

Abstract
Acute lung injury (ALI) leading to respiratory distress is a common sequela of shock/trauma, however, modeling this process in mice with a single shock or septic event is inconsistent. One explanation is that hemorrhage is often just a “priming insult,” thus, secondary stimuli may be required to “trigger” ALI. To test this we carried out studies in which we assessed the capacity of hemorrhage alone or hemorrhage followed by septic challenge (CLP) to induce ALI. Lung edema, bronchoalveolar lavage interleukin (IL)-6, alveolar congestion, as well as lung IL-6, macrophage inflammatory protein (MIP)-2, and myeloperoxidase (MPO) activity were all increased in mice subjected to CLP at 24 but not 72 hours following hemorrhage. This was associated with a marked increase in the susceptibility of these mice to septic mortality. Peripheral blood neutrophils derived from 24 hours post-hemorrhage, but not Sham animals, exhibited an ex vivo decrease in apoptotic frequency and an increase in respiratory burst capacity, consistent with in vivo “priming.” Subsequently, we observed that adoptive transfer of neutrophils from hemorrhaged but not sham-hemorrhage animals to neutropenic recipients reproduce ALI when subsequently septically challenged, implying that this priming was mediated by neutrophils. We also found marked general increases in lung IL-6, MIP-2, and MPO in mice deficient for toll-like receptor (TLR-4) or the combined lack of TLR-4/FasL. However, the TLR-4 defect markedly attenuated neutrophil influx into the lung while not altering the change in local cytokine/chemokine expression. Alternatively, the combined loss of FasL and TLR-4 did not inhibit the increase in MPO and exacerbated lung IL-6/MIP-2 levels even further.
Reports indicate that upward of 50% of those trauma victims that survive the initial period of shock following severe injury develop some form of multiple organ failure (MODS). 1,2 A similar scenario of events is frequently encountered in patients in the intensive care unit where acute lung injury is often the sequela of elective surgical procedures. 3-5 This occurs despite the provision of apparently adequate fluid resuscitation, specific antibiotics, aggressive operative intervention, nutritional support, and recently, antibodies against endotoxin [lipopolysaccharide (LPS)] as well as anti-cytokine therapies in these patients. 2,6,7 In this respect, acute lung injury, resulting from hypotensive insult and/or sepsis, leading to adult respiratory distress syndrome (ARDS) is reported to be one of the most common forms of organ dysfunction encountered in these critically ill trauma patients. 4 Thus, it is paramount to understand the pathophysiological mechanisms and the contributions made by various immune cell components to the development of acute lung injury.
While our basic biological understanding of the neutrophil or monocyte/macrophage inflammatory/apoptotic response in vitro/ex vivo has broadened, it still remains difficult to establish the contribution of the neutrophil/monocyte/macrophage in vivo to the induction of acute lung injury induced by the traumatic shock antecedent to inflammatory or septic challenge. This is, in part, due to the limitations inherent in the use of phagocytes typically obtained from normal healthy human volunteers. Also, while it is possible to design in vitro experiments which begin to approach the in vivo pathological status associated with hypotensive shock or septic insult, these experiments cannot replicate the complexity of the environment within the tissues of intact animals. Similarly, while materials from traumatized human subjects offer some insight into the changes induced in these cells, there is marked heterogeneity in the extent and nature of the injury, variation in the time from injury, significant differences in the medical treatments provided, and relatively limited access to vital tissue types. 8 Complications also arise from the nature of the patient population, as they can be heterogenous with respect to dietary status, sex, genetic background/predisposition (heredity), or age. 8,9 Thus, animal models offer a potentially useful, controllable, alternate scenario in which to address the pathobiology of acute lung injury induced by hemorrhage shock followed by subsequent septic challenge. 10,11
There is a body of literature from ex vivo/in vitro studies suggesting that prior injury can have a “priming” effect on the phagocytic components of the immune system. 12-16 Once this “primed” phagocytic system, eg, neutrophils/macrophage/monocytes, is challenged (“triggered”) by sepsis, the host would respond with an exaggerated inflammatory mediator release, which would, in turn, induce a scenario of events leading to septic shock, multiple organ dysfunction, and death. Immediately following hemorrhagic shock (the first 24 hours), 17,18 it is possible to detect elevated systemic release of pro-inflammatory mediators thought to play a role in the activation of macrophages/neutrophils. However, a clear causal link between this early pro-inflammatory event and phagocyte “priming” leading to organ injury and/or death has been difficult to document in vivo. These difficulties may, in part, reflect temporal differences between the development of phagocyte “priming” versus the onset of immune dysfunction. Phagocyte “priming” is thought to be transient in nature and most evident during the early period following injury. 16,19 In light of this, one would anticipate that the phagocyte’s responsiveness as well as its ability to contribute to acute lung injury might change over time after the initial trauma. However, to our knowledge, only a few other groups 14,20-22 have assessed phagocyte responses and their contributions to acute lung injury (ALI) in mice (an important genetic model system), let alone examined acute lung injury in mice subjected to a salient two-hit model of hemorrhagic shock (as a priming event) followed by septic challenge (triggering). Thus, this study sets out to test the hypothesis that hypotensive shock produces transient priming of the phagocytic arm of the innate immune response, which consequently leaves the mouse more susceptible to acute lung injury as a result of subsequent infectious challenge. Further, that such “priming” for acute lung injury would be effected by cognate genes involved in the regulation of the response to microbes and cell survival.
The aim of this study was, first, to determine whether hemorrhage alone (“priming”) or hemorrhage followed by secondary septic challenge (as a “trigger”) can induce evidence of ALI in mice. The second was to determine to what extent does this hemorrhage-induced priming increase the animal’s susceptibility to the lethal effects of septic challenge. Third, we wished to establish the degree to which induction of ALI in this model is mediated by the neutrophil. Fourth, we examined whether the loss of endotoxin sensitivity/signaling via toll-like receptor (TLR)-4 and/or Fas ligand (FasL) death receptor-driven apoptotic processes have an effect on the hemorrhage-induced changes in the lungs of septically challenged mice.
Materials and Methods
Animals
Male inbred C3H/HeN mice (endotoxin-sensitive) (Charles River, Wilmington, MA), C3H/HeOuJ (endotoxin-sensitive), C3H/HeJ (endotoxin-tolerant; TLR-4 −/−) or C3H/HeJ-FasLgld mice (endotoxin-tolerant; TLR-4 −/−, FasL −/−) (Jackson Laboratory, Bar Harbor, ME), 6 to 8 weeks of age, which had been acclimated in our animal facility no less than 7 to 10 days, were used for the study described here. All of the experiments performed here were carried out in accordance with the National Institutes of Health Guidelines on Laboratory Animals and were approved by the Rhode Island Hospital Committee on Animal Use and Care.
Mouse Model of Hemorrhagic Shock and Resuscitation
For these studies we used a mouse model of non-lethal hemorrhagic shock and resuscitation which we previously described. 10,23,24 Briefly, following overnight fasting (12 to 14 hours) the animals were lightly anesthetized with methoxyflurane, restrained in a supine position, and both femoral arteries were catheterized under aseptic conditions with polyethylene no. 10 tubing. The mice were allowed to awaken, blood pressure (BP) was monitored continuously by attaching one of the catheters to a strain gauge pressure transducer/polygraph, while the other catheter was used for bleeding. The mice were bled to a mean BP of 30 ± 5 mm Hg (pre-hemorrhage BP of ∼95 mm Hg) and maintained at that level for 90 minutes. Ringer’s lactate, equivalent to four times the volume of shed blood, was provided as fluid resuscitation. The catheters were then removed, the vessels ligated, the groin incisions closed, and the mice returned to their cages. The sham animals were anesthetized, restrained in a supine position for the same duration, and blood vessels were catheterized, but not bled.
Mouse Model of Polymicrobial Sepsis
To produce subsequent septic insult in these mice, animals were anesthetized a second time (24 or 72 hours post-hemorrhage) with methoxyflurane, and following a 1-cm midline incision, the cecum was exposed, ligated with 4–0 silk so as to prevent intestinal obstruction, and punctured twice with a 22-gauge needle. 25 The cecum was then returned to the peritoneal cavity and the abdominal incision was closed in two layers (all incisions were bathed in xylocaine). Saline (4 ml/100 × g body weight (BW)) was given subcutaneously to resuscitate the animal. The mice were then returned to their cages and provided food and water ad libitum. 26,27
Blood and Tissue Samples
Heparinized blood samples were obtained by cardiac puncture and processed for plasma at 24 hours post-CLP. The lungs were then lavaged (the fluids clarified and retained) and/or lung tissue harvested (snap-frozen).
It should be noted for some studies in which whole blood was processed using Coulter’s Lysis reagent (Coulter Immunol. Corp., Hialeah, FL) whole blood lysis procedure to remove contaminating red blood cells as previously described. 28 The percentage of viable cells was determined by Trypan blue exclusion. The number of neutrophils and the extent of ex vivo apoptosis induced 24 hours following Sham-Hem or Hem, but before CLP, was determined flow cytometrically using combined Grl (anti-mouse-neutrophil marker clone RB6–8C5, rat IgG2b, BD Pharmingen Inc., San Diego, CA) and the terminal transfer of fluorescein deoxyuridine diphosphate to end terminal site (TUNEL) staining, as previously described in our laboratory. 28
Granulocyte Myeloperoxidase Assessment
Myeloperoxidase (MPO) activity was determined using a method described by Koike et al. 29 Typically, lung tissue was removed from the metofane-euthanized animal, snap-frozen in liquid nitrogen and stored at −70°C until assay. The frozen tissue samples were then thawed, homogenized in 0.2 mmol/L potassium phosphate buffer (PPB, pH 7.4) with a Wheaton Teflon homogenizer (∼3400 rpm on ice) (Wheaton Sci. Prod., Millville, NJ), and centrifuged at 4,000 × g for 30 minutes at 4°C. Pellets were resuspended in 10 volumes of 50 mmol/L PPB (pH 6.0) containing 0.5% hexadecyltrimethyl ammonium bromide and sonicated for 90 seconds. Samples were then centrifuged at 12,000 × g for 10 minutes at 4°C. The supernatants (5 μl/well) and reaction buffer (195 μl), 50 mmol/L PPB (pH 6.0) containing 530 nmol/L o-dianisidine and 150 nmol/L H2O2 were added to a 96-well plate. The plates were read spectrophotometrically at 460 nm for 3 minutes on CERES UV900C Microplate reader (BioTek Inst. Inc., Winoski, VT). MPO activity (U/g) = A460 × 13.5/g, where A460 = rate of change in absorbance between 1 and 3 minutes.
Assessment of Lung Water Content (Edema)
In some animals following exsanguination, the lungs were harvested, rinsed in saline, and weighed immediately (g, wet weight) then dried at 80°C in a Thelco Lab oven and re-weighed until the weight became constant (g, dry weight). The change in the ratio of wet weight to dry weight (Wet wt./Dry wt.) was taken as indicator of organ edema. 30
Assessment of Cytokine Production
IL-6 and MIP-2 levels present in lung tissue homogenates were determined by using the “sandwich ELISA” technique described previously in our laboratory. 31,32 Antibody pairs and cytokine standards for IL-6 and MIP-2 were obtained from BD Pharmingen and R & D Systems (Minneapolis, MN), respectively. Minimum detectable cytokine (sensitivity) was ∼0.03 ng IL-6/ml or ∼0.008 ng MIP-2/ml.
Assessment of Lung Morphology
The left lobe of the mouse lung was inflated at ∼15 mm Hg and fixed with 4% formalin. In some cases, the specimens were then processed, stained with hematoxylin and eosin, and examined by light microscopy. Alternatively, neutrophil tissue infiltration and their in situ morphology was assessed using the Leder stain for granulocyte-specific naphthol AS-D chloroacetate esterase using a kit from Sigma Chemical Company. 33 For some studies alveolar septal thickening was also morphometrically quantitated as an index of tissue injury using MOCHA Image Analysis software (Jandel Sci., San Rafael, CA). In brief, slides were screened in a blinded fashion by the operator and 12 images (magnification, × 200; ∼500:m2/image) were acquired per lung section exclusive of terminal bronchi or major pulmonary vasculature. The percentage of each image that was open alveolar space was determined following thresholding-out the open (bright area) alveolar space and then establishing the pixel number filled by the tissue (septae) using “flood-object measurements” command. This value was, in turn, subtracted from the total pixel number per field, and multiplied by 100 to give the % alveolar space/field.
Neutrophil Depletion
Mice were depleted of neutrophils/polymorphonuclear leukocytes (PMN) by i.v. tail injection of 500 μg of rat anti-mouse PMN antibody (clone RB6–8C5, rat IgG2b) 48 hours before CLP. 28 To the extent that these animals were neutropenic, we observed that circulating blood neutrophil number (which is typically ∼36% PMN in this C3H strain [or ∼4.0 × 106 PMN/2 ml mouse blood]-Hematology/Mouse Phenome Database, Jackson Laboratories, Bar Harbor, ME) was decreased to less than 5% (∼0.57 × 106 total blood PMN) that of untreated mice at 48 hours following antibody administration.
Mouse Blood Neutrophil Isolation
For experiments where purified mouse neutrophils (donor cells) were needed, blood PMN isolation was performed as per manufacturer’s protocol using the NIM·2 Neutrophil Isolation Medium kit (Cardinal Associates, Inc., Santa Fe, NM). Fresh heparinized blood was collected via cardiac puncture and layered over a two-component density gradient system and centrifuged 45 minutes at 900 × g at 20°C. The band formed at the interface of the two-gradient steps containing neutrophils was collected and washed. In our experience, this yields ∼3 to 5 × 106 PMN/ml of blood (∼95% PMN by morphology and 98% viable by Tryphan blue exclusion). To the extent that this protocol leads to overt activation of these cells we found, in preliminary studies, 34 that ex vivo culture of these isolated cells induced no chemokine (MIP-2) release in the absence of a stimulant, such as tumor necrosis factor (TNF)-α. However, the inclusion of TNF-α with PMN- induced release of MIP-2, further implies preservation of the functional capacity of these isolated cells.
Neutrophil Adoptive Transfer Experiments
One × 106 total donor blood PMN isolated from either mice subjected to hemorrhage 24 hours earlier (primed), sham-hemorrhage (unprimed), or naive mice were injected into the tail vein of anesthetized syngeneic C3H/HeN mouse with a 27-gauge needle (0.1 ml) immediately before undergoing CLP. Twenty-four hours later the recipient animals were euthanized and the extent of neutrophil influx (lung MPO and lung morphology) and lung cytokine content was assessed.
Neutrophil Respiratory Burst Capacity
To assess the capacity of isolated mouse blood PMNs to produce a respiratory burst, cells were stimulated using a bovine serum albumin (BSA) immune complex (IC) according to the methods of Jones et al. 35 Briefly, a high binding polystyrene microtiter plate was coated with 100 μl of 10 mg/ml BSA in phosphate-buffered saline (PBS) and incubated one hour at 37°C. The plate was washed three times with PBS, coated with 100 μl rabbit anti-BSA antibody at 10 μg/ml (Sigma Diagnostics, St. Louis, MO) in PBS and incubated 1 hour at 37°C. The plate was again washed three times with PBS. PMN from 3 mice/group, 3 × 105 cells/well in triplicate were mixed with a 2X concentration of Cytochrome C (Sigma Diagnostics, St. Louis, MO) solution in RPMI without phenol red (0.0199 × g cytochrome C/10 ml RPMI plus 10 μl MOPS). The plate was covered with plastic film and placed in a Microplate Bio Kinetics reader (BIOTEK Instruments, Winooski, VT) at 37°C. Optical density readings at dual wavelengths of 550/630 nm were made every 10 minutes for a total of 90 minutes using BIOTEK DeltaSoft3 software. Superoxide production was calculated using the absorbance at (90 minutes-zero) × 15.238 (a predetermined absorbance constant) = nmol superoxide/3 × 105 PMN.
Statistical Analysis
The data are presented as a mean ± SE for each group (n = 4 to 6/group). Differences for cytokine yields and MPO were considered to be significant if P < 0.05, as determined by analysis of variance and a Tukey’s test. Significant changes (at P < 0.05) in percentile or ratio data were established by a Mann-Whitney U-test. Significant difference (P < 0.05) in survival rates were established by a Fisher exact test (n = 18/group).
Results
Hemorrhage Induces Transient Priming for ALI in Septic Mice
In our initial series of studies, we set out to determine whether hemorrhage by itself, sepsis (CLP) alone, or the combination of hemorrhage followed by subsequent septic challenge were sufficient to produce marked evidence of acute lung injury (ALI) as well as pulmonary inflammation. Figure 1 ▶ illustrates that the inflammatory indices such as tissue levels of the pro-inflammatory cytokine IL-6, the chemokine MIP-2, and MPO activity (an index of PMN tissue infiltration) were all increased only in the lungs of mice subjected to both hemorrhage and CLP (24 hours post-hem) (Figure 1) ▶ , but not sham-hemorrhage/sham-CLP, sham-hemorrhage/CLP, or hemorrhage/sham-CLP. Interestingly, this effect on the lungs was lost (or markedly reduced in the case of MIP-2) when sepsis (CLP) was induced 72 hours after hemorrhage as opposed to 24 hours (Figure 1) ▶ .
Figure 1.
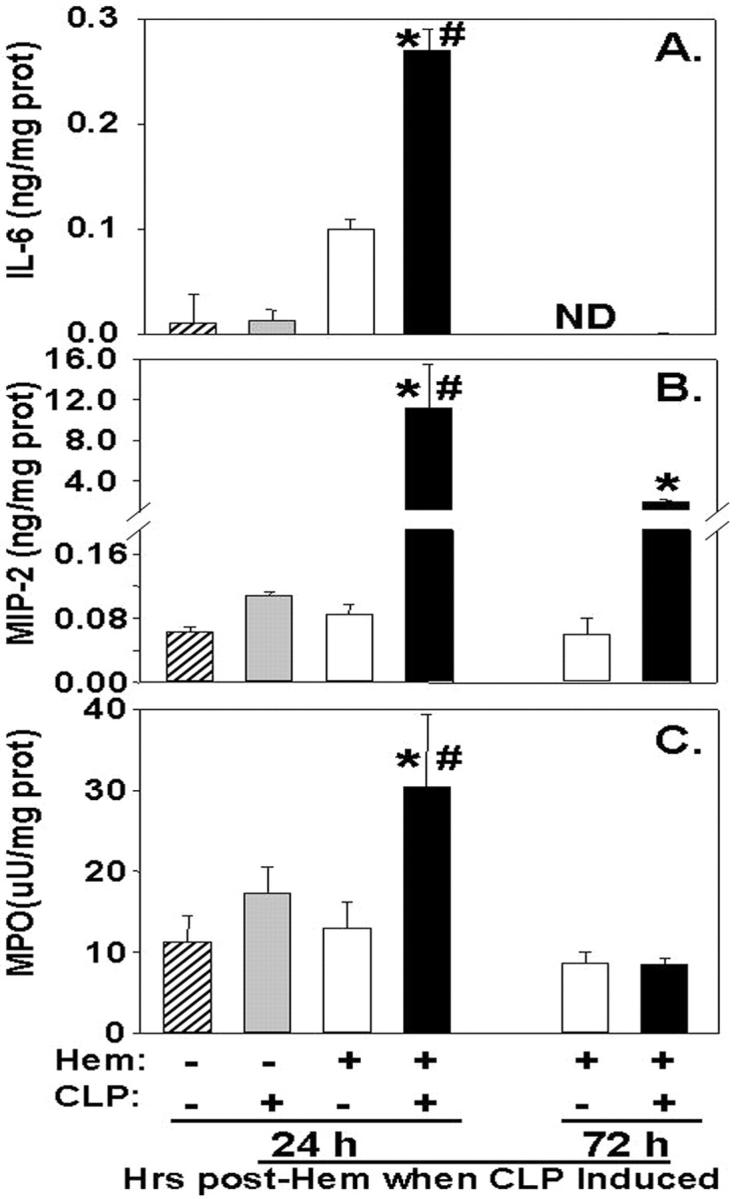
IL-6 (A), MIP-2 (B) and MPO (C) activity in the lung homogenates of C3H/HeN mice subjected to CLP 24 hours following hemorrhage (Hem) as compared to animals simply subjected to Hem alone/Sham-CLP, Sham-Hem/CLP, or Sham/Sham. All 72 hours post-Hem/CLP levels were markedly lower than those seen at 24 hours post-Hem. *, P < 0.05 vs. equivalent Hem alone group; #, P < 0.05 vs. 72 hours Hem before CLP group; n = 4 to 6/group.
To the extent this transient period of susceptibility to lung neutrophil influx and local tissue cytokine/chemokine increase was associated with lung injury, we found, as shown in Figure 2 ▶ , that there was a marked increase in both bronchoalveolar lavage fluid IL-6 levels and lung water content (edema) when animals subjected to hemorrhage 24 hours earlier were subsequently exposed to CLP. Concordant histological examination of caudal lobe lung section(s) of mice subjected to both hemorrhage and sepsis (Figure 3C) ▶ illustrated a consistent increase in alveolar septal thickening, associated with marked cellular infiltrates and alveolar congestion/collapse that was not observed in sham (Figure 3A) ▶ or hemorrhage alone (Figure 3B) ▶ .
Figure 2.

A: IL-6 levels in bronchoalveolar lavages are significantly elevated when compared to Hem/Sham-CLP, Sham-hem/CLP, or Sham-hem/Sham-CLP. B: Lung edema (Wet wt./Dry wt. [g/g]) as an index of organ injury is markedly increased in C3H/HeN mice subjected to CLP 24 hours following hemorrhage (Hem/CLP) as compared to animals simply subjected to Hem/Sham-CLP (Hem), Sham-Hem/CLP (CLP), and Sham-Hem/Sham-CLP (Sham). *, P < 0.05 vs. all other groups; n = 4/group.
Figure 3.
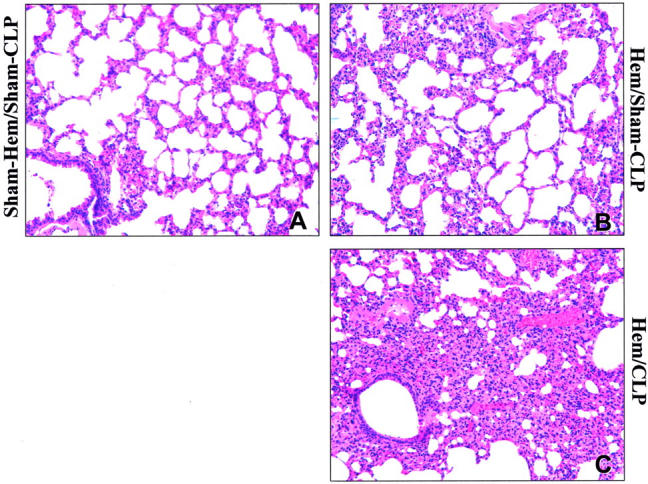
Hematoxylin and eosin stain of representative sections of the caudal lobe of the lung of C3H/HeN mice subjected to Sham-Hem/Sham-CLP (A), Hem/Sham-CLP (B), or Hem/CLP (C) (magnification, ×200). Note: Hemorrhage followed 24 hours later by CLP produced an increase in lung cellularity, septal thickening, and alveolar congestion (C). Such changes are less evident in the Hem/Sham-CLP mouse sections (B) and are largely absent in the Sham-Hem/Sham-CLP animals lung (A).
Having seen this, a question arises as to whether this transient increase in acute lung injury seen in these mice subjected to combined insults of hemorrhage following septic challenge 24 hours after is associated with a change in the animals’ ability to survive the septic challenge. To assess this, we determined the mortality rates of animals subjected to either sham-hemorrhage or hemorrhage followed at 24 hours by CLP. The animals subjected to both hypotensive and septic insult showed a much greater decline in survival than that observed with sham-hemorrhage plus CLP (Figure 4) ▶ .
Figure 4.

Survival of C3H/HeN mice subjected to hemorrhage (Hem) or Sham-Hem 24 hours before CLP. *, P < 0.05 vs. Sham-Hem group; n = 18/group.
Hemorrhage Induces Ex Vivo Suppression of Peripheral Blood Neutrophil Apoptosis and Priming for Enhanced Respiratory Capacity
Having observed that hemorrhage induced an early transient period of priming for increased lung injury on subsequent septic challenge, we attempted to assess the extent to which neutrophils in circulation of hemorrhaged animal exhibit changes indicative of functional priming and/or change in constitutive apoptotic rate. Figure 5 ▶ indicates that ex vivo assessment of peripheral blood from 24 hours post-hemorrhage (but not subjected to CLP) mice while exhibiting a trend toward an increase in the number of leukocytes (WBC), showed a significant increase in the % of leukocytes that were neutrophils, however, the extent of neutrophils that were apoptotic, while low in overall frequency, was markedly decreased. Repeated in vitro assessment of the capacity of purified peripheral blood PMN from these hemorrhaged mice to produce a respiratory burst in response to immune complex stimulation, was consistently greater than that seen from sham-hemorrhaged animals’ cells (Figure 6) ▶ .
Figure 5.
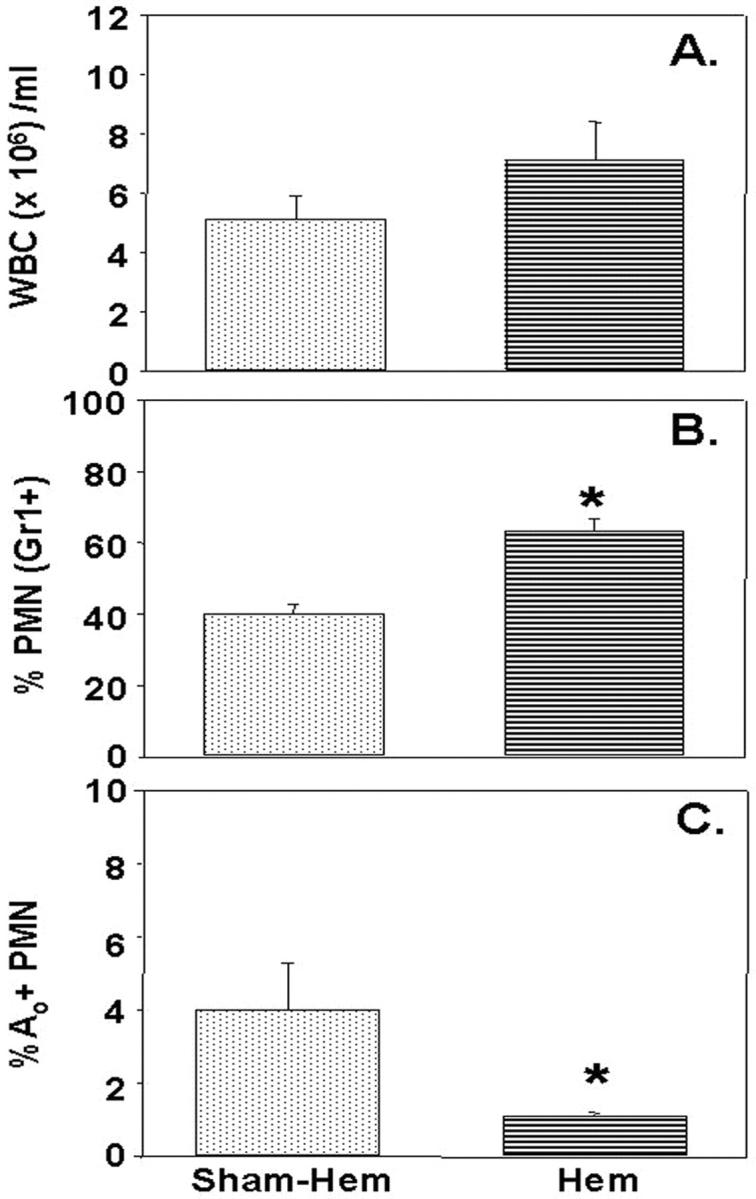
While total leukocyte (WBC) number/ml blood (A) did not significantly change 24 hours post-Hem, there was a marked increase in the % of cells that were neutrophils (PMN; based on Gr1+ staining) (B), however, the frequency of apoptotic (Ao) PMNs markedly declined (C). *, P < 0.05 vs. all other groups; n = 4/group.
Figure 6.

Respiratory burst capacity of C3H/HeN mouse blood neutrophils isolated 24 hours post-Hem was consistently lower than that observed from Sham-Hem animals’ cells. A typical result of four independent experiments is provided.
Neutrophils Serve as the Effector/Mediator of ALI
Having documented that hemorrhage produces a transient period of priming for acute lung injury which appears to be associated with phenotypic and functional changes in the hemorrhage mouse neutrophil population, we asked if this lung injury was the result of a neutrophil mediated process. To address the question, we carried out a series of experiments in which we adoptively transferred blood neutrophils isolated from normal untreated mice, or animals subjected 24 hours earlier to either hemorrhage or sham-hemorrhage, to neutropenic (anti-Gr1 antibody-treated mice) syngeneic recipient mice which were immediately subjected to CLP. When the lungs were harvested 24 hours post-CLP, there was a significant increase in the lung MPO, as well as the tissue IL-6 and MIP-2 levels, in animals having received neutrophils from hemorrhaged donor mice compared to all other groups (Figure 7) ▶ . Morphological examination of representative sections derived from the recipient animals illustrated that only the animals that had received blood neutrophils from a hemorrhaged donor mouse exhibited marked cellular infiltration and alveolar congestion/collapse following septic challenge (see Figure 8 ▶ ). This was morphometrically confirmed by the observation that the alveolar space occupied in the images of septic recipient mouse lungs section that received hemorrhage mouse donor neutrophils was only 53.1 ± 1.36% of the field, compared to either septic animals receiving sham-hemorrhage donor cells (63.8 ± 2.16), sham-septic mice that received hemorrhaged donor animal cells (62.5 ± 1.83%), or a sham-septic mouse that received sham-hemorrhaged donor mouse cells (72.7 ± 1.63%).
Figure 7.
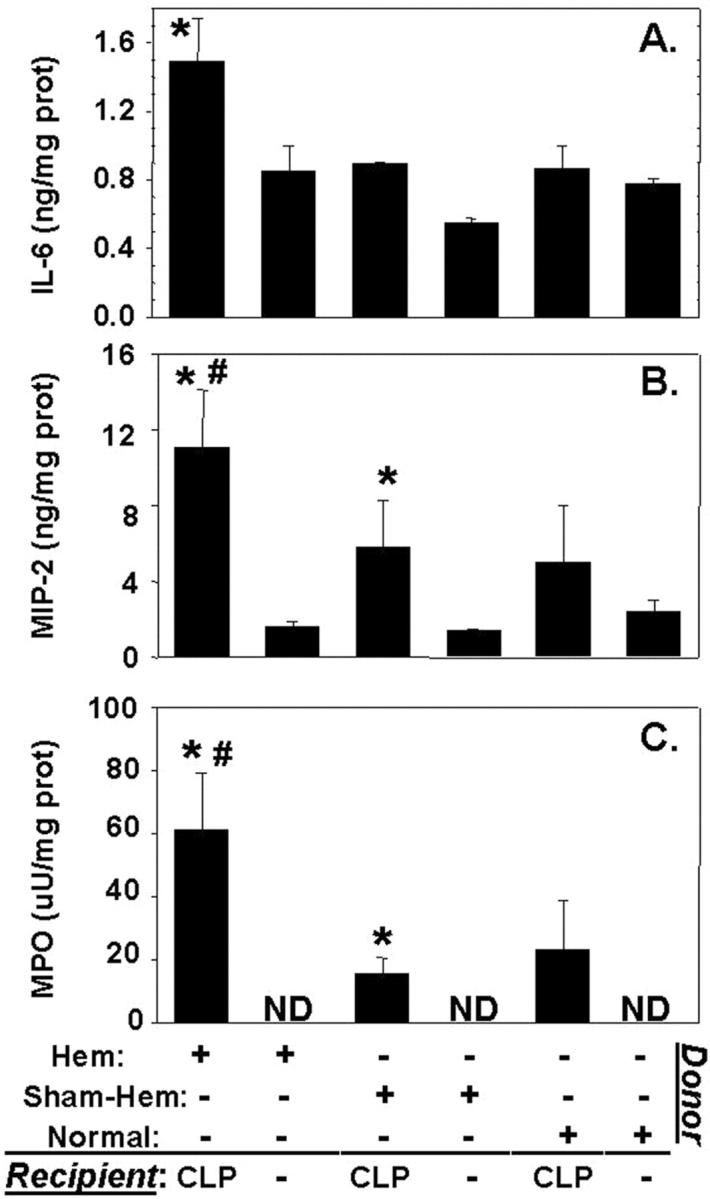
Changes in IL-6 (A), MIP-2 (B), and MPO (C) seen following adoptive transfer of peripheral blood neutrophils isolated from C3H/HeN donor mice which had been hemorrhaged (Hem) (24 hours earlier), Sham-Hem, or were untreated (normal) to neutropenic recipient, C3H/HeN mice that were subsequently subjected to CLP. Values are mean ± SE, n = 4 mice/group. *, P < 0.05 vs. equivalent group not subjected to CLP, while #, P < 0.05 vs. all other groups. ND, not detected.
Figure 8.
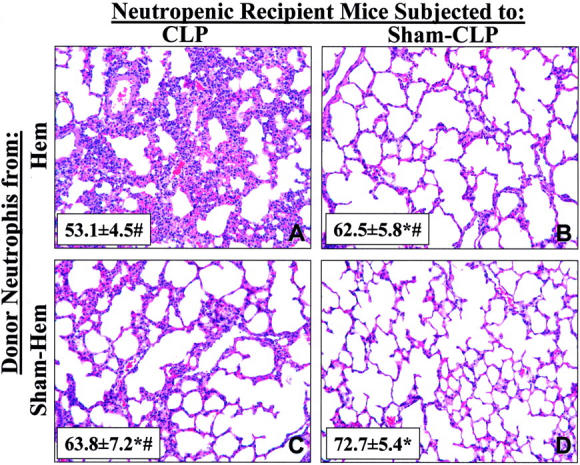
Hematoxylin and eosin stain of representative sections of the left lobe of the lung of recipient neutropenic mice 24 hours post-CLP (A, C) or Sham-CLP (B, D) that had received either hemorrhage (Hem; A, B) or Sham-hemorrhage (Sham-Hem; C, D) donor mouse blood neutrophils (magnification, ×200). Inset, represents the cumulative morphometric determination of % alveolar space present per field from 12 images taken per lung tissue section. Values given as mean ± SEM, n = 3/group. *, P < 0.05 vs. CLP neutropenic mouse receiving PMN from Hem mouse. #, P < 0.05 vs. Sham-CLP neutropenic mouse receiving Sham-hemorrhage mouse donor PMN.
Inability to Signal through TLR-4 and/or FasL Deficiency Has Diverse Effects on Hemorrhage-Induced Priming
Since it has been documented in vitro that the priming of neutrophil function is affected by endotoxin, directly or indirectly, through a number of inflammatory agents, 14,19,36 as well as by death receptor family of proteins (Fas/FasL), 36-38 we wanted to assess the contribution of FasL and/or endotoxin signaling through TLR-4 to the changes in lung inflammatory indices seen here. Figure 9 ▶ illustrates that while the levels of lung IL-6, MIP-2, and MPO were markedly increased in all animals compared to hemorrhage alone, FasL gene deficiency and TLR-4 −/− had a divergent effect on lung cytokine/chemokine expression and lung MPO. FasL deficiency augmented the expression of MIP-2 and IL-6 compared to either the C3H/HeOuJ or C3H/HeJ mice. Alternatively, deficiency of a functional TLR-4 gene in the C3H/HeJ mouse markedly attenuated the influx of neutrophils to the lung as indicated by the reduction in MPO when compared to the C3H/HeJ FasLgld, FasL −/−/TLR-4 −/− mouse or the HeOuJ background control animal. Histological examination suggested that while cellularity and the extent of alveolar congestion/collapse was greatest in the C3H/HeOuJ mouse lungs following combined insults, this was partially attenuated in both the C3H/HeJ and C3H/HeJ-FasLgld mouse lungs (Figure 10) ▶ .
Figure 9.
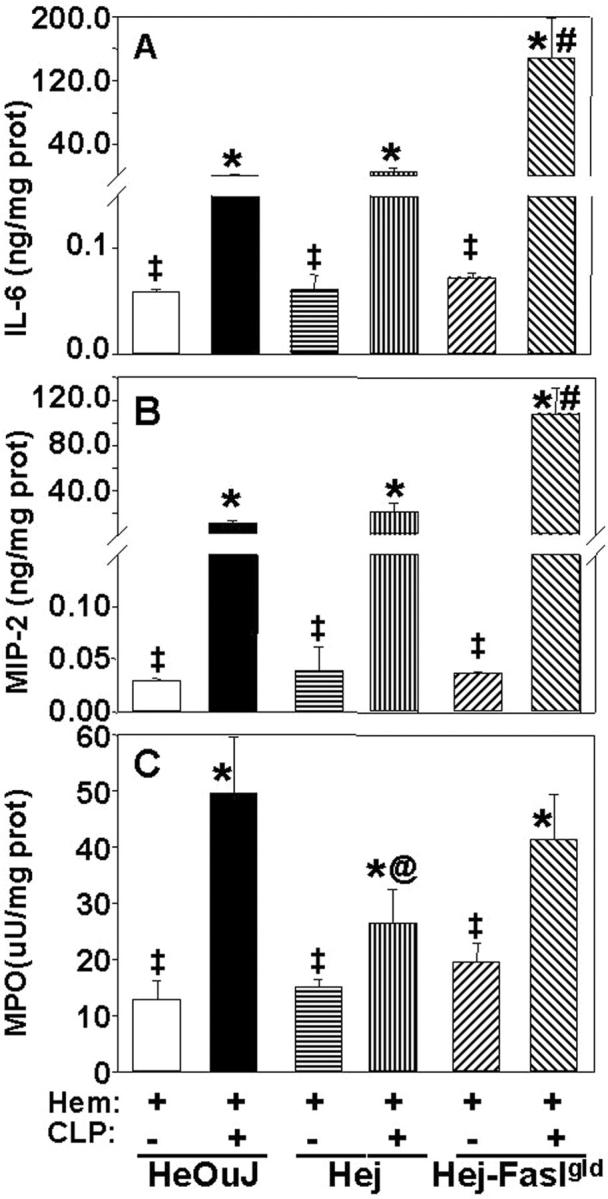
IL-6 (A), MIP-2 (B), and MPO (C) levels in the lung homogenates of C3H/HeOuJ mice hemorrhaged (Hem) 24 hours before CLP compared Hem/Sham-CLP or compared to Hem/CLP, C3H/HeJ (TLR-4 −/−) or Hem/CLP C3H/HeJ-FasLgld (TLR-4 −/−, FasL −/−). Values given as mean ± SE, n = 5 to 6 mice/group. *, P < 0.05 vs. Hem/Sham-CLP, C3H/HeOuJ (HeOuJ), #, P < 0.05 vs. Hem/CLP C3H/HeOuJ or C3H/HeJ (HeJ). @, P < 0.05 vs. Hem/CLP C3H/HeOuJ or C3H/HeJ-FasLgld (gld). ‡, P < 0.05 vs. the equivalent Hem/CLP group.
Figure 10.
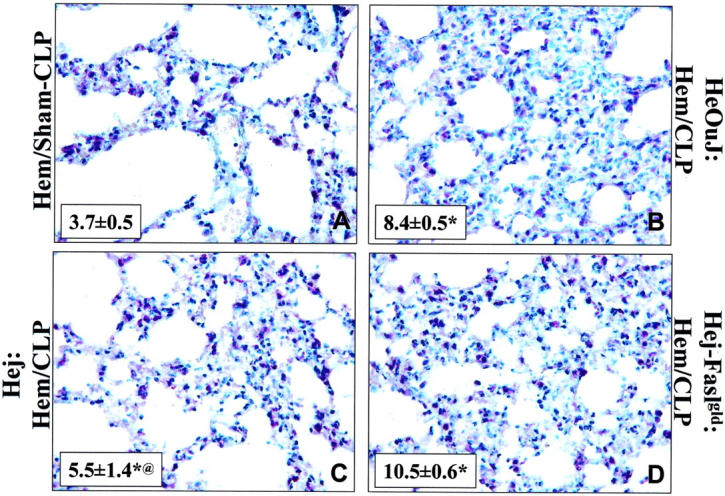
Hematoxylin stain of representative sections of the lung of Hem/Sham-CLP (A); Hem/CLP, C3H/HeOuJ mouse (B); Hem/CLP, C3H/HeJ (C); or Hem/CLP, C3H/HeJ-FasLgld (D) mouse (magnification, ×400). Note: Hemorrhage followed 24 hours later by CLP produced an increase in lung cellularity, septal thickening, and alveolar congestion in all mouse strains, however, the extent of these changes was typically greater in endotoxin-sensitive (TLR+/+)/FasL+/+ mice (A, B) then either the C3H/HeJ (C) or C3H/HeJ-FasLgld (D). Inset, represents cumulative determination of the % PMN (napthol AS-D chloroacetate esterase+ cells) per total number cells counted in 7 to 8 field, 25:m2; magnification, ×400. Values given as mean ± SEM, n = 3/group. *, P < 0.05 vs. Hem/Sham-CLP and @, P < 0.05 vs. HeOuJ:Hem/CLP or Hej-Faslgld:Hem/CLP.
Discussion and Conclusions
Most experimental models trying to address the pathobiology of organ injury start with a healthy animal which is either subjected to septic challenge, eg, microbe/microbial product, or to shock alone, eg, hemorrhage, burn, etc. However, typically the patient in the intensive care unit, besides being subjected to their initial trauma, faces an array of potential secondary stimuli, ie, microbial challenge, hypoxic events, toxic stimuli, repetitive surgical trauma, etc, which are suggested to serve as a trigger for acute lung injury precipitating ARDS and/or MODS. 16,39
There is a body of literature suggesting that prior injury can have a “priming” effect on the phagocytic components of the immune system as well as non-immune cells such as epithelia, endothelia, etc. 12-14,39-41 This “primed” phagocytic system, eg, neutrophils/macrophage/monocytes and/or the surrounding pulmonary endothelia/epithelia, etc, when challenged (“triggered”) by sepsis, is thought to respond with an exaggerated inflammatory mediator release, which would, in turn, induce the scenario described earlier leading to septic shock, multiple organ dysfunction, and death. In this regard, immediately following hemorrhagic shock (the first 24 hours) we have shown that it is possible to detect elevated systemic as well as local tissue release of pro-inflammatory mediators associated with activation of macrophages/neutrophils during the subsequent response to septic challenge. 17,18 Phagocyte “priming” is also suggested to be transient in nature and most evident during this early period following injury. 12,42,43 Thus, one would anticipate that the phagocyte’s responsiveness as well as its ability to contribute to acute lung injury might change over time after the initial trauma. Here we document that this does indeed appear to be the case as the priming response evident at 24 hours following the initial hypotensive insult, as evidenced by marked increase in lung IL-6, MIP-2, and MPO, are lost (or markedly reduced for MIP-2) by 72 hours post-shock. It is worth noting that while a number of cytokines could have been chosen as our indices of local inflammation, due to limitations in available material/sample sizes/volumes, we chose to assess the pleotropic pro-inflammatory cytokine IL-6, as it is one of the most consistently detected cytokines associated with shock/traumatic injury. 6 We also chose to examine the mouse chemokine MIP-2 (one of the homologues of human IL-8) as it is thought to be a critical mediator of neutrophil recruitment/activation. 44 With respect to changes in neutrophil influx in the lung and associated injury, the small size of the mouse as a model restricted our primary examination to changes in tissue MPO, morphological assessment of lung specimen, bronchoalveolar lavage cytokine level IL-6, and change in lung tissue wet to dry weight ratio as an index of edema. The observation of priming in this mouse model is also in keeping with the studies of Fan et al 14 who demonstrated that hemorrhage in a rat can produce priming for lung injury following subsequent intratracheal exposure to endotoxin. This increase in susceptibility to lung injury 24 hours following hemorrhage is also associated with an increase in mortality resultant from sepsis. This suggests that the lung injury produced here, along with other organ dysfunction/damage, contributes to reducing the host’s ability to ward off the lethal effects of the septic challenge. In this respect, while it was not our objective to examine other organ systems in this study, we previously demonstrated that, in response to CLP alone, there is a marked decrease in cardiac output and an increase in liver damage, 45 on which prior hemorrhage, as produced here, would appear to add the further complication of lung injury.
Numerous studies looking at various inflammatory mediators have suggested that neutrophils are a primary mediator/effector cell involved in producing ALI. However, often one of the more difficult hurdles to overcome with human materials has been the ability to verify in vivo that inflammatory agents or conditions that produce what appears to be “priming” in vitro exhibit such a potential/capacity in intact animals. Similarly, while neutrophils derived from patients’ systemic blood appear to have augmented inflammatory/cytotoxic capacity, we typically only are able to extrapolate on the ability of such “priming” of these cells to actually contribute to tissue injury in the patient.
In animal models, we can begin to address issues such as the capacity of neutrophils from animals primed by hemorrhagic shock to mediate changes in the lungs of these animals following septic insult. However, as the effect of hemorrhage on mouse blood PMN has not been examined, we initially set out to determine whether hypotensive shock could induce changes in mouse neutrophil apoptosis (ex vivo) and respiratory capacity (in vitro) that were consistent with functional priming seen in injured patient’s cells. 12,39,46 Similar to the types of observations that have been made in patients, 12,39,46 we found there was a marked decline in ex vivo peripheral blood mouse neutrophil apoptosis in hemorrhaged mice. In vitro assessment of these isolated blood neutrophils from hemorrhaged mice consistently exhibited a higher respiratory burst capacity.
To the extent that these cells, present in circulation 24 hours post-hemorrhage (just before CLP), retain an enhanced ability to induce subsequent lung inflammation/injury, we chose to compare the capacity of isolated hemorrhage mouse blood neutrophils (donor) to those from Sham protocol or unmanipulated mice to produce ALI. To assess this potential, we adoptively transferred the donor mouse neutrophils to recipients depleted of neutrophils (48 hours prior with anti-Gr1 [mouse PMN] Ab). Following this, mice were challenged with CLP. Neutrophils from hemorrhaged animals produced a marked increase in CLP-induced lung MIP-2 and MPO levels indicating that priming is transferable by transfer of neutrophils. This observation is supported, in part, by the recent findings of Moxley et al 47 in rats, in which they showed that cells isolated from bronchoalveolar lavage fluids of endotoxin-challenged animals could produce ALI when re-instilled into a donor rat’s lungs. However, their model differs significantly from the model used here. First, their model was essentially a single high dose lethal endotoxin insult model. Second, the cells used for transfer represent a mixed population that includes a large percentage of macrophages, lymphocytes, and neutrophils. Third, the transferred cells do not transmigrate from circulation as they are instilled intratracheally into the lungs (and thus, are not naturally recruited from the blood). Fourth, the presence of a normal functional recipient neutrophil pool is a potential confounding effect. Here we have used sub-lethal hypotensive shock (hemorrhage) as opposed to lethal endotoxic shock to prime the donors’ cells. The neutrophil population was purified (98%) from blood and then transferred, intravenously to a recipient whose own neutrophils had been previously depleted, so as to minimize the recipient granulocyte contributions. Thus, unlike the study of Moxley et al, 47 these results indicate that purified donor neutrophils, as opposed to other possible contaminant populations, serve as the effector/mediator of hemorrhage induce priming for ALI. However, we realize our results do not address the nature of the processes, the mediators and/or the type of cell-cell interactions (autocrine and/or paracrine) which are induced initially by hemorrhage in the donor which then produce this functional change (priming) in the peripheral blood neutrophil pool.
It has been reported that exposure to endotoxin can induce the release of a variety of inflammatory agents, such as IL-1β, TNFα, IL-6, IL-8, MIP-2, soluble FasL, etc, which are thought to induce in vitro priming in granulocytes. 14,19,36 It has also been shown that the constitutive process of neutrophil apoptosis is altered by priming stimuli. 48-51 Neutrophils, as well as other phagocytes, and endothelial and epithelial cells in the lung all possess death receptors for the Fas pathway. This pathway has also been implicated as a contributor to pathology of lung injury both in animal models of acute lung injury and ARDS patients. 36-38 In light of this, we attempted to assess whether deficiency in the capacity to respond to endotoxin due to gene deficiency in the toll-like receptor (TLR-4) 52 and/or the loss of the death receptor gene product for FasL 53 would alter the extent of lung injury produced in mice by the combined insult of hemorrhage and CLP.
Interestingly, we found that irrespective of the inability to respond to endotoxin via TLR-4 alone or the combined TLR-4 deficiency and the lack of a functional FasL gene product, a marked increase in lung IL-6, MIP-2, and MPO levels were still seen when compared to hemorrhage alone. This indicates that all these animals are susceptible to hemorrhage-induced priming. However, the extent of local inflammation observed was divergent in these animals. While deficiency of the TLR-4 gene product in the HeJ markedly suppressed neutrophil influx, as indicated by the drop in MPO levels, lung neutrophil number (%), and reduced alveolar collapse, it did not alter the extent of local cytokine/chemokine expression. This suggests that signaling through TLR-4 is a necessary component in maximizing neutrophil recruitment and/or migration to the lung but does not markedly effect priming for the cytokine response to sepsis.
Such an observation is in keeping with the finding that neutrophils, unlike other granulocyte populations, but similar to macrophage/monocytes, express functional TLR-4 and TLR-2 receptors. 54 How TLR-4 loss might alter mouse neutrophil function remains to be established in this setting. These divergent findings also suggest that suppression in neutrophil recruitment is not related to a lack of chemokine expression. This finding of reduced neutrophil recruitment also differs from the work of Hazoit et al 55 who indicate that neutrophil recruitment increased in animals subjected to gram-negative microbial challenge alone which were deficient in either CD14 or TLR-4 gene products. One possible explanation is that the two-hit process in which hemorrhage-induced priming when followed by polymicrobial septic challenge is distinct from those changes seen with simple mono-specific bacterial/toxin challenge.
Stimulation of Fas/FasL pathway is primarily thought to be involved with the induction of apoptotic cell death. With respect to most immune cells, beside the suggested developmental role of apoptosis, the induction of such FasL-mediated cell death is thought to be an important component in controlling the extent of the inflammatory process. 56,57 This is based on the observation that activated immune cells typically increase their sensitivity to Fas-mediated cell death through increase of Fas on these cells. Thus, FasL serves as an anti-inflammatory agent involved in the resolution of an immune response. Alternatively, a number of recent studies 58,59 have also documented that ligation of the Fas receptor can also signal the increased release of various inflammatory mediators, implying that the Fas/FasL system can serve as a pro-inflammatory mechanism in certain cell types or tissues. The observation made here of increased lung cytokine/chemokine levels and the inability to alter neutrophil influx (as indicated by MPO) implies that the loss of FasL has removed a potential anti-inflammatory pathway in these animals. We would speculate that this is due to the inability to delete, via apoptosis, activated macrophages, neutrophils, or other immune and possibly non-immune cells that are contributing to the release of these cytokines and the neutrophil influx. Further studies with possible targeted depletion of some of these cell populations would be needed to establish this.
Acknowledgments
We thank Ms. Ginny Hovanesian and Mr. Paul Monfils at the Core Laboratories Facilities at Rhode Island Hospital for the assistance with the histological preparation/imaging done in this study.
Footnotes
Address reprint requests to Alfred Ayala, Aldrich 227, Division of Surgical Research, Rhode Island Hospital, 593 Eddy Street, Providence, RI 02903. E-mail: aayala@lifespan.org.
Supported in part by research funds from Lifespan-Rhode Island Hospital and National Institutes of Health grant RO1-GM53209 (to A.A.).
References
- 1.Longnecker DE: Introduction. Fluid Resuscitation. Pope A French G Longnecker DE eds. State of the Science for Testing Combat Casualties and Civilian Injuries. 1999:9-16 National Academy Press, Washington DC [PubMed]
- 2.Baue AE, Durham R, Faist E: Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure: are we winning the battle? Shock 1998, 10:79-89 [DOI] [PubMed] [Google Scholar]
- 3.Murray JF, Matthay MA, Luce JM, Flick MR: An expanded definition of adult respiratory distress syndrome. Am J Respir Crit Care Med 1988, 138:720-723 [DOI] [PubMed] [Google Scholar]
- 4.Bernard GR, Artigas A, Brigham KL: The American-European Consensus Conference on ARDS: definitions, mechanisms, relevant outcomes, and clinical trails coordination. Am J Respir Crit Care Med 1994, 149:818-824 [DOI] [PubMed] [Google Scholar]
- 5.Rinaldo JE, Rogers RM: Adult respiratory distress syndrome: changing concepts of lung injury and repair. N Engl J Med 1982, 306:900-910 [DOI] [PubMed] [Google Scholar]
- 6.Barriere SL, Lowry SF: An overview of mortality risk prediction in sepsis. Crit Care Med 1995, 23:376-393 [DOI] [PubMed] [Google Scholar]
- 7.Natanson C, Esposito CJ, Banks SM: The sirens’ songs of confirmatory sepsis trails: selection bias and sampling error. Crit Care Med 1998, 26:1927-1931 [DOI] [PubMed] [Google Scholar]
- 8.Zellweger R, Ayala A, Chaudry IH: Predisposing factors: effects of sex, nutritional factors, and age on immunity following shock and sepsis. Redl H Schlag G eds. Cytokines in Severe Sepsis and Septic Shock. 1998:57-77 Birkhauser Publishing, Ltd, Basel, Switzerland
- 9.Angele MK, Schwacha MG, Ayala A, Chaudry IH: Effect of gender and sex hormones on immune responses following shock. Shock 2000, 14:81-90 [DOI] [PubMed] [Google Scholar]
- 10.Ayala A, Wang P, Chaudry IH: Shock models: hemorrhage. Souba W Wilmore D eds. Surgical Research. 2001:317-330 Academic Press, San Diego
- 11.Simms HH, Souba WW, Wilmore D: Models of ARDS, aspiration. Souba W Wilmore D eds. Surgical Research. 2000:317-330 Academic Press, San Diego
- 12.Friese RS, Rehring TF, Wollmering M, Moore EE, Ketch LL, Banerjee A, Harken AH: Trauma primes cells: editorial review. Shock 1994, 1:388-394 [DOI] [PubMed] [Google Scholar]
- 13.Bankey PE, Hill S, Geldon D: Sequential insult enhances liver macrophage-signaled hepatocyte dysfunction. J Surg Res 1994, 57:185-191 [DOI] [PubMed] [Google Scholar]
- 14.Fan J, Marshall JC, Jimenez M, Shek PN, Zagorski J, Rotstein OD: Hemorrhagic shock primes for increased expression of cytokine-induced neutrophil chemoattractant in the lung: role in pulmonary inflammation following lipopolysaccaride. J Immunol 1998, 161:440-447 [PubMed] [Google Scholar]
- 15.Hauser CJ, Fekete Z, Goodman ER, Kleinstein E, Livingston DH, Deitch EA: CXCR2 stimulation primes CXCR1 [Ca2+]i responses to IL-8 in human neutrophils. Shock 1999, 12:428-437 [DOI] [PubMed] [Google Scholar]
- 16.Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA: Post-injury multiple organ failure: the role of the gut. Shock 2001, 15:1-10 [DOI] [PubMed] [Google Scholar]
- 17.Ayala A, Perrin MM, Meldrum DR, Ertel W, Chaudry IH: Hemorrhage induces an increase in serum TNF which is not associated with elevated levels of endotoxin. Cytokine 1990, 2:170-174 [DOI] [PubMed] [Google Scholar]
- 18.Ayala A, Perrin MM, Ertel W, Chaudry IH: Differential effects of haemorrhage on Kupffer cells: decreased antigen presentation despite increased inflammatory cytokine (IL-1, IL-6, and TNF) release. Cytokine 1992, 4:66-75 [DOI] [PubMed] [Google Scholar]
- 19.Abraham E, Carmody A, Shenkar R, Arcaroli J: Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol 2000, 279:L1137-L1145 [DOI] [PubMed] [Google Scholar]
- 20.Hierholzer C, Harbrecht B, Menezes JM, Kane J, MacMicking J, Nathan CF, Peitzman AB, Billiar TR, Tweardy DJ: Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med 1998, 187:917-928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shenkar R, Abraham E: Mechanisms of neutrophil activation after hemorrhage or endotoxemia: roles of reactive oxygen intermediates, NF-κ B, and cyclic AMP response element binding protein. J Immunol 1999, 163:954-962 [PubMed] [Google Scholar]
- 22.Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, Standiford TJ: IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J Immunol 1999, 162:392-399 [PubMed] [Google Scholar]
- 23.Ayala A, Perrin MM, Chaudry IH: Defective macrophage antigen presentation following haemorrhage is associated with the loss of MHC class II (Ia) antigens. Immunology 1990, 70:33-39 [PMC free article] [PubMed] [Google Scholar]
- 24.Ayala A, Perrin MM, Wang P, Ertel W, Chaudry IH: Hemorrhage induces enhanced Kupffer cell cytotoxicity while decreasing peritoneal or splenic macrophage capacity: involvement of cell-associated TNF and reactive nitrogen. J Immunol 1991, 147:4147-4154 [PubMed] [Google Scholar]
- 25.Ayala A, Perrin MM, Kisala JM, Ertel W, Chaudry IH: Polymicrobial sepsis selectively activates peritoneal but not alveolar macrophage to release inflammatory mediators (IL-1, IL-6, and TNF) Circ Shock 1992, 36:191-199 [PubMed] [Google Scholar]
- 26.Fink MP, Heard SO: Laboratory models of sepsis and septic shock. J Surg Res 1990, 49:186-196 [DOI] [PubMed] [Google Scholar]
- 27.Chaudry IH, Ayala A, Singh G, Wang P, Hauptman JG: Rodent models of endotoxemia and sepsis. Schlag G Redl H eds. Pathophysiology of Shock, Sepsis, and Organ Failure. 1993:1048-1059 Springer-Verlag, Berlin
- 28.Ayala A, Karr SM, Evans TA, Chaudry IH: Factors responsible for peritoneal granulocyte apoptosis during sepsis. J Surg Res 1997, 69:67-75 [DOI] [PubMed] [Google Scholar]
- 29.Koike K, Moore EE, Moore FA, Read RA, Carl VS, Banerjee A: Gut ischemia/reperfusion produces injury independent of endotoxin. Crit Care Med 1994, 22:1438-1444 [DOI] [PubMed] [Google Scholar]
- 30.Wang P, Ba ZF, Lu M-C, Ayala A, Harkema JM, Chaudry IH: Measurement of circulating blood volume in vivo after trauma-hemorrhage and hemodilution. Am J Physiol 1994, 266:R368-R374 [DOI] [PubMed] [Google Scholar]
- 31.Song GY, Chung CS, Jarrar D, Chaudry IH, Ayala A: Evolution of an immune suppressive macrophage phenotype as a product of p38 MAPK activation in polymicrobial sepsis. Shock 2001, 15:42-48 [DOI] [PubMed] [Google Scholar]
- 32.Angele MK, Knoferl MW, Schwacha MG, Ayala A, Bland KI, Cioffi WG, Josephson SL, Chaudry IH: Hemorrhage decreases macrophage inflammatory protein 2 and interleukin-6 release. Ann Surg 1999, 229:651-661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burstone BA: Enzyme Histochemistry and Its Application in the Study of Neoplasms. 1962:88-113 Academic Press, New York
- 34.Lomas J, Chung CS, Song GY, Grutkoski PS, Dunican AL, Simms HH, Ayala A: Role of MIP-2 in suppression of neutrophil apoptosis. Shock 2001, 15:74S [Google Scholar]
- 35.Jones SL, Sharief Y, Chilcoat CD: Signaling mechanism for equine neutrophil activation by immune complexes. Veterinary Immunol Immunopathol 2001, 82:87-100 [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H: Up-regulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med 2000, 161:237-243 [DOI] [PubMed] [Google Scholar]
- 37.Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC: Fas (CD95) induces alveolar epithelial cell apoptosis in vivo. Am J Pathol 2001, 158:153-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kunihiko K, Fujiwara I, Nakajima H: Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am J Respir Crit Care Med 2001, 163:762-769 [DOI] [PubMed] [Google Scholar]
- 39.Meldrum DR, Cleveland JC, Jr, Moore EE, Patrick DA, Banerjee A, Harken AH: Adaptive and maladaptive mechanisms of cellular priming. Ann Surg 1997, 226:587-598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooug-Holub E, Toews GB, Van Iwaarden JF, Strieter RM, Kunkel SL, Paine R, Standiford TJ: Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect Immun 1997, 65:1139-1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ: CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun 2000, 68:4289-4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore FA, Moore EE, Poggetti RS, McAnena OJ: Does early portal bacteremia/endotoxemia prime for post-injury multiple organ failure? Faist E Meakins J Schildberg FW eds. Host Defense Dysfunction in Trauma, Shock, and Sepsis. 1993:899-902 Springer-Verlag, Berlin-Heidelberg
- 43.Lederer JA, Rodrick ML, Mannick JA: The effects of injury on the adaptive immune response. Shock 1999, 11:153-159 [DOI] [PubMed] [Google Scholar]
- 44.Call DR, Nemzek JA, Ebong SJ, Bolgos GR, Newcomb DE, Wollenberg GK, Remick DG: Differential local and systemic regulation of the murine chemokines KC and MIP2. Shock 2001, 15:278-284 [DOI] [PubMed] [Google Scholar]
- 45.Chung CS, Yang SL, Song GY, Lomas J, Wang P, Simms HH, Chaudry IH, Ayala A: Inhibition of Fas signaling prevents hepatic injury and improves organ blood flow. Surgery 2001, 130:339-345 [DOI] [PubMed] [Google Scholar]
- 46.Biffl WL, Moore EE, Zallen G, Johnson JL, Gabriel J, Offner PJ, Silliman CC: Neutrophils are primed for cytotoxicity and resist apoptosis in injured patients at risk for multiple organ failure. Surgery 1999, 126:198-202 [PubMed] [Google Scholar]
- 47.Moxley MA, Baird TL, Corbett JA: Adoptive transfer of acute lung injury. Am J Physiol 2000, 279:L985-L993 [DOI] [PubMed] [Google Scholar]
- 48.Dunican AL, Leuenroth SJ, Grutkoski P, Ayala A, Simms HH: TNF-α-induced suppression of PMN apoptosis is mediated through IL-8 production. Shock 2000, 14:284-289 [DOI] [PubMed] [Google Scholar]
- 49.Watson WG, Rotstein OD, Paraodo J, Jimenez M, Soric I, Bitar R, Marshall JC: Impaired apoptotic death signaling in inflammatory lung neutrophils is associated with decreased expression of interleukin-1β converting enzyme family proteases (caspases). Surgery 1997, 122:163-172 [DOI] [PubMed] [Google Scholar]
- 50.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A: Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 1992, 80:2012-2020 [PubMed] [Google Scholar]
- 51.Grutkoski PS, D’Amico R, Ayala A, Simms HH: IL-1β stimulation induces paracrine regulation of PMN function and apoptosis. Shock 1999, 12:373-381 [DOI] [PubMed] [Google Scholar]
- 52.Poltorak A, He X, Smironva I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B: Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998, 282:2085-2088 [DOI] [PubMed] [Google Scholar]
- 53.Nagata S, Suda T: Fas and Fas ligand: lpr and gld mutations. Immunol Today 1995, 16:39-43 [DOI] [PubMed] [Google Scholar]
- 54.Sabroe I, Jones EC, Usher LR, Whyte MKB, Dower SK: Toll-like receptor (TLR)2 and TLR4 in human peripheral blood granulocytes: a critical role for monocytes in leukocyte lipopolysaccharide responses. J Immunol 2002, 168:4701-4710 [DOI] [PubMed] [Google Scholar]
- 55.Haziot A, Hijiya N, Gangloff SC, Silver J, Goyert SM: Induction of a novel mechanism of accelerated bacterial clearance by lipopolysaccharide in CD14-deficient and toll-like receptor 4-deficient mice. J Immunol 2001, 166:1075-1078 [DOI] [PubMed] [Google Scholar]
- 56.Lynch DH, Ramsdell F, Alderson MR: Fas and FasL in the homeostatic regulation of immune responses. Immunol Today 1995, 16:569-574 [DOI] [PubMed] [Google Scholar]
- 57.Van Parijs L, Biuckians A, Abbas AK: Functional roles of Fas and Bcl-2-regulated apoptosis of T lymphocytes. J Immunol 1998, 160:2065-2071 [PubMed] [Google Scholar]
- 58.Hohlbaum AM, Moe S, Marshak-Rothstein A: Opposing effects of transmembrane and soluble Fas ligand expression on inflammation and tumor survival. J Exp Med 2001, 191:1209-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kell MR, Shelley O, Mannick JA, Guo Z, Lederer JA: A central role of CD95 (Fas) in T-cell reactivity after injury. Surgery 2000, 128:159-164 [DOI] [PubMed] [Google Scholar]