Nipah virus inclusions as seen in the CNS. A-D: Characteristic intracytoplasmic nucleocapsid viral inclusions (arrows) within infected neuronal (A, B) and endothelial (C, D) cells. Details of filamentous nucleocapsids (arrowheads) and associated dense material are seen in high-power magnifications (B, D). E: Unusual viral inclusions composed of aggregates of curvilinear membranes (curved arrow) in close association with typical nucleocapsids (arrow). Original magnifications: ×12,000 (A, C); ×115,000 (B, D); ×42,000 (E).
IHC
Antibody Apecificity
Anti-Hendra and anti-Nipah antibodies showed similar specificity reacting with viral antigens in formalin-fixed infected Vero E6 cells. After absorption with excess viral antigen, these antibodies failed to react with infected cells. No staining was seen when uninfected cell controls were reacted with either antibody. Tissues from other types of viral encephalitides were negative when tested with anti-Hendra and anti-Nipah antibodies, and all Nipah cases were negative when tested with anti-flavivirus antibody.
Cellular Targets and Distribution of Nipah Viral Antigen
Blood Vessels: Immunostaining of Nipah viral antigens was seen in blood vessels of most organs, particularly in those with vasculitis. No vascular staining was found in spleen or liver. Vascular staining was seen mainly in endothelium, endothelial syncytia (Figure 1, F and G ▶ , and Figure 8, B and F ▶ ), and smooth muscle of the tunica media (Figure 6, E and F) ▶ . No staining was present in medium-size or larger arteries.
CNS: In the CNS, apart from blood vessels, the most prominent staining was seen in neurons associated with necrotic plaques or vasculitis. However, not all plaques were associated with viral antigen staining. Neuronal staining at the periphery of necrotic plaques was usually in the form of either concentric or eccentric rings (Figure 4C) ▶ . The occasional remaining neurons in plaques and microcystic areas showed positive staining (Figure 4F) ▶ . Occasionally, areas of neuronal staining with no demonstrable adjacent plaque or vasculitis were seen. Cytoplasmic and nuclear staining were observed as granular or homogeneous in the neurons and peripheral processes of the perikaryon (Figure 3, B and D ▶ ; and Figure 4, D, E, and F ▶ ). Immunostained round or oval granules (Figure 3B) ▶ corresponded to cytoplasmic viral inclusions as seen with H&E (Figure 3A) ▶ . Immunostaining was seen in three of eight of the spinal cords examined and was localized in areas with parenchymal necrosis, inflammation, and syncytial cell formation.
Immunostaining of glial cells (astrocytes or oligodendrocytes) was seen in rare cells (Figure 4H) ▶ . This relative sparing of glial cells was clearly evident in the putamen, where pencil bundles of Wilson (white matter tracts) immediately adjacent to positively stained neurons showed no immunostaining (Figure 4G) ▶ . Similarly, there was general sparing of the white matter tracts in the anterior pons in contrast to abundant pontine nuclear staining. Focal immunostaining of the ependyma (Figure 5D) ▶ was seen in several cases; the choroid plexus was negative for viral antigen in all cases.
In the case of relapse encephalitis (case 32; Table 1 ▶ ), glial and neuronal staining was more diffuse and prominent compared with that for the other cases (Figure 5; B, C, and D) ▶ . In contrast to findings for the acute cases, the blood vessels of case 32 were negative for Nipah viral antigens.
Non-CNS Organs: Localization of viral antigen was clearly seen in the non-CNS tissues, although to a lesser extent (Table 3) ▶ . Twenty-four percent of cases were positive by IHC in the lung and kidney, compared with 84% in the CNS. In the lung, viral antigens were usually found in areas of fibrinoid necrosis and in blood vessels (Figure 6; B, E, and F) ▶ . Viral antigens were also noted in multinucleated giant cells in or lining the alveolar space in three cases (Figure 6D) ▶ . Only one case had bronchial inflammation and unequivocal staining of the bronchiolar epithelium (case 22; Table 1 ▶ and Figure 6, G and H ▶ ).
In the kidney, glomerular capillaries, small blood vessels, and syncytial cells in the periphery of the glomerulus exhibited viral antigen staining (Figure 8B) ▶ . In the heart, IHC staining was found mainly in the blood vessels (Figure 8F) ▶ . In one case, there was focal staining of cardiac myocytes (Figure 8G) ▶ . Nipah virus antigen was identified in macrophages and multinucleated giant cells in the spleen and lymph nodes (Figure 7C) ▶ . No immunostaining of viral antigens was seen in the liver. In the relapse case, immunostaining was not seen outside of the CNS.
Nipah Antibody Detection Serological Assays
Nipah IgM antibodies were more often detected than IgG in serum and CSF of patients (Table 1) ▶ . In the single largest group of 18 patients with duration of illness of 6 to 10 days, IgM was found in the serum of 94% and the CSF of 64%, and IgG was found in 12% and 9%, respectively. IgM antibodies generally appeared earlier than IgG and in the serum before CSF (Figure 10) ▶ .
Figure 10.

Temporal relationship of IgM and IgG in CSF and serum in fatal Nipah virus infection. Cases were grouped into four subgroups by duration of illness: 0 to 5 days (n = 6), 6 to 10 days (n = 18), 11 to 15 days (n = 3), and 16 to 35 days (n = 4). The percentage of positive cases for each type of immunoglobulin was calculated using as numerator the number of patients within each subgroup with a positive antibody level. Because the duration of illness for case 32 (Table 1) ▶ could not be determined with accuracy, it was excluded.
Correlation of IHC and Serological Test Results
All cases in this study were examined by IHC, and serology was performed on serum or CSF of all but one case. IHC analysis showed 87% of cases to be positive for viral antigen, and 94% of cases had positive Nipah antibody results in the serum and/or CSF (Table 5) ▶ . There was an 81% correlation between positive IHC and serology. Discrepancies between IHC and serological assays were found in six cases; four of these cases were IHC-negative and serology-positive, and two cases were IHC-positive and serology-negative. Another IHC (case 15) had no serology available (Table 5) ▶ .
Table 5.
Correlation of Immunohistochemistry and Serological Assays as Diagnostic Tests in 31 Fatal Cases of Nipah Virus Infection*
| Test results | Positive cases/total tested | % Positive cases |
|---|---|---|
| Serology (either serum or CSF) | ||
| IgM-positive | 28/31 | 90 |
| IgG-positive | 9/31 | 29 |
| IgM- and/or IgG-positive | 29/31 | 94 |
| Immunohistochemistry (IHC) | 27/31 | 87 |
| Combined Serology and IHC | ||
| Both serology and IHC-negative | 0/31 | 0 |
| Both serology and IHC-positive | 25/31† | 81 |
| Either serology or IHC-positive | 31/31 | 100 |
*Correlation of IHC and serology was done in 31 cases; one IHC-positive case (case 15) was excluded because serology was not done.
†In four cases (cases 14, 19, 20, 31), the IHC was negative and serology was positive. In two cases, the IHC was positive with negative serology (cases 16 and 27).
Virus Isolation
Virus isolation from the CSF was attempted for eight cases and successful for all but one, which was also IHC-negative (case 31; Table 1 ▶ ).
Temporal Evolution of Lesions and Presence of Viral Antigens in the CNS
The temporal evolution of histopathological lesions and viral antigens in the brain in acute fatal Nipah infection is shown in Figure 2 ▶ . Vasculitis, thrombosis, necrotic plaques, and syncytia peaked between 6 and 10 days after fever onset. On the other hand, parenchymal inflammation and perivascular cuffing were most severe 11 to 15 days after fever onset and were found in >60% of tissue sections of cases with duration of illness >16 days.
Viral antigens were more commonly detected in tissues 6 to 10 days after fever onset and gradually decreased throughout time. Tissues of all 18 patients with a 6 to 10 day duration were positive by IHC. In contrast, among the four patients whose duration of illness was >16 days, only one (case 26) was IHC-positive (Table 1) ▶ .
Discussion
The outbreak of Nipah virus infection in Southeast Asia is a prime example of emerging infectious diseases that continue to occur worldwide. Other notable recent outbreaks of emerging infectious diseases include hantavirus pulmonary syndrome, 11 ehrlichiosis, 15,16 and West Nile virus encephalitis 17,18 in North America; Ebola hemorrhagic fever in Africa; 13,19,20 Hendra virus encephalitis in Australia; 21 leptospirosis in Central and South America; 22 variant Creutzfeldt-Jakob disease in Europe; 23 and enterovirus71-associated hand, foot, and mouth disease in Asia. 24,25 In many of these outbreaks, pathologists, as part of a multidisciplinary team, played key roles in identifying the causative agent or describing the pathogenic processes. 26-28 In the Nipah virus outbreak, the clinical and laboratory speculation initially centered on JE virus as the causative agent; however, several features of the outbreak argued against this possibility. First, JE virus appeared an unlikely agent in an outbreak that affected mostly adults rather than children in an area of endemic JE virus transmission. Second, most, if not all, patients had a history of direct contact with pigs. Additionally, there was clustering of cases with higher infection rates in the same households than would be expected with JE. Finally, many patients had been previously vaccinated against JE with a standard vaccine and were therefore likely to have developed protective immunity against the virus. 2 Our study was instrumental in excluding JE virus and in identifying Nipah virus as the etiological agent of the outbreak. The following discussion focuses on the pathological changes and cellular distribution of viral antigens as they relate to the diagnosis, pathogenesis, and clinical presentation of Nipah virus infection.
The diagnosis of Nipah virus infection, suspected by history and clinical manifestations, can be supported by characteristic histopathological findings. These histopathological findings include syncytial giant cell formation, vasculitis, and viral inclusions. Other CNS changes observed, including perivascular cuffing, parenchymal inflammation, and neuronophagia, are rather nonspecific features and can be found in other acute viral encephalitides. 29-31 From a diagnostic standpoint, perhaps the most unique histopathological finding is the presence of syncytial multinucleated endothelial cells. To our knowledge, this feature has not been described in other infective encephalitides other than Hendra virus encephalitis. 32 However, this feature occurred in only approximately one-fourth of the cases and cannot be used as a sensitive criterion for the diagnosis of Nipah virus infection. There is a higher likelihood of identifying these cells in small blood vessels of the CNS of patients who succumb early to the disease. The presence of multinucleated giant cells of nonendothelial origin found in the kidney at the edge of the glomerulus also seems to be a unique, albeit relatively rare, feature of Nipah virus infection. Characteristic multinucleated giant cells were also seen in the lung, spleen, and lymph nodes; however, these cells can also be seen in measles virus, respiratory syncytial virus, parainfluenza virus, herpesvirus, and other infections and therefore are not unique to Nipah virus infection. 33-37
Widespread vasculitis with predominant CNS involvement is a useful histopathological feature that may suggest the diagnosis of Nipah virus infection. Many infectious agents can cause vasculitis, including herpesviruses, rickettsiae, and Neisseria. 38 However, the histopathological features are somewhat different in these infections. In rickettsial encephalitis, 39 vasculitis is usually more subtle and CNS necrosis less prominent than seen in Nipah virus encephalitis. Varicella-zoster and herpes simplex infections may be associated with granulomatous angiitis, a feature not seen in Nipah virus infection. 40,41
Finally, the diagnosis of Nipah virus infection can be supported by the histopathological and ultrastructural appearance and distribution of characteristic viral inclusions. However, similar inclusions can be seen in other paromyxoviral infections, 33,42 and therefore unequivocal diagnosis can be made only by laboratory tests such as IHC, virus isolation, PCR, and serology.
The utility of IHC as a diagnostic modality was established by correlation with results of virus isolation and serological assays. Virus can be isolated from the CSF, respiratory secretions, and urine of patients 14 but should be done in biosafety level 4 laboratory facilities. The combination of IHC and serological tests established the diagnosis for all cases regardless of duration of illness. Four seropositive and IHC-negative cases had duration of illness of ≥14 days, emphasizing the important role of serology in the diagnosis and suggesting that in most cases viral antigens are cleared by 2 weeks after infection. Conversely, the three IHC-positive cases with negative or unavailable serological results underscore the importance of IHC in the diagnosis of fatal Nipah virus infection. CNS is the optimal tissue for IHC analysis because it was three to four times more likely to be positive for Nipah virus antigens than were lung or kidney tissues, the next most likely organs to be positive (Table 3) ▶ .
In this study, several lines of evidence allowed for precise characterization of viral tropism and its consequences. Epithelial cell involvement, although not common, included bronchiolar mucosa, renal tubules, and podocytes at the edge of the glomerulus. Endothelial cells and neurons had a remarkably high viral load, as evidenced by immunostaining of Nipah virus antigens. The ultrastructural finding of inclusions in endothelial and neuronal cells is consistent with replication in these sites. 43,44 In situ hybridization studies also confirm involvement of these cells in viral replication (data not shown). There is considerable diversity and heterogeneity of endothelial cells. Our data showed that small vessels, such as small arteries, arterioles, venules, and capillaries, were more prone to vasculitis and thrombosis than were larger vessels. Differences in endothelial susceptibility to viral infection could certainly account for the different frequencies of vasculitis in various organs: CNS, 80% of cases, followed by the lung, heart, and kidney with 62%, 31%, and 24%, respectively.
Widespread vasculitis, a key event in the pathogenesis of Nipah virus infection, seems to be a consequence of infection of the vascular endothelial and smooth muscle cells. Overall, the frequency of vasculitis seemed to be proportional to necrosis and necrotic plaques, particularly in the CNS and lung. The necrotic plaques and the acute encephalitic syndrome may stem from both direct neuronal infection and ischemic injury (Figure 11) ▶ . This sequence of pathological events is supported by the concomitant increase in frequency of syncytia, vasculitis, thrombosis, necrotic plaques, and viral antigen in the CNS (Figure 2) ▶ .
Figure 11.

Pathogenesis of Nipah virus infection.
The pattern of vasculitis and viral antigen distribution suggests that endothelial infection occurs before transmural spread to underlying smooth muscle in the tunica media. Finding of numerous infected neurons in association with necrotic plaques and vasculitic vessels suggests that a breach of the blood-brain-barrier may facilitate virus escape and extravascular CNS infection (Figure 11) ▶ . In subacute sclerosing panencephalitis caused by measles virus, endothelial infection is also thought to facilitate initial virus entry into the brain. 45 Once neurons are infected, interneuronal spread along neuronal pathways may contribute to viral dissemination. Viral spread through damaged vessels may also explain non-CNS parenchymal infection.
The appearance of similar pathological lesions in several organs at the same time suggest an early viremic phase that follows primary viral replication. In measles, another paramyxoviral infection, primary viral replication occurs in respiratory tract mucosa and lymphoid organs and is followed by a cell-associated viremia. 46 In this series of fatal Nipah virus cases, the extensive lymphoid necrosis and immunostaining of lymphoid and respiratory tissues suggest that these tissues can also be similarly involved in primary replication. Endothelium, although unlikely to be a primary replication site, may act as a site of secondary viral replication and amplification of viremia. The temporal sequence of antibody rise first in serum and then in the CSF provides indirect evidence that viremia occurs before CNS infection and probably reflects the fact that induction of antigen-specific, antibody-secreting B cells first occurs in the peripheral lymphoid tissues (Figure 10) ▶ .
CNS predilection, as evidenced by pathological changes and viral immunolocalization, correlates well with the encephalitic syndrome reported in fatal and nonfatal cases. 2,3,6 In this series of fatal cases, predominant clinical features included drowsiness, disorientation, confusion, segmental myoclonus, and areflexia. Clinically, nonfatal cases also undergo a similar, less-severe acute encephalitic syndrome. 6 Brain magnetic resonance imaging studies of nonfatal and fatal cases show similar scattered, small, and discrete lesions that probably represent necrotic plaques. 47-50 In our study, CNS pathological changes were more severe than histopathological changes in other organs. This finding also correlates with the lower frequency of non-CNS clinical manifestations (Table 2) ▶ . A pulmonary syndrome was reported in some cases from Singapore, 3 and 40% of cases described in this study had cough or respiratory symptoms. These symptoms could be related to pulmonary lesions, such as necrosis, edema, and hemorrhage.
The sequelae of acute Nipah virus infection are still unclear. Acute encephalitis probably occurs in a significant proportion of cases after exposure to virus. Mortality from acute Nipah encephalitis may be as high as 30 to 40% and is more likely in patients with brainstem signs and virus isolated from CSF. 6,51 Approximately 15% of patients with acute encephalitic syndrome develop residual neurological deficits. 6 Asymptomatic cases have also been reported with rare late-onset encephalitis ∼10 weeks after exposure. 6,50,52 Overall, the prevalence of relapse Nipah encephalitis and late-onset encephalitis in initially asymptomatic patients is estimated to be ∼7.5% and 3.4%, respectively (CT Tan, personal communication). Case 32 is likely a relapse case because of the clinical course and pathological findings of excessive CNS viral inclusions and confluent necrotic plaques. This diagnosis is also supported by results showing elevated IgG, but not IgM, and the histopathological absence of vasculitis. The clinical, radiological, and pathological features of this case are similar to those of a patient with Hendra virus infection who initially had aseptic meningitis and recovered, only to suffer a fatal meningoencephalitis 13 months later. 6,32,50 This clinical spectrum is also somewhat similar to that of other paramyxoviruses, such as measles virus, that can cause both acute and subacute progressive encephalitides. 46
The emergence of novel paramyxoviruses, such as Hendra and Nipah, throughout a short span underscores the importance of these viruses as zoonotic agents. 53,54 Human Nipah virus infection most likely originated from direct contact with pigs that also acted as intermediate and amplifying hosts rather than as natural hosts. 5,8,55,56 Studies indicate that virus transmission from pig to humans resulted from direct contact with these animals and that the risk of person-to-person transmission is extremely low. 5,14,57,58 Although the natural host for Nipah virus appears to be the fruit bat from the Pteropus family, 59 the mode of virus transmission to pigs is still under investigation.
A fundamental principle in the concept of emerging infectious diseases is recognition of disease. As illustrated in this report, recognition and description of a disease by pathologists as part of a multidisciplinary team plays a key role in furthering the understanding of emerging infectious diseases. This investigation also highlights the important role of autopsies in combination with contemporary pathological methods in providing insights into the pathogenesis of new clinical entities.
Acknowledgments
We thank P. Stockton, K. Veilleux, L. Pezzanite, and T. Stevens for serological assay work at the Centers for Disease Control; C. Nadarajah, M. N. Zainal, A. R. Ali, K. Kamalasothy, O. Noridah, A. H. Marina, and L. C. Chong for assistance in Malaysia and Singapore; J. O’Connor for editorial assistance; and S. Grimes for administrative assistance in preparing the manuscript.
Footnotes
Address reprint requests to Sherif R. Zaki, M.D., Ph.D., Chief, Infectious Disease Pathology Activity, National Center for Infectious Diseases, Centers for Disease Control and Prevention, 1600 Clifton Rd. N. E., MS-G32, Atlanta, GA 30333. E-mail: szaki@cdc.gov.
Supported by the Ministry of Health, Malaysia, the University of Malaya Medical Center, and the Malaysian Government (research grant 06-02-03-0743.
The Nipah Virus Pathology Working Group at CDC, Atlanta, consists of Jeanine Bartlett, Tara Ferebee-Harris, Patricia Greer, Lisa M. Harper, Jeltley Montague, Tim Morken, and Chalanda Smith; and in Malaysia, Sharifah Safoorah Syed Alwee, Thuaibah Hashim, Khairul Azman Ibrahim, Fauziah Kassim, Lily Manoramah, George Paul, Norraha Abdul Rahman, Kalyani Supramaniam, Thayaparan Tarmizi, Nor Yatizah Mohd Yatim, Rosna Yunus, and Suryati Yusuf.
References
- 1.: Centers for Disease Control and Prevention: Outbreak of Hendra-like virus-Malaysia and Singapore, 1998–1999. MMWR Morb Mortal Wkly Rep 1999, 48:265-269 [PubMed] [Google Scholar]
- 2.Chua KB, Goh KJ, Wong KT, Kamarulzaman A, Tan PS, Ksiazek TG, Zaki SR, Paul G, Lam SK, Tan CT: Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 1999, 354:1257-1259 [DOI] [PubMed] [Google Scholar]
- 3.Paton NI, Leo YS, Zaki SR, Auchus AP, Lee KE, Ling AE, Chew SK, Ang B, Rollin PE, Umapathi T, Sng I, Lee CC, Lim E, Ksiazek TG: Outbreak of Nipah-virus infection among abattoir workers in Singapore. Lancet 1999, 354:1253-1256 [DOI] [PubMed] [Google Scholar]
- 4.Tambyah PA, Tan JH, Ong BK, Ho KH, Chan KP: First case of Nipah virus encephalitis in Singapore. Intern Med J 2001, 31:132-133 [PubMed] [Google Scholar]
- 5.Parashar UD, Sunn LM, Ong F, Mounts AW, Arif MT, Ksiazek TG, Kamaluddin MA, Mustafa AN, Kaur H, Ding LM, Othman G, Radzi HM, Kitsutani PT, Stockton PC, Arokiasamy J, Gary HE, Jr, Anderson LJ: Case-control study of risk factors for human infection with a new zoonotic paramyxovirus, Nipah virus, during a 1998–1999 outbreak of severe encephalitis in Malaysia. J Infect Dis 2000, 181:1755-1759 [DOI] [PubMed] [Google Scholar]
- 6.Goh KJ, Tan CT, Chew NK, Tan PS, Kamarulzaman A, Sarji SA, Wong KT, Abdullah BJ, Chua KB, Lam SK: Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N Engl J Med 2000, 342:1229-1235 [DOI] [PubMed] [Google Scholar]
- 7.Lee KE, Umapathi T, Tan CB, Tjia HT, Chua TS, Oh HM, Fock KM, Kurup A, Das A, Tan AK, Lee WL: The neurological manifestations of Nipah virus encephalitis, a novel paramyxovirus. Ann Neurol 1999, 46:428-432 [PubMed] [Google Scholar]
- 8.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW: Nipah virus: a recently emergent deadly paramyxovirus. Science 2000, 288:1432-1435 [DOI] [PubMed] [Google Scholar]
- 9.Harcourt BH, Tamin A, Ksiazek TG, Rollin PE, Anderson LJ, Bellini WJ, Rota PA: Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 2000, 271:334-349 [DOI] [PubMed] [Google Scholar]
- 10.: Centers for Disease Control and Prevention: Update: outbreak of Nipah virus—Malaysia and Singapore, 1999. MMWR Morb Mortal Wkly Rep 1999, 48:335-337 [PubMed] [Google Scholar]
- 11.Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, Feddersen RM, Zumwalt RE, Miller GL, Khan AS: Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am J Pathol 1995, 146:552-579 [PMC free article] [PubMed] [Google Scholar]
- 12.Mollenhauer HH: Plastic embedding mixtures for use in electron microscopy. Stain Technol 1964, 39:111-114 [PubMed] [Google Scholar]
- 13.Ksiazek TG, Rollin PE, Williams AJ, Bressler DS, Martin ML, Swanepoel R, Burt FJ, Leman PA, Khan AS, Rowe AK, Mukunu R, Sanchez A, Peters CJ: Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis 1999, 179(Suppl 1):S177-S187 [DOI] [PubMed] [Google Scholar]
- 14.Chua KB, Lam SK, Goh KJ, Hooi PS, Ksiazek TG, Kamarulzaman A, Olson J, Tan CT: The presence of Nipah virus in respiratory secretions and urine of patients during an outbreak of Nipah virus encephalitis in Malaysia. J Infect 2001, 42:40-43 [DOI] [PubMed] [Google Scholar]
- 15.Bakken JS, Dumler JS, Chen SM, Eckman MR, Van Etta LL, Walker DH: Human granulocytic ehrlichiosis in the upper Midwest United States. A new species emerging? JAMA 1994, 272:212-218 [PubMed] [Google Scholar]
- 16.Walker DH, Dumler JS: Human monocytic and granulocytic ehrlichioses. Discovery and diagnosis of emerging tick-borne infections and the critical role of the pathologist. Arch Pathol Lab Med 1997, 121:785-791 [PubMed] [Google Scholar]
- 17.: Centers for Disease Control and Prevention: Outbreak of West Nile-like viral encephalitis—New York, 1999. MMWR Morb Mortal Wkly Rep 1999, 48:845-849 [PubMed] [Google Scholar]
- 18.Shieh WJ, Guarner J, Layton M, Fine A, Miller J, Nash D, Campbell GL, Roehrig JT, Gubler DJ, Zaki SR: The role of pathology in an investigation of an outbreak of West Nile encephalitis in New York, 1999. Emerg Infect Dis 2000, 6:370-372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan AS, Tshioko FK, Heymann DL, Le Guenno B, Nabeth P, Kerstiens B, Fleerackers Y, Kilmarx PH, Rodier GR, Nkuku O, Rollin PE, Sanchez A, Zaki SR, Swanepoel R, Tomori O, Nichol ST, Peters CJ, Muyembe-Tamfum JJ, Ksiazek TG: The reemergence of Ebola hemorrhagic fever, Democratic Republic of the Congo, 1995. J Infect Dis 1999, 179(Suppl 1):S76-S86 [DOI] [PubMed] [Google Scholar]
- 20.Zaki SR, Shieh WJ, Greer PW, Goldsmith CS, Ferebee T, Katshitshi J, Tshioko FK, Bwaka MA, Swanepoel R, Calain P, Khan AS, Lloyd E, Rollin PE, Ksiazek TG, Peters CJ: A novel immunohistochemical assay for the detection of Ebola virus in skin: implications for diagnosis, spread, and surveillance of Ebola hemorrhagic fever. J Infect Dis 1999, 179(Suppl 1):S36-S47 [DOI] [PubMed] [Google Scholar]
- 21.Murray K, Selleck P, Hooper P, Hyatt A, Gould A, Gleeson L, Westbury H, Hiley L, Selvey L, Rodwell B: A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268:94-97 [DOI] [PubMed] [Google Scholar]
- 22.Zaki SR, Shieh WJ, : The Epidemic Working Group at Ministry of Health in Nicaragua: Leptospirosis associated with outbreak of acute febrile illness and pulmonary haemorrhage, Nicaragua, 1995. Lancet 1996, 347:535-536 [DOI] [PubMed] [Google Scholar]
- 23.Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG: A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347:921-925 [DOI] [PubMed] [Google Scholar]
- 24.Shieh WJ, Jung SM, Hsueh C, Kuo TT, Mounts A, Parashar U, Yang CF, Guarner J, Ksiazek TG, Dawson J, Goldsmith C, Chang GJ, Oberste SM, Pallansch MA, Anderson LJ, Zaki SR: Pathologic studies of fatal cases in outbreak of hand, foot, and mouth disease, Taiwan. Emerg Infect Dis 2001, 7:146-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackenzie JS, Chua KB, Daniels PW, Eaton BT, Field HE, Hall RA, Halpin K, Johansen CA, Kirkland PD, Lam SK, McMinn P, Nisbet DJ, Paru R, Pyke AT, Ritchie SA, Siba P, Smith DW, Smith GA, van den Hurk AF, Wang LF, Williams DT: Emerging viral diseases of Southeast Asia and the Western Pacific. Emerg Infect Dis 2001, 7:497-504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaki SR, Paddock C: The emerging role of pathology in infectious diseases. Scheld WM Craig WA Hughes JM eds. Emerging Infections III. 1999:pp 181-200 ASM Press, Washington DC
- 27.Wong KT: Emerging and re-emerging epidemic encephalitis: a tale of two viruses. Neuropathol Appl Neurobiol 2000, 26:313-318 [DOI] [PubMed] [Google Scholar]
- 28.Schwartz DA, Bryan RT, Hughes JM: Pathology and emerging infections—quo vadimus? Am J Pathol 1995, 147:1525-1533 [PMC free article] [PubMed] [Google Scholar]
- 29.Esiri MM, Kennedy PGE: Viral diseases. Graham DI Lantos PL eds. Greenfield’s Neuropathology. 1997:pp 3-63 Arnold, London
- 30.Hamilton RL, Wiley CA: Neuropathology of viral infections of the nervous system. Davis RL Robertson DM eds. Textbook of Neuropathology. 1997:pp 927-1061 Williams and Wilkins, Baltimore
- 31.Manz HJ: Arboviral encephalitides. Connor DH Chandler FW Schwartz DA Manz HJ Lack EE eds. Pathology of Infectious Diseases. 1997:pp 71-83 Appleton and Lange, Stamford
- 32.O’Sullivan JD, Allworth AM, Paterson DL, Snow TM, Boots R, Gleeson LJ, Gould AR, Hyatt AD, Bradfield J: Fatal encephalitis due to novel paramyxovirus transmitted from horses. Lancet 1997, 349:93-95 [DOI] [PubMed] [Google Scholar]
- 33.Zaki SR, Bellini WJ: Measles. Connor DH Chandler FW Schwartz DA Manz HJ Lack EE eds. Pathology of Infectious Diseases. 1997:pp 233-244 Appleton and Lange, Stamford
- 34.Delage G, Brochu P, Robillard L, Jasmin G, Joncas JH, Lapointe N: Giant cell pneumonia due to respiratory syncytial virus. Occurrence in severe combined immunodeficiency syndrome. Arch Pathol Lab Med 1984, 108:623-625 [PubMed] [Google Scholar]
- 35.Delage G, Brochu P, Pelletier M, Jasmin G, Lapointe N: Giant-cell pneumonia caused by parainfluenza virus. J Pediatr 1979, 94:426-429 [DOI] [PubMed] [Google Scholar]
- 36.Read GS, Person S, Keller PM: Genetic studies of cell fusion induced by herpes simplex virus type 1. J Virol 1980, 35:105-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Worrell JT, Cockerell CJ: Histopathology of peripheral nerves in cutaneous herpesvirus infection. Am J Dermatopathol 1997, 19:133-137 [DOI] [PubMed] [Google Scholar]
- 38.Somer T, Finegold SM: Vasculitides associated with infections, immunization, and antimicrobial drugs. Clin Infect Dis 1995, 20:1010-1036 [DOI] [PubMed] [Google Scholar]
- 39.Walker DH, Dumler JS: Rickettsial infections. Connor DH Chandler FW Schwartz DA Manz HJ Lack EE eds. Pathology of Infectious Diseases. 1997:pp 789-799 Appleton and Lange, Stamford
- 40.Fukumoto S, Kinjo M, Hokamura K, Tanaka K: Subarachnoid hemorrhage and granulomatous angiitis of the basilar artery: demonstration of the varicella-zoster-virus in the basilar artery lesions. Stroke 1986, 17:1024-1028 [DOI] [PubMed] [Google Scholar]
- 41.Schmitt JA, Dietzmann K, Muller U, Krause P: Granulomatous vasculitis—an uncommon manifestation of herpes simplex infection of the central nervous system. Zentralbl Pathol 1992, 138:298-302 [PubMed] [Google Scholar]
- 42.O’Neil SP, Shieh WJ, Zaki SR: Pathology and pathogenesis of virus infections. Kaufman SH Sher A Ahmed R eds. Immunology of Infectious Diseases. 2002:pp 307-328 ASM Press, Washington DC
- 43.Goldsmith CS, Whistler T, Rollin PE, Ksiazek TG, Rota PA, Bellini WJ, Daszak P, Wong KT, Shieh WJ, Zaki SR: Elucidation of Nipah virus morphogenesis and replication using ultrastructural and molecular approaches. Virus Res , (in press) [DOI] [PubMed]
- 44.Hyatt AD, Zaki SR, Goldsmith CS, Wise TG, Hengstberger SG: Ultrastructure of Hendra virus and Nipah virus within cultured cells and host animals. Microbes Infect 2001, 3:297-306 [DOI] [PubMed] [Google Scholar]
- 45.Cosby SL, Brankin B: Measles virus infection of cerebral endothelial cells and effect on their adhesive properties. Vet Microbiol 1995, 44:135-139 [DOI] [PubMed] [Google Scholar]
- 46.Griffin DE, Bellini WJ: Measles virus. Fields BN Knipe DM Howley PM Chanock RM Melnick JL Monath TP Roizman B Straus SE eds. Fields Virology. 1996:pp 1267-1312 Lippincott-Raven, Philadelphia
- 47.Lim CC, Sitoh YY, Lee KE, Kurup A, Hui F: Meningoencephalitis caused by a novel paramyxovirus: an advanced MRI case report in an emerging disease. Singapore Med J 1999, 40:356-358 [PubMed] [Google Scholar]
- 48.Lim CC, Sitoh YY, Hui F, Lee KE, Ang BS, Lim E, Lim WE, Oh HM, Tambyah PA, Wong JS, Tan CB, Chee TS: Nipah viral encephalitis or Japanese encephalitis? MR findings in a new zoonotic disease. Am J Neuroradiol 2000, 21:455-461 [PMC free article] [PubMed] [Google Scholar]
- 49.Lim CC, Lee KE, Lee WL, Tambyah PA, Lee CC, Sitoh YY, Auchus AP, Lin BK, Hui F: Nipah virus encephalitis: serial MR study of an emerging disease. Radiology 2002, 222:219-226 [DOI] [PubMed] [Google Scholar]
- 50.Sarji SA, Abdullah BJ, Goh KJ, Tan CT, Wong KT: MR imaging features of Nipah encephalitis. Am J Roentgenol 2000, 175:437-442 [DOI] [PubMed] [Google Scholar]
- 51.Chua KB, Lam SK, Tan CT, Hooi PS, Goh KJ, Chew NK, Tan KS, Kamarulzaman A, Wong KT: High mortality in Nipah encephalitis is associated with presence of virus in cerebrospinal fluid. Ann Neurol 2000, 48:802-805 [PubMed] [Google Scholar]
- 52.Chan KP, Rollin PE, Ksiazek TG, Leo YS, Goh KT, Paton N, I, Sng EH, Ling AE: A survey of Nipah virus infection among various risk groups in Singapore. Epidemiol Infect 2002, 128:93-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hooper P, Zaki S, Daniels P, Middleton D: Comparative pathology of the diseases caused by Hendra and Nipah viruses. Microbes Infect 2001, 3:315-322 [DOI] [PubMed] [Google Scholar]
- 54.Mahy BW, Brown CC: Emerging zoonoses: crossing the species barrier. Rev Sci Tech 2000, 19:33-40 [DOI] [PubMed] [Google Scholar]
- 55.Chew MH, Arguin PM, Shay DK, Goh KT, Rollin PE, Shieh WJ, Zaki SR, Rota PA, Ling AE, Ksiazek TG, Chew SK, Anderson LJ: Risk factors for Nipah virus infection among abattoir workers in Singapore. J Infect Dis 2000, 181:1760-1763 [DOI] [PubMed] [Google Scholar]
- 56.Sahani M, Parashar UD, Ali R, Das P, Lye MS, Isa MM, Arif MT, Ksiazek TG, Sivamoorthy M: Nipah virus infection among abattoir workers in Malaysia, 1998–1999. Int J Epidemiol 2001, 30:1017-1020 [DOI] [PubMed] [Google Scholar]
- 57.Mounts AW, Kaur H, Parashar UD, Ksiazek TG, Cannon D, Arokiasamy JT, Anderson LJ, Lye MS: A cohort study of health care workers to assess nosocomial transmissibility of Nipah virus, Malaysia, 1999. J Infect Dis 2001, 183:810-813 [DOI] [PubMed] [Google Scholar]
- 58.Tan CT, Tan KS: Nosocomial transmissibility of Nipah virus. J Infect Dis 2001, 184:1367. [DOI] [PubMed] [Google Scholar]
- 59.Chua KB, Lek KC, Hooi PS, Wee KF, Khong JH, Chua BH, Chan YP, Lim ME, Lam SK: Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect 2002, 4:145-151 [DOI] [PubMed] [Google Scholar]