

Abstract
Chronic diseases of the kidney have a progressive course toward organ failure. Common pathway mechanisms of progressive injury, irrespectively of the etiology of the underlying diseases, include glomerular capillary hypertension and enhanced passage of plasma proteins across the glomerular capillary barrier because of impaired permselective function. These changes are associated with podocyte injury and glomerular sclerosis. Direct evidence for causal roles is lacking, particularly for the link between intraglomerular protein deposition and sclerosing reaction. Because transforming growth factor-β1 (TGF-β1) is the putative central mediator of scarring, we hypothesized that TGF-β1 can be up-regulated by protein overload of podocytes thereby contributing to sclerosis. In rats with renal mass reduction, protein accumulation in podocytes as a consequence of enhanced transcapillary passage preceded podocyte dedifferentiation and injury, increase in TGF-β1 expression in podocytes, and TGF-β1-dependent activation of mesangial cells. Angiotensin-converting enzyme inhibitor prevented both accumulation of plasma proteins and TGF-β1 overexpression in podocytes and sclerosis. Albumin load on podocytes in vitro caused loss of the synaptopodin differentiation marker and enhanced TGF-β1 mRNA and protein. Conditioned medium of albumin-stimulated podocytes induced a sclerosing phenotype in mesangial cells, an effect mimicked by TGF-β1 and blocked by anti-TGF-β1 antibodies. Thus, the passage of excess plasma proteins across the glomerular capillary wall is the trigger of podocyte dysfunction and of a TGF-β1-mediated mechanism underlying sclerosis. Agents to reduce TGF-β1, possibly combined with angiotensin blockade, should have priority in novel approaches to treatment of progressive nephropathies.
Progression of kidney disease is a major health care problem in the United States and worldwide, such that the provision of adequate treatment to all patients is absorbing a large proportion of the health care budget and is being looked at with enormous concern by policymakers. The key lesion in glomeruli is sclerosis, consisting in the accumulation of extracellular matrix material and obliteration of the capillary filter that contribute to the loss of renal function. Putative factors that underlie sclerosis include high intraglomerular capillary pressure, 1,2 glomerular stretching and hypertrophy, 3 and the passage of excess amounts of plasma proteins across the glomerular capillary filter. 4 These factors by yet undefined cellular mechanisms in vivo may lead to synthesis of transforming growth factor (TGF)-β1 and other mediators of injury possibly amenable to pharmacological manipulation. Plasma proteins are also ultrafiltered in excess amounts and thus may promote glomerular cell dysfunction in settings of high intraglomerular capillary pressure, 4,5 a maladaptive response to any loss of critical amounts of functioning nephrons. Proteinuria is a potent predictor of progression in humans 6 and precedes sclerosis in virtually all models of diseases of the glomerular filtering barrier. 4 However, direct evidence for the causal role of enhanced passage of proteins in the induction of a prosclerosing response is lacking.
Podocyte dysfunction 4,7-12 and local production of TGF-β1 13,14 have been tightly implicated in the pathogenesis of glomerulosclerosis. The highly specialized podocyte is endowed with foot processes that provide support and permselective function to the filtering barrier. It is also the primary target of factors that may perpetuate injury. The systemic injection of albumin to rats caused podocyte abnormalities, possibly via protein overload of the cell. 15-19 Despite its relevance to sclerosis, this mechanism remained controversial, partly because of the lack of available models using characterized podocytes in vitro. Likewise elusive are the common pathway stimuli leading to the exuberant synthesis of TGF-β1, which was consistently associated with abnormal matrix accumulation in experimental models 20-22 and human renal diseases. 23-25 The role of TGF-β1 is attributable to its ability to promote sclerosing phenotypes, namely in mesangial cells and fibroblasts, 20,21 to the extent that both the transfection of the TGF-β1 gene into normal kidneys in rats and the transgenic TGF-β1 expression in mice induced an increase in glomerular TGF-β1 production and extracellular matrix accumulation. 22 Here we tested the hypothesis that TGF-β1 can be up-regulated in dysfunctional podocytes as a consequence of the enhanced passage of plasma proteins through the glomerular barrier, thereby activating a reaction that contributes to sclerosis. We chose the remnant kidney model in rats with extensive reduction of the renal mass (RMR) and an in vitro approach using differentiated podocytes. Because angiotensin-converting enzyme (ACE) inhibitors have the peculiar property of limiting the passage of proteins across the barrier, 26-29 we investigated whether lisinopril by this action could prevent TGF-β1 synthesis, activation of mesangial cells, and glomerulosclerosis.
Materials and Methods
Animals
Studies were conducted in male Sprague Dawley, CD-COBS rats (275 to 300 g initial body weight) obtained from Charles River SpA (Calco, Italy). The animals were housed in a constant temperature room with a 12-hour dark/12-hour light cycle and fed a standard diet. Animal care and treatment were conducted in conformity with the institutional guidelines that are in compliance with national (D.L. n.116, G.U., suppl 40, 18 Febbraio 1992, Circolare No 8, G.U., 14 Luglio 1994) and international laws and policies (EEC Council Directive 86/609, OJL 358, Dec 1987; Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996).
Disease Model and Protocol
Five-sixths of renal mass ablation was accomplished by surgical removal of the right kidney and ligation of two or three extrarenal branches of the left renal artery 5 in anesthetized rats. Age-matched rats were used as controls after sham operation, consisting of a laparotomy and manipulation of renal pedicles. Three groups of rats with renal mass reduction (n = 7 each group) were sacrificed at 7, 14, and 30 days after surgery, respectively; sham-operated controls were sacrificed at day 30 (n = 7). To assess the effects of ACE inhibitor, rats with renal mass reduction received lisinopril (25 mg/L in the drinking water) 30,31 starting from 1 day after surgery and then sacrificed at 30 days (n = 7). In all groups 24-hour urines were collected in metabolic cages both before the time of disease induction and at sacrifice for the determination of urinary protein excretion levels. Systolic blood pressure and serum creatinine were assessed at 30 days in RMR rats either untreated or treated with lisinopril.
Systolic Blood Pressure
Systolic blood pressure was recorded by tail plethysmography in conscious rats. 32
Analytic Methods
Proteinuria was determined by the modified Coomassie blue G dye-binding assay for proteins with bovine serum albumin as standard. 33 Serum creatinine was measured by the alkaline picrate method. 34
Tissue Preparation
At the end of the study, the animals were anesthetized with sodium pentobarbital intraperitoneally (0.1 ml/100 g body wt of a 60 mg/ml solution in saline) and the kidneys were fixed by perfusion via abdominal aorta. 35 Kidneys were perfused with Hanks’ solution for 5 minutes and then fixed with periodate-lysine paraformaldehyde fixative 36 for 10 minutes, followed by overnight fixation in the same fixative at 4°C. The tissue fragments from remnant kidneys were taken from the center of noninfarcted areas. Fixed tissue specimens were extensively washed with phosphate-buffered saline (PBS) (0.9% NaCl in 10 mmol/L of sodium phosphate buffer, pH 7.4) and stored in the same buffer.
Immunohistochemistry
The tissue specimens were infiltrated by immersion in 30% sucrose/PBS for at least 1 hour at room temperature, embedded in OCT medium, and frozen in liquid nitrogen. Tissue sections were cut at 5-μm thickness using a Mikrom 500 O cryostat (Mikrom, Walldorf, Germany) and either stained immediately or stored at −20°C until further processing. Nonspecific binding of antibodies was blocked either with PBS/1% bovine serum albumin or, for the detection of albumin, with PBS/1% gelatin for 15 minutes (room temperature). Sections were incubated for direct immunofluorescence with fluorescein isothiocyanate (FITC)-conjugated goat anti-rat IgG (30 μg/ml in PBS; Jackson ImmunoResearch Laboratories, West Grove, PA) or FITC goat anti-rat C3 (whole C3) (20 μg/ml; Cappel Laboratories, Durham, NC), for 1 hour at room temperature. Other sections were stained for albumin by indirect immunofluorescence using rabbit anti-rat albumin (2.5 μg/ml in PBS; Cappel) overnight at 4°C, followed by three washes in PBS and incubation with tetramethylrhodamine isothiocyanate-conjugated goat anti-rabbit IgG (affinity purified, 25 μg/ml; Jackson ImmunoResearch Laboratories). Sections were also stained for both albumin and IgG by sequential incubations as described above. After the final washes in PBS, all slides were mounted using 100 mmol/L of Tris-HCl:glycerol, 50:50, 2% N-propyl gallate, pH 8. Sections were examined with a Leika DM-R microscope equipped with epifluorescence and appropriate filters.
Mouse monoclonal antibodies were used for the detection of α-smooth muscle actin (α-SMA) (1A4; Sigma Chemical Co., St. Louis, MO), rat macrophages (ED1; Serotec, Oxford, UK), rat MHC-class II antigen (MHC-II) (OX-6; Sera-Lab Ltd., Crawley Down, Sussex, UK), desmin (D33; DAKO, Glostrup, Denmark), and synaptopodin (cell culture supernatant; Progen Immunodiagnostica, Heidelberg, Germany). Tissue sections were blocked with PBS/1% bovine serum albumin, incubated overnight at 4°C with the primary antibody in PBS (1A4, ascites fluid, 1:400; ED1, 10 μg/ml; OX-6, 10 μg/ml; anti-synaptopodin monoclonal antibody, undiluted). After washes in PBS, they were incubated with Cy-3-conjugated donkey anti-mouse IgG antibodies [affinity purified, absorbed with rat IgG (Jackson Laboratories), 5 μg/ml in PBS] for 1 hour at room temperature. Double labeling was performed by overnight incubation with mouse monoclonal antibody to α-SMA, desmin, or synaptopodin, followed by Cy-3 anti-mouse IgG, and then with FITC anti-rat C3 as described above. In control experiments primary antibodies were either omitted or substituted with nonimmune mouse serum or rabbit serum.
For the evaluation of immunofluorescence staining for IgG, C3, or albumin, each consecutive glomerulus was graded semiquantitatively as follows: 0, absent staining of the glomerular tuft; 0.5, trace staining; 1+ granular staining in <25% of the glomerular tuft showing segmental reactivity; 2+, granular staining in 25 to 50% of the glomerular tuft showing a predominant segmental increase in reactivity; 3+, granular staining in >50% of the tuft; 4+, diffuse staining of the tuft with or without segmental granular pattern. For the evaluation of staining for α-SMA, each glomerulus was also assigned a score accordingly to the following patterns: 37 0, absent or very weak staining of the glomerular tuft; 1+, weak staining with 1 to 25% of the glomerular tuft showing increased reactivity; 2+, 25 to 50% of the glomerular tuft demonstrating a focal strong staining; 3+, 50 to 75% of the glomerular tuft showing increased reactivity; 4+, >75% of the glomerular tuft staining strongly. Glomerular ED1-positive cells and MHC-II-positive cells in each section were also counted. The evaluation was performed on at least 20 consecutive glomerular cross sections by an observer who was blinded to the experimental groups and the mean values were calculated for each specimen.
Light Microscopy
Small fragments of kidney cortex adjacent to those used for the immunohistochemical analysis were dehydrated and embedded in paraffin. Sections (4 μm thick) were stained using the periodic acid-Schiff method. Glomerulosclerosis was defined as glomeruli showing evidence of segmental or global collapse of capillaries, increase in glomerular matrix, and adhesions of the capillary tuft to the Bowman’s capsule. The extent of glomerulosclerosis was expressed as a percentage of the number of glomeruli counted for each animal.
In Situ Hybridization
The rat TGF-β1 anti-sense and sense RNA probes were prepared and labeled by in vitro transcription using digoxigenin-labeled uridine triphosphate (Boehringer Mannheim Biochemica, Mannheim, Germany). 38 A 700-bp rat TGF-β cDNA was cloned into the EcoRI/HindIII sites of the pBluescript vector between T7 and T3 promoters. Fragments of renal cortex of three rats from each group were fixed in Dubosq-Brazil, dehydrated in alcohol, and embedded in paraffin. Sections were cut at 4 μm and processed as described. Briefly, after permeabilization with proteinase K (40 μg/ml, Sigma Chemical Co.), the sections were hybridized with the RNA probes at the final concentrations of 0.1 to 0.5 ng/μl in 2× standard saline citrate, 10% dextran sulfate, 1× Denhardt’s solution, 20 mmol/L vanadyl ribonucleoside complex (Life Technologies, Inc., Gaithersburg, MD), and 0.1 mol/L sodium phosphate, and incubated overnight in a moist chamber at 45°C. After being washed in 0.2× standard saline citrate and blocked with a buffer-blocking solution (50 mg/ml skimmed dried milk, 150 mmol/L NaCl in 100 mmol/L Tris HCl, pH 7.8) at room temperature for 30 minutes, the sections were incubated with anti-digoxigenin antibody conjugated with alkaline phosphatase (Boehringer Mannheim Biochemica) at the dilution of 1:1000 for 45 minutes at 37°C. Colorimetric detection with nitro blue tetrazolium salt and 5-bromo-4-chloro-3-indolyl phosphate (Boehringer Mannheim Biochemica) was then performed and the sections were mounted in 60% glycerol and examined by light microscopy. The negative control included the hybridization step with the sense probe. For semiquantitative evaluation of the podocyte-associated signal at least 10 glomeruli were graded in each section as follows: 0, no staining; 1+, trace staining; 2+, strong staining in at least one podocyte/epithelial cell area; 3+, strong staining of epithelial areas with segmental distribution or at sites of adhesion.
In Vitro Studies
Cell Culture and Incubation
Immortalized mouse podocytes (a kind gift from Dr. Peter Mundel, Albert Einstein College of Medicine, Bronx, NY) were grown in permissive conditions, at 33°C in RPMI 1640 medium (Life Technologies, Inc.) supplemented with 10% fetal bovine serum (Life Technologies, Inc.), 10 U/ml of mouse recombinant γ-interferon (Sigma Chemical Co.), and 100 U/ml of penicillin plus 0.1 mg/ml of streptomycin (Sigma Chemical Co.). 39 To induce differentiation podocytes were maintained in nonpermissive conditions at 37°C without γ-interferon for 14 days. Confluent cells were used for experiments.
SV40 MES 13, a well-characterized mouse glomerular mesangial cell line 40 was obtained from the American Type Culture Collection (Rockville, MD). Mesangial cells were grown in a 3:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F12 medium (Sigma Chemical Co.), 5% fetal bovine serum (Life Technologies, Inc.) and 14 mmol/L HEPES (Sigma Chemical Co.).
To investigate the effect of protein overload on TGF-β1 mRNA expression, differentiated podocytes were exposed to 10 mg/ml of human serum albumin (HSA) (endotoxin content <0.05 EU/ml, using Limulus amoebocyte lysate assay; Sigma Chemical Co.) for 6, 24, and 48 hours. At the end of the incubation, total RNA was obtained for Northern blot analysis. The production of TGF-β1 was quantified by enzyme-linked immunosorbent assay in conditioned supernatant of podocytes treated with HSA (10 mg/ml) or medium for 15, 24, and 48 hours. Synaptopodin expression in podocytes was also studied by fluorescence confocal microscopy at 15, 24, and 48 hours.
To assess whether TGF-β1 produced by podocytes on albumin stimulation affected mesangial cell phenotype by increasing α-SMA expression, mesangial cells grown on coverslips and kept in serum-free condition for 48 hours were exposed to conditioned supernatant of podocytes treated or not with HSA (10 mg/ml) for 48 hours. Human recombinant TGF-β1 (10 ng/ml, Sigma Chemical Co.) was used as positive control. Additionally, the effect of supernatant of HSA-stimulated podocytes was tested in the presence of neutralizing mouse monoclonal anti-TGF-β1 antibody (100 μg/ml; Genzyme, Cambridge, MA). Then mesangial cells were fixed and α-SMA staining was evaluated by fluorescence confocal microscopy.
Northern Blot Analysis
Total RNA was isolated from podocytes by the guanidium isothiocyanate/cesium chloride procedure. Fifteen μg of total RNA was then fractionated on 1.6% agarose gel and blotted onto synthetic membranes (Zeta-probe; Bio-Rad, Richmond, CA). A 0.45-kb EcoRI/HindIII fragment of human TGF-β1 cDNA from plasmid pUC18 (British-Biotechnology, Abingdon, Oxon, UK) was used to detect the 2.5-kb transcript. The probe was labeled with α-32P dCTP by the random-primed method. Hybridization was performed overnight in 0.5 mol/L of Na2HPO4, pH 7.2, 7% sodium dodecyl sulfate. Filters were washed twice for 30 minutes with 40 mmol/L Na2HPO4, pH 7.2 and 5% sodium dodecyl sulfate, and twice for 10 minutes with 40 mmol/L Na2HPO4, pH 7.2 and 1% sodium dodecyl sulfate at 65°C. Membranes were subsequently probed with a β-actin cDNA, taken as internal standard of equal loading of the samples on the membrane. TGF-β1 mRNA optical density was normalized to that of the constituently released β-actin gene expression.
Enzyme-Linked Immunosorbent Assay
Measurement of TGF-β1 in the cell supernatant was performed by using a commercially available enzyme-linked immunosorbent assay kit (Promega, Madison, WI) following the manufacturer’s instructions. TGF-β1 antibody crossreactivity with other TGF-β isoforms was as follows: TGF-β1.2 heterodimer, 11.5%; TGF-β2, 1.6%; and TGF-β3, 0.7%.
Fluorescence Confocal Microscopy
Podocytes or mesangial cells grown on coverslips after appropriate incubations were fixed in 2% paraformaldehyde plus 4% sucrose in PBS, pH 7.4, for 10 minutes at 37°C. After three washings (5 minutes) with PBS, the cells were permeabilized with 0.1% Triton X-100 in PBS for 3 minutes. Cells were rinsed with PBS and nonspecific binding sites were saturated in blocking solution (2% fetal bovine serum, 2% bovine serum albumin, and 0.2% bovine gelatin in PBS) for 30 minutes. Podocytes were incubated with mouse monoclonal antibody to synaptopodin for 1 hour at room temperature, washed three times with PBS, and then incubated with FITC-conjugated goat anti-mouse antibody (30 μg/ml, Jackson Laboratories). Mesangial cells were incubated with mouse monoclonal antibody anti-α-SMA (clone 1A4, ascites fluid diluted in PBS 1:300; Sigma) overnight at 4°C, washed three times with PBS, and finally incubated with Cy3-conjugated donkey anti-mouse IgG antibodies [affinity purified, absorbed with rat IgG (Jackson Laboratories), 15 μg/ml in PBS] for 1 hour at room temperature. Negative control experiments with secondary antibody Cy3-conjugated donkey anti-mouse IgG alone were performed. Coverslips were washed, mounted in 1% N-propyl-gallate in 50% glycerol and 0.1 mol/L Tris-HCl, pH 8, and examined under confocal inverted laser microscope (Insight plus; Meridian Instruments, Inc., Okemos, MI). Representative fields were digitized with millions of colors and printed.
Statistical Analysis
The results are expressed as mean ± SD. Analysis of variance with Tukey test for multiple comparisons was used to analyze proteinuria, serum creatinine, and systolic blood pressure data. Immunohistochemistry and histology data were analyzed using the nonparametric Kruskal-Wallis test. Statistical significance was defined as P < 0.05.
Results
Abnormal Accumulation of Plasma Proteins in Podocytes Precedes and Is Tightly Associated with Sclerosing Activation of Mesangial Cells
After extensive RMR, compensatory increases in remnant nephron function are associated with maladaptive intraglomerular processes that eventually prove detrimental. 1 The increase in glomerular capillary hydraulic pressure serving to maintain glomerular filtration is associated with impaired glomerular permselectivity to proteins and consequent proteinuria. We first determined whether plasma proteins accumulate locally as a consequence of their enhanced intraglomerular passage, and whether protein accumulation may co-localize with a prosclerosing phenotypic change. Abnormal accumulation of IgG and C3 was detected in the glomeruli of RMR rats sacrificed at various intervals after surgery (Figure 1) ▶ . In contrast to sham controls (Figure 1, a and b) ▶ , kidneys at 7 days after RMR showed focal and segmental distribution of IgG and C3 (Figure 1, c and d) ▶ in glomeruli, with a granular staining that concentrated in podocytes and to a lesser extent in mesangial areas. In kidneys at 14 days (Figure 1, e and f) ▶ or 30 days after RMR (Figure 1, g and h) ▶ the podocyte-associated IgG and C3 staining was more diffuse, in addition to segmental patterns. The underlying mechanism was related to enhanced intraglomerular passage of proteins rather than reduced clearance, as albumin (molecular weight, 69,000) revealed similar changes. By double staining for albumin and IgG, the marker proteins were detected within the same glomerular areas in a granular pattern within podocytes (Figure 1, i and j) ▶ . Evidence of significant accumulation of proteins starting from 7 days after surgery was confirmed by semiquantitative analysis (Figure 2) ▶ . The excess protein uptake was associated with increasingly high levels of urinary protein excretion (mg/24 hours, RMR: 7 days, 46 ± 20; 14 days, 101 ± 24; 30 days, 225 ± 89; sham-operated rats: 27 ± 5; P < 0.01 RMR at 14 days and 30 days versus sham control).
Figure 1.
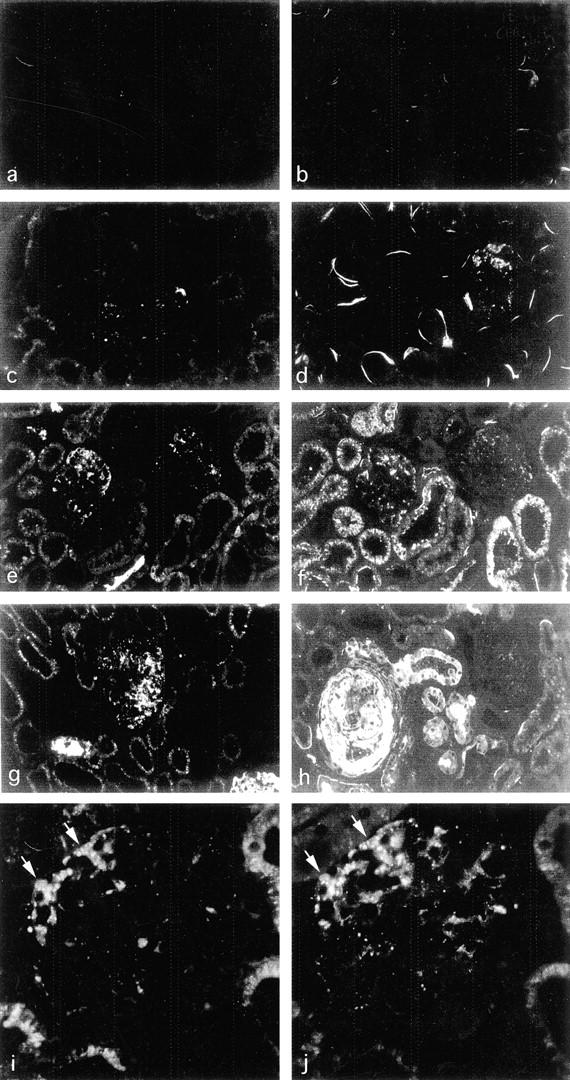
Enhanced intraglomerular passage and accumulation of plasma proteins in RMR, a rat model of progressive nephropathy. a–h: Sections of kidneys after sham operation or RMR stained by immunofluorescence for IgG (left) or C3 (right). a and b, sham-operated control; c and d, RMR at 7 days, focal staining; e and f, RMR at 14 days, staining more evident both in glomeruli and in proximal tubuli; g and h, RMR at 30 days, heterogeneous patterns with strong irregular staining, or minimal amount of staining in one glomerulus in h. i and j: Abnormal albumin (i) and IgG staining (j) co-localize to the same podocytes (arrows) and mesangial areas of RMR kidney at 14 days. Note the glomerular intracellular staining of podocytes that reflects excess protein uptake. Direct immunofluorescence using FITC-conjugated antibodies. Original magnifications: ×125 (a–h); ×500 (i and j).
Figure 2.

Semiquantitative analysis of IgG, C3, and albumin staining in glomeruli. °, P < 0.01 versus control; *, P < 0.01 versus control and RMR at 7 days; #, P < 0.05 versus 7 days RMR.
The phenotypic change to express α-SMA in mesangial cells is typically associated with sclerosis that develops in weeks in RMR and in other rat models, 37,41 and it is a feature of myofibroblast transformation contributing to the production of abnormal extracellular matrix. 20,42-44 Therefore, we studied the phenotypic change in relation to the onset of enhanced passage of proteins. RMR kidneys at 7 days showed no changes in α-SMA staining in glomeruli except for a minimal and focal increase (Figure 3, a and b) ▶ . Strong α-SMA expression in mesangial areas was found at subsequent time points (Figure 3, c and d) ▶ . Mean scores were the following: RMR at 7 days, 0.40 ± 0.08; at 14 days, 1.21 ± 0.25; at 30 days, 1.16 ± 0.50; sham, 0 (P < 0.01 RMR at 14 days and 30 days versus sham, and P < 0.05 at 7 days versus other groups). Dual immunofluorescence analysis revealed a distinctive relation between sites of protein accumulation and mesangial α-SMA expression (Figure 4; a to h) ▶ . Thus, protein accumulation in podocytes preceded myofibroblast transformation of adjacent cells, as reflected by the evidence that in RMR at 7 days the segmental IgG (Figure 4c) ▶ or C3 (Figure 4d) ▶ deposition (arrowheads) was invariably associated with no or minimal increase in glomerular α-SMA staining, and that the increase in mesangial α-SMA at 14 days was confined to glomeruli showing positive staining for the protein markers whereas the latter could be found in glomeruli showing no α-SMA expression. In addition, within glomeruli α-SMA was localized to IgG-positive (Figure 4e) ▶ and C3-positive (Figure 4f ▶ , arrows) portions of the tuft. These findings were in agreement with the likelihood that the enhanced passage and intracellular accumulation of proteins might play role in the segmental adhesion of the capillary tuft to the Bowman’s capsule, morphological manifestation of initial sclerosis. The pattern evolved consistently toward segmental deposition of proteinaceous material associated with α-SMA expression at the peripheral edge of the lesion (Figure 4, g and h) ▶ . These changes were paralleled by development of sclerosis. By light microscopy, in RMR rats sclerotic lesions were present in a significant percentage of glomeruli at 14 days, and they further increased at 30 days (percent of glomeruli: RMR at: 7 days, 0.8 ± 1.0; 14 days, 6.6 ± 5.7; 30 days, 18.9 ± 9.5; sham: 1.0 ± 0.8; P < 0.05 RMR at 14 days and P < 0.01 RMR at 30 days versus sham and RMR at 7 days).
Figure 3.
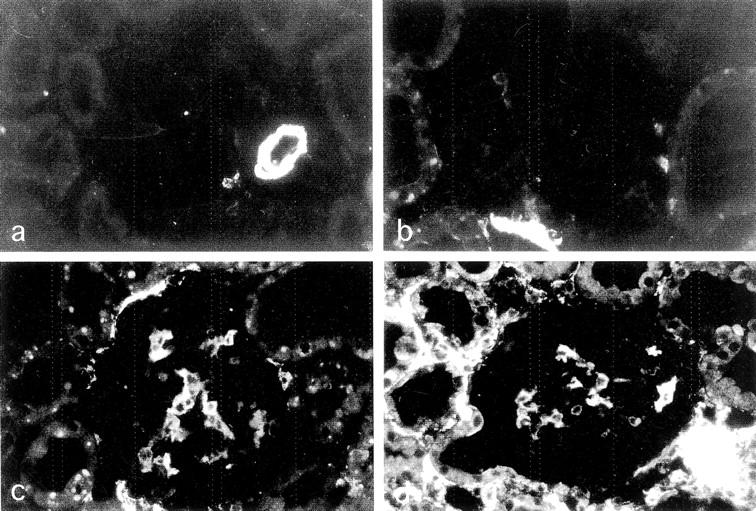
Expression of α-SMA in glomeruli in progressive nephropathy. a: Sham control, α-SMA staining confined to vessel wall and absent or minimal in glomeruli. b–d: RMR, b, at 7 days, absent or very weak α-SMA expression; c, at 14 days, strong focal staining and initial periglomerular expression; d, at 30 days, further increased expression in periglomerular tubulointerstitial areas. Original magnifications, ×250.
Figure 4.
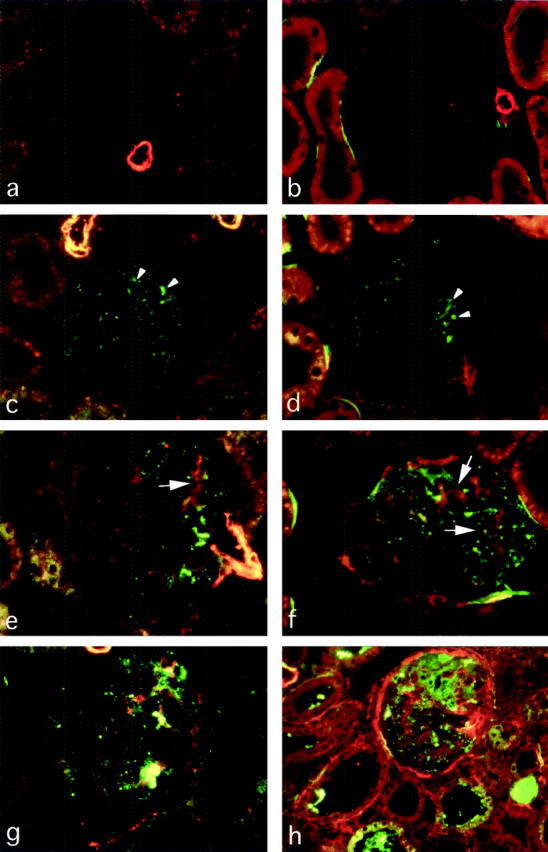
a–h: Double-immunofluorescence detection of α-SMA and IgG (left) or α-SMA and C3 (right) in RMR kidneys. α-SMA is stained in red (Cy-3-conjugated secondary antibody), IgG and C3 are stained in green (FITC-conjugated antibodies). a and b, sham control; c and d, RMR at 7 days; e and f, RMR at 14 days; g and h, RMR at 30 days. The comparison of α-SMA expression with both IgG and C3 deposition shows that the abnormal accumulation of plasma proteins (arrowheads) is detected both in advance of and in close association with the mesangial (arrows) and periglomerular myofibroblast transformation. Original magnifications, ×250.
Because previous studies found that macrophages accumulate into glomeruli during the development of sclerosis, 37,45 we also analyzed for comparison the time course of glomerular ED1 macrophage accumulation and, additionally, changes in MHC-II expression in RMR. Marked and significant increases in numbers of both ED-1-positive cells and of cells expressing MHC class II were found in RMR glomeruli at 14 days and 30 days after RMR (Figure 5) ▶ . The cells showing positive staining for either marker did not show segmental patterns of distribution.
Figure 5.

Numbers of ED1 macrophages and MHC class II-positive cells in glomeruli. °, P < 0.01 versus control; *, P < 0.01 versus control and RMR at 7 days.
Protein-Laden Podocytes Express Desmin, Lose Synaptopodin, and Up-Regulate TGF-β1 Gene in RMR
Next, given that intraglomerular protein deposition is a very early event, and podocyte dysfunction and TGF-β1 up-regulation are theoretically central factors in sclerosis, we assessed whether the latter abnormalities can be related spatially or temporally with protein accumulation in podocytes in RMR. The intermediate filament protein, desmin, present in mesangial cells in the normal glomerulus, is highly expressed by podocytes in vivo exclusively in the setting of glomerular injury and thus it is studied as a general marker of cell function perturbation and injury. 44,46,47 Podocytes in kidneys after sham operation (Figure 6a) ▶ or RMR at 7 days (Figure 6b) ▶ typically had no or faint desmin expression. In contrast, in RMR both at 14 days (Figure 6c ▶ , arrowheads) and 30 days after surgery (Figure 6d) ▶ podocytes became strongly desmin-positive, particularly those located at the periphery of the capillary tuft. The enhanced passage and intracellular accumulation of proteins in podocytes preceded the appearance of high expression of desmin, as IgG was detectable in podocytes in the absence of desmin by double staining of sections of RMR kidneys at 7 days (Figure 7b ▶ , compare with sham kidney in Figure 7a ▶ ), this despite the persistence of desmin in adjacent mesangial cells providing internal positive control. At subsequent intervals, newly expressed desmin co-localized with IgG in podocytes either in glomerular segments (14 days, Figure 7c ▶ ) or in more diffuse patterns (30 days, Figure 7d ▶ ). Co-localization was also found in areas of tuft adhesion containing extracellular C3- or IgG-positive material. These findings suggest that the exposure to high protein load may cause podocyte dysfunction contributing to sclerosis. Such possibility was strengthened by analysis of synaptopodin, an actin-associated protein specifically expressed by podocytes and a unique differentiation marker that appears in tight coincidence with the developing podocyte foot processes. 48,49 Thus, a diminution of synaptopodin expression first became apparent in localized areas of the glomeruli in RMR kidneys at 14 days (Figure 6g ▶ , arrowheads), and then more evident at 30 days (Figure 6h) ▶ . Double staining for synaptopodin and IgG revealed that the loss of synaptopodin both at 14 days and at 30 days was associated with accumulation of plasma proteins in the podocytes (Figure 7, e and f) ▶ . The disappearance of the foot process differentiation marker mirrored the appearance of strong expression of desmin, indicating that the loss of synaptopodin is associated with cell dysfunction in protein-laden podocytes in this model.
Figure 6.
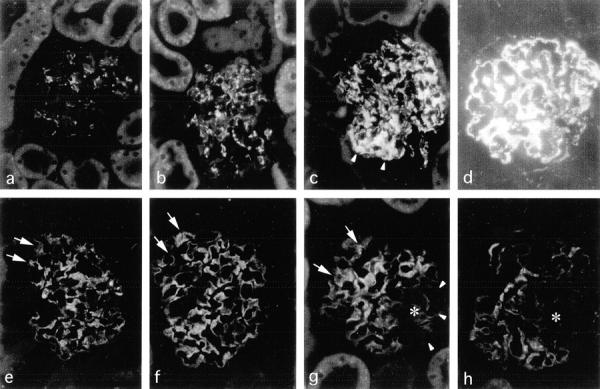
Immunofluorescence detection of desmin (a–d) and synaptopodin (e–h) in RMR glomeruli. In contrast to normal patterns revealed in sham control (a and e) and RMR at 7 days (b and f), high desmin expression and loss of synaptopodin in podocytes are visualized in RMR at 14 days (c and g, arrowheads) and 30 days (d and h) after surgery. In respect to areas in which synaptopodin is preserved (arrows), the asterisks indicate well-defined areas in which the foot process-associated differentiation marker is lost. Original magnifications, ×250.
Figure 7.
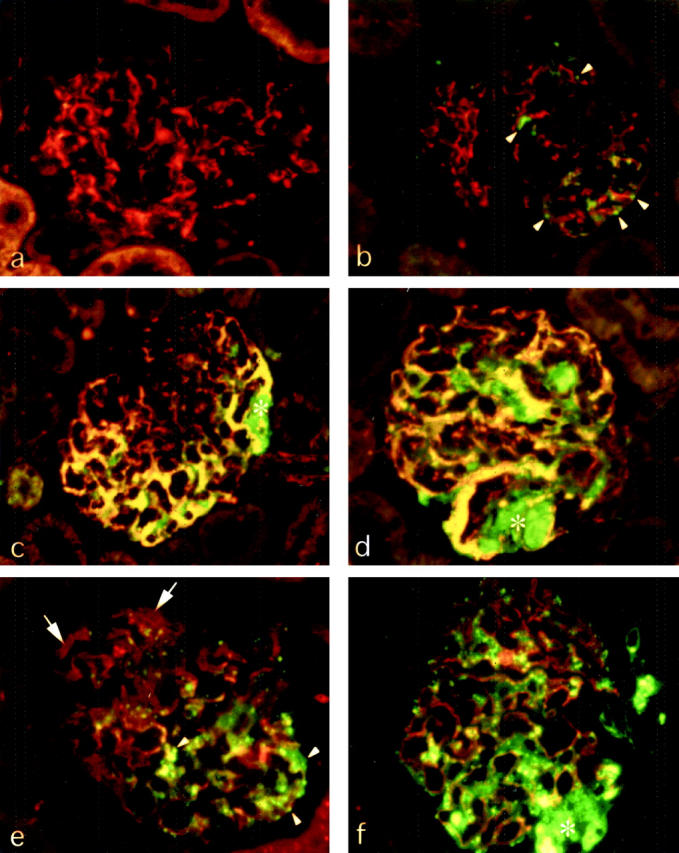
Comparison of the glomerular sites of abnormal deposition of protein and altered expression of desmin and synaptopodin. a–d: Dual labeling of desmin and IgG. In a glomerulus of sham control (a) desmin (red) is confined to mesangial cells and IgG (green) is not detectable. A glomerulus of RMR kidney at 7 days (b) shows granular IgG staining (arrowheads) in podocytes, associated with no or very weak desmin staining, consistent with IgG accumulation in advance of high desmin expression. In sections of RMR kidneys at 14 days (c) and at 30 days (d) both the high expression of desmin and the IgG staining co-localize mainly to peripheral podocytes (yellow). e and f: By dual labeling for synaptopodin and IgG in RMR at 14 days (e) and at 30 (f), IgG-positive podocytes (arrowheads), in contrast to IgG-negative podocytes (arrows), show diminution or loss of synaptopodin (red). Severe podocyte damage and possibly detachment and loss are detectable in association with extracellular IgG deposition in areas of segmental adhesion and sclerosis (asterisks). Original magnifications, ×250.
In situ hybridization analysis revealed that TGF-β1 up-regulation in RMR occurred concomitantly to podocyte abnormalities after the onset of enhanced transglomerular passage of proteins. As compared to sham controls (Figure 8a) ▶ glomeruli of RMR kidneys at 7 days showed no or very little increases in the intensity of TGF-β1 mRNA signal (Figure 8b) ▶ . In contrast at 14 days (Figure 8c) ▶ and 30 days (Figure 8d) ▶ TGF-β1 mRNA expression increased markedly in podocytes. An increase in TGF-β1 mRNA hybridization signal was also detected in endothelial cell areas. Mean scores were the following: RMR: at 7 days, 0.61 ± 0.12; 14 days, 1.39 ± 0.35; 30 days, 1.40 ± 0.36; sham, 0.63 ± 0.09 (P < 0.01 RMR at 14 days and 30 days versus sham). Sclerotic areas in advanced stage of injury exhibited strong diminution or loss of TGF-β1 mRNA signal (not shown). Sections stained with sense probe did not show staining (Figure 8f) ▶ .
Figure 8.
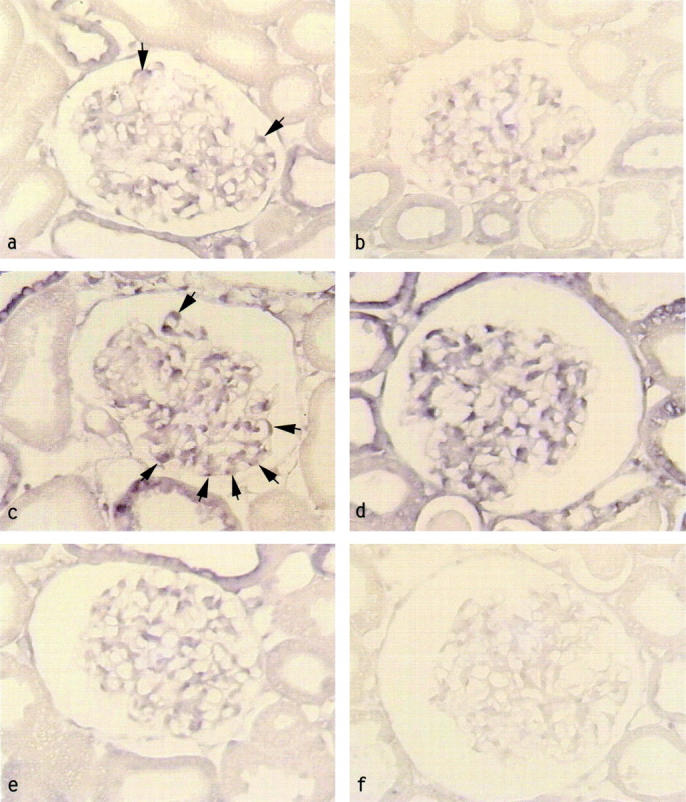
Glomerular TGF-β1 up-regulation in RMR and effect of ACE inhibitor. TGF-β1 mRNA expression by in situ hybridization in glomeruli of a, sham control revealing weak signal in podocytes, b, RMR at 7 days; c, RMR at 14 days; d, RMR at 30 days; e, RMR at 30 days plus ACE inhibitor. TGF-β1 mRNA signal is increased in RMR at 14 days (c) in podocytes (arrows) and intracapillary areas, and it becomes more diffuse at 30 days (d). The ACE inhibitor prevents TGF-β1 up-regulation (e). f, RMR at 30 days, sense probe. Original magnifications, ×375.
Protection Against Sclerosis by ACE Inhibitor Is Associated with Abrogation of Intraglomerular Protein Load and of TGF-β1 Overexpression
Because ACE inhibitors have potent protective effects against renal structural and functional damage, 50,51 we determined whether lisinopril may limit both TGF-β1 up-regulation, potentially responsible for myofibroblast-mediated scarring, and the intraglomerular protein deposition that may act as the common pathway stimulatory factor. In fact, the increase in TGF-β1 mRNA signal was fully suppressed in glomeruli of RMR rats at 30 days given lisinopril from 1 day after surgery, which showed a pattern comparable to that found in sham-operated controls (Figure 8e) ▶ . This effect was associated with protection against proteinuria, α-SMA overexpression, and glomerulosclerosis, and with significant reductions in the numbers of ED-1 macrophages and MHC-II-positive cells in the glomeruli as compared to untreated RMR rats (Table 1) ▶ . Serum creatinine levels measured at 30 days were significantly lower than in untreated RMR rats (0.98 ± 0.15 versus 1.41 ± 0.18 mg/dl, P < 0.01), and the systolic blood pressure was maintained at 93 ± 15 mmHg, a mean value significantly lower than that recorded in untreated RMR rats (170 ± 11 mmHg).
Table 1.
Effects of Lisinopril on Urinary Protein Levels, Intraglomerular Protein and Inflammatory Cell Accumulation, α-SMA Expression, and Glomerulosclerosis
| Group | Proteinuria (mg/day) | IgG (score) | C3 (score) | Alb (score) | ED-1 | OX-6 (MHC 11) | α-SMA (score) | Glomerulosclerosis (%) |
|---|---|---|---|---|---|---|---|---|
| (cells/glom section) | ||||||||
| 30-day RMR | 225 ± 89* | 1.30 ± 0.34* | 0.73 ± 0.18* | 1.80 ± 0.38* | 12.51 ± 2.73* | 12.09 ± 4.04* | 1.16 ± 0.46* | 18.9 ± 9.5* |
| 30-day RMR+ lisinopril | 28 ± 8† | 0.36 ± 0.35† | 0.18 ± 0.17† | 0.30 ± 0.20† | 7.20 ± 1.65† | 5.96 ± 1.77† | 0.34 ± 0.26† | 1.5 ± 2.3† |
| 30-day control | 27 ± 5 | 0.26 ± 0.12 | 0.03 ± 0.05 | 0.19 ± 0.07 | 2.93 ± 0.70 | 3.09 ± 0.87 | 0 | 1.05 ± 0.8 |
*, P < 0.01 versus control;
†, P < 0.01 versus RMR at 30 days.
In addition to inhibition of the renin angiotensin system and reduction of intraglomerular capillary pressure, ACE inhibitor’s renoprotective effects can be attributed to preservation of glomerular permselectivity to plasma proteins. 26-29 It was unanswered whether such action may account for the protective effect against glomerulosclerosis. Like for TGF-β1, lisinopril here prevented the abnormal intraglomerular accumulation of plasma proteins in RMR kidneys (Table 1 ▶ , and Figure 9b ▶ , compare with sham kidney, Figure 9a ▶ ). Together with the effect of ACE inhibitor on TGF-β1 overexpression, these findings are in agreement with a pathogenic role for the underlying protein influx, possibly via abnormal uptake of plasma proteins and activation of a TGF-β1 pathway in podocytes.
Figure 9.
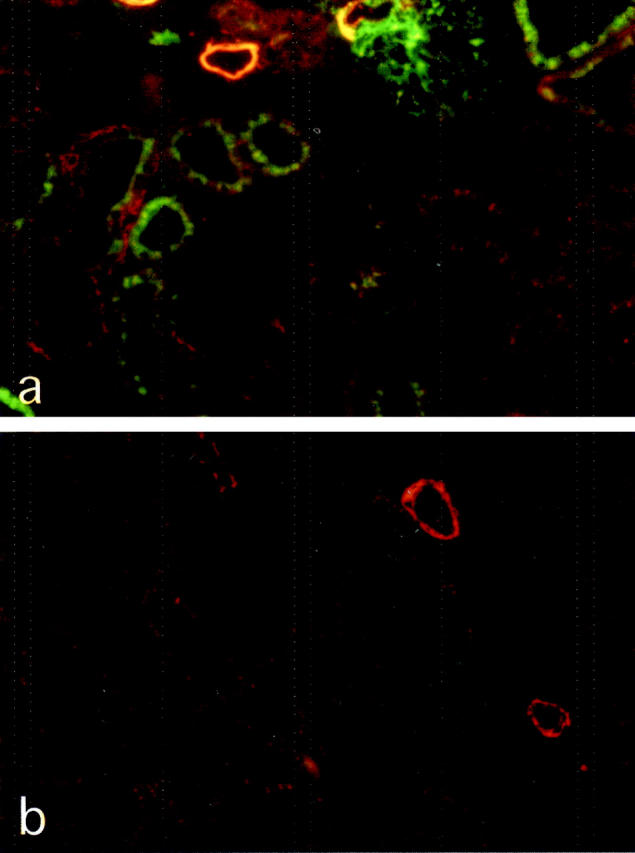
Sections of RMR kidney at 30 days and RMR kidney of an ACEi-treated rat, stained for IgG (green) and α-SMA (red). Compared to untreated RMR (a), the ACE inhibitor prevents abnormal accumulation of IgG (b). Original magnifications, ×125.
Protein Overload Stimulates Podocytes to Up-Regulate TGF-β1 Gene, Promoting TGF-β1-Mediated Phenotypic Change of Mesangial Cells
To assess whether protein load may play a direct role in TGF-β1 gene up-regulation in podocytes, we evaluated the effect of exposure of differentiated podocytes in culture to HSA (Figure 10a) ▶ . By Northern blot, a weak 2.5-kb transcript specific for TGF-β1 was observed in unstimulated control podocytes. Albumin induced a twofold increase in TGF-β1 mRNA over control (P < 0.01) after 6 hours of incubation. Transcript levels remained high at 24 and 48 hours (1.6- to 1.7-fold, P < 0.05) (Figure 10b) ▶ .
Figure 10.

Synthesis of TGF-β1, induction of downstream phenotypic change of mesangial cells, and loss of synaptopodin by podocytes in response to protein load. a–c: Effects of HSA on TGF-β1 mRNA expression by Northern blot analysis (a and b) and on TGF-β1 production by enzyme-linked immunosorbent assay (c) in cultured podocytes. *, P < 0.05 and **, P < 0.01 versus control. d–h: Expression of α-SMA in mesangial cells exposed to conditioned medium of unstimulated podocytes (d) or of HSA-stimulated podocytes alone (e) or in the presence of anti-TGF-β1 antibody (f), as assessed by fluorescence confocal microscopy. g and h: α-SMA in control mesangial cells either unstimulated (g) or exposed to TGF-β1 (h). i–n: Confocal microscopy analysis of the effect of HSA on podocyte synaptopodin. Podocytes exhibit marked reduction of synaptopodin staining after exposure to HSA for 6 hours (i) and 24 hours (l), and partial recovery at 48 hours (n) as compared to unstimulated control at the corresponding time points (i, k, and m). Original magnifications, ×1500.
Albumin-induced TGF-β1 mRNA up-regulation was associated with the increase of protein release into the cell supernatants (Figure 10c) ▶ . After 15 hours of incubation with HSA, TGF-β1 production increased (150%) in respect to unstimulated cells, with values reaching statistical significance (P < 0.01) at 24 and 48 hours (177% and 180% increase over control, respectively). In light of the in vivo findings of podocyte-mesangial cell interaction and high α-SMA expression at sites of protein accumulation, we assessed whether the protein-induced stimulation of podocytes in vitro may cause downstream activation of mesangial cells, by experiments using conditioned medium of HSA-stimulated podocytes. No α-SMA expression was detectable by fluorescence confocal microscopy in unstimulated mesangial cells in culture (Figure 10, d and g) ▶ . The conditioned medium of HSA-stimulated podocytes induced the expression of α-SMA in mesangial cells (Figure 10e) ▶ , mimicking the effect of the exposure of mesangial cells to TGF-β1 (Figure 10h) ▶ . The stimulatory effect of conditioned medium of HSA-stimulated podocytes on α-SMA expression was abrogated by the addition of anti-TGF-β1-neutralizing antibody (Figure 10f) ▶ .
Because TGF-β1 up-regulation was associated with enhanced protein uptake and podocyte dysfunction in RMR, we also determined whether protein load may cause loss of synaptopodin expression in podocytes in vitro. Synaptopodin staining as visualized by confocal microscopy was markedly reduced with respect to unstimulated cells (Figure 10; h, j, and l) ▶ after either 6 hours (Figure 10i) ▶ and 24 hours (Figure 10k) ▶ of incubation with HSA, and partially recovered at 48 hours (Figure 10m) ▶ .
Discussion
The current study explored TGF-β1 up-regulation by glomerular podocytes in response to enhanced intraglomerular passage of proteins as a potential trigger of glomerular scarring. Three different approaches were adopted with the following results. First, in vivo, abnormal uptake of plasma proteins by podocytes preceded and then was spatially associated with podocyte injury and sclerosing events including TGF-β1 overexpression. Second, exposure of cultured podocytes to albumin induced both the loss of a highly specialized molecule and TGF-β1 up-regulation with downstream sclerosing phenotype of mesangial cells, suggesting that the changes found in kidney were causally linked, at least in part, to the effect of protein load of inducing podocyte dysfunction. Third, back to the in vivo model, TGF-β1 overexpression and the sclerosing reaction were prevented in animals in which the enhanced passage of proteins was pharmacologically limited. This mechanism as unraveled in the RMR model may operate irrespective of etiology, be it a gene abnormality, immune disease, or diabetes that share in common permselective defects of the glomerular barrier. Remarkably, to our knowledge, no data are available providing comparable evidence for the association of other major factors, specifically glomerular hypertension, cell stretching, or angiotensin II, with the activation in vivo of a given cell type that could be detected, as shown here, at the level of individual glomeruli by double-staining techniques.
Hemodynamic adaptive changes have been implicated in promoting progressive sclerosis in RMR and diabetes. 1,52 The glomerulus is a specialized filtering network, and the presence of a fenestrated endothelium may further render the podocyte at the urinary aspect of the barrier intrinsically susceptible to increases in protein load associated with high glomerular capillary hydraulic pressure. Studies in rats with renal vein constriction 53 or passive Heymann nephritis 54 indeed showed that enhancing intraglomerular capillary pressure disrupts the glomerular barrier to circulating proteins. Glomerular permselectivity is lost in RMR as a consequence of altered size-selective function of the glomerular capillary wall, as shown by evidence that the fractional clearance of neutral dextrans >38-Å molecular radius is enhanced. 5 Here, the segmental accumulation of albumin, IgG, and C3 in the initial stage of disease at 7 days clearly reflects increased influx of proteins because of impairment of permselective function, in agreement with studies using dextran or other macromolecular probes. 5 We cannot exclude that a reduced mesangial clearance of IgG and C3 may contribute at later time points. The passage of macromolecules through the mesangium has been documented, 55 and a hemodynamic challenge may cause macromolecular overload of the mesangium as revealed by detection of ferritin used as a tracer. 5,56
Despite multifactorial features of glomerular sclerosis, studies have collectively identified podocyte injury as a central cellular event, 4,7,8 thus we elected to examine whether the enhanced intraglomerular passage of proteins may promote podocyte dysfunction pari passu with high TGF-β1 synthesis. Changes in intermediate filament and cytoskeletal-associated components, desmin and synaptopodin, are associated with progressive injury both in experimental and human nephropathies. 44,46,47,57-59 Findings that the enhanced transglomerular passage and podocyte uptake of proteins, in advance of structural injury in RMR, precedes both high desmin expression and concomitant loss of synaptopodin in protein-laden podocytes were consistent with a causal role of protein overload. In this respect, TGF-β1 up-regulation in response to albumin can be seen as one functional aspect of the podocyte reaction to excess protein uptake. Although mechanisms are not defined, signal transduction events appear to be similar to those induced by known stimuli of TGF-β1 synthesis (via AP-1, MAP kinases, smad, PKC). 60-62 Preliminary data indeed indicate that protein overload may act by promoting podocyte actin-cytoskeleton rearrangement and up-regulation of AP-1-dependent gene expression. 63
The effect of inducing the loss of actin-associated synaptopodin in differentiated podocytes in vitro further directly implicates protein accumulation as the trigger of podocyte dysfunction. Synaptopodin is a unique differentiation molecule perhaps instrumental in foot process formation during nephrogenesis. 48 Its loss is one indicator for loss of cell differentiation in areas of segmental sclerosis. 57-59 Until now, there was indirect evidence to suggest that the protein load may promote injury including the effacement of highly differentiated foot processes. In RMR, protein droplet formation also precedes the loss of podocyte architecture that manifests with effacement of foot processes followed by cell detachment from the basement membrane and tuft adhesion to the adjacent parietal layer. 5,47 Together, our data revealing the loss of a specific differentiation molecule of foot processes in a mirror-like pattern with high desmin expression, and TGF-β1 induction by protein load in early stage of injury are consistent with roles in adhesion, and they hint to self-sustained effects of enhanced transglomerular passage of proteins in continuing perturbation of foot process function and eventual detachment.
Podocyte dysfunction and TGF-β1 up-regulation in RMR were associated with phenotypic transformation of mesangial cells. Podocytes on protein challenge effectively promote mesangial cell activation, as reflected by the effect of conditioned medium of albumin-stimulated podocytes, like TGF-β1 itself, to induce α-SMA expression in cultured mesangial cells. A causal link for responses of such a type cannot be usually documented in vivo. However, the co-localization of granular staining for albumin and other plasma proteins, unequivocal sign of excess uptake by podocytes, and mesangial α-SMA can only be interpreted to indicate that the response elicited by the protein stimulus exactly duplicates interactions that actually occur in the kidney. Furthermore, experiments to address whether the enhanced passage of proteins by up-regulating TGF-β1 may lead to the relevant mesangial cell response in RMR also showed that abrogating the permselective defect by lisinopril prevents the abnormal accumulation of proteins into glomeruli and the associated TGF-β1 up-regulation, as well as α-SMA expression and sclerosis. Multiple targets may account for the drug’s action to limit glomerular influx of proteins and proteinuria. Limiting angiotensin II-dependent increases in intraglomerular capillary pressure and/or permselective dysfunction may explain the advantage offered against other anti-hypertensive agents shown to have minimal effects against albumin and IgG deposition 64 and, consistently, glomerular injury. 64-66 However, results of recent studies in spontaneous or immune-mediated models of sclerosis indicate that preservation of permselectivity by lisinopril can be also explained by prevention of altered expression of nephrin 67 and ZO-1 68 that normally serve to maintain functional properties and architecture of the slit diaphragm. Our provisional interpretation is that angiotensin II blockers act by preserving the slit diaphragm integrity, either by reducing angiotensin II or by preserving podocyte foot process architecture.
TGF-β1 is a mediator that lies in a final common pathway for multiple causes of glomerular sclerosis, and the enhanced passage of proteins are likely to stimulate TGF-β1 synthesis both independently and in association with other stimuli such as glomerular angiotensin II, mechanical stress, or high glucose. Angiotensin II 69 and cell stretch 70 were indeed found to stimulate TGF-β synthesis, and our data certainly do not discount the importance of their inhibition as a major mechanism underlying the ACEi’s protective effect. The emerging role of podocyte synthesis of TGF-β1 has been recently credited by the evidence that cultured glomerular epithelial cells can up-regulate TGF-β1 synthesis in response to high glucose. 71 It is in the more general framework of pathways independent of immune and metabolic factors that we have examined podocyte activation and the relevant stimulus. It is mandatory to identify key targets of therapies preventing the need of dialysis and renal transplantation before the costs of unrestricted care become unsustainable. TGF-β1 blocking reagents should be tested urgently for combined therapies with drugs that act on the upstream stimuli, to minimize both proteinuria and local consequences on glomerular structure and function.
Acknowledgments
We thank Dr. Ariela Benigni, Head Department of Molecular Medicine, for her contributions to this study, and Dr. Peter Mundel, Albert Einstein College of Medicine, Bronx, NY, for providing the murine podocytes.
Footnotes
Address reprint requests to Mauro Abbate, M.D., Via Gavazzeni 11 24125, Bergamo, Italy. E-mail: abbate@marionegri.it.
Part of this work was presented at the American Society of Nephrology/International Society of Nephrology World Congress of Nephrology (San Francisco, California, October 10 to 17, 2001).
References
- 1.Brenner BM, Meyer TW, Hostetter TH: Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med 1982, 307:652-659 [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, Meyer TW, Rennke HG, Brenner BM: Control of glomerular hypertension limits glomerular injury in rats with reduced renal mass. J Clin Invest 1985, 76:612-619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fries JWU, Sandstrom DJ, Meyer TW, Rennke HG: Glomerular hypertrophy and epithelial cell injury modulate progressive glomerulosclerosis in the rat. Lab Invest 1989, 60:205-218 [PubMed] [Google Scholar]
- 4.Remuzzi G, Bertani T: Is glomerulosclerosis a consequence of altered glomerular permeability to macromolecules? Kidney Int 1990, 38:384-394 [DOI] [PubMed] [Google Scholar]
- 5.Olson JL, Hostetter TH, Rennke HG, Brenner BM, Venkatachalam MA: Altered glomerular permselectivity and progressive sclerosis following extreme ablation of renal mass. Kidney Int 1982, 22:112-126 [DOI] [PubMed] [Google Scholar]
- 6.Remuzzi G, Bertani T: Pathophysiology of progressive nephropathies. N Engl J Med 1998, 339:1448-1456 [DOI] [PubMed] [Google Scholar]
- 7.Rennke HG: How does glomerular epithelial cell injury contribute to progressive glomerular damage? Kidney Int 1994, 45:S58-S63 [PubMed] [Google Scholar]
- 8.Kriz W, Gretz N, Lemley KV: Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998, 54:687-697 [DOI] [PubMed] [Google Scholar]
- 9.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS: Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 1999, 286:312-315 [DOI] [PubMed] [Google Scholar]
- 10.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K: Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1998, 1:575-582 [DOI] [PubMed] [Google Scholar]
- 11.Somlo S, Mundel P: Getting a foothold in nephrotic syndrome. Nat Genet 2000, 24:333-335 [DOI] [PubMed] [Google Scholar]
- 12.Wickelgren I: First components found for new kidney filter. Science 1999, 286:225-226 [DOI] [PubMed] [Google Scholar]
- 13.Border WA, Noble NA, Jamamoto T, Harper JR, Yamaguchi Y, Pierschbacher MD, Ruoslahti E: Natural inhibitor of transforming growth factor-β protects against scarring in experimental kidney disease. Nature 1992, 360:361-364 [DOI] [PubMed] [Google Scholar]
- 14.Okuda S, Languino LR, Ruoslahti E, Border WA: Elevated expression of transforming growth factor-β and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J Clin Invest 1990, 86:453-462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies DJ, Messina A, Thumwood CM, Ryan GB: Glomerular podocyte injury in protein overload proteinuria. Pathology 1985, 17:412-419 [DOI] [PubMed] [Google Scholar]
- 16.Davies DJ, Brewer DB: Irreversible glomerular damage following heterologous serum albumin overload. J Pathol 1977, 123:45-52 [DOI] [PubMed] [Google Scholar]
- 17.Davies DJ, Brewer DB, Hardwicke J: Urinary proteins and glomerular morphometry in protein overload proteinuria. Lab Invest 1978, 38:232-243 [PubMed] [Google Scholar]
- 18.Brewer DB, Filip O: The morphometry of the glomerular epithelial cell and its foot processes after the injection of bovine serum albumin or egg albumin. J Pathol 1976, 120:209-220 [DOI] [PubMed] [Google Scholar]
- 19.Marks MI, Drummond KN: Nephropathy and persistent proteinuria after albumin administration in the rat. Lab Invest 1970, 23:416-420 [PubMed] [Google Scholar]
- 20.Stephenson LA, Haney LB, Hussaini IM, Karns LR, Glass WF, II: Regulation of smooth muscle α-actin expression and hypertrophy in cultured mesangial cells. Kidney Int 1998, 54:1175-1187 [DOI] [PubMed] [Google Scholar]
- 21.Desmoulière A, Geinoz A, Gabbiani F, Gabbiani G: Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 1993, 122:103-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isaka Y, Fujiwara Y, Ueda N, Kaneda Y, Kamada T, Imai E: Glomerulosclerosis induced by in vivo transfection of transforming growth factor-β or platelet-derived growth factor gene into the rat kidney. J Clin Invest 1993, 92:2597-2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA: Expression of transforming growth factor β is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA 1993, 90:1814-1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto T, Noble NA, Cohen AH, Nast CC, Hishida A, Gold LI, Border WA: Expression of transforming growth factor-β isoforms in human glomerular diseases. Kidney Int 1996, 49:461-469 [DOI] [PubMed] [Google Scholar]
- 25.Niemir ZI, Stein H, Noronha IL, Kruger C, Andrassy K, Ritz E, Waldherr R: PDGF and TGF-β contribute to the natural course of human IgA glomerulonephritis. Kidney Int 1995, 48:1530-1541 [DOI] [PubMed] [Google Scholar]
- 26.Remuzzi A, Puntorieri S, Battaglia C, Bertani T, Remuzzi G: Angiotensin converting enzyme inhibition ameliorates glomerular filtration of macromolecules and water and lessens glomerular injury in the rat. J Clin Invest 1990, 85:541-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Remuzzi A, Perticucci E, Ruggenenti P, Mosconi L, Limonta M, Remuzzi G: Angiotensin converting enzyme inhibition improves glomerular size-selectivity in IgA nephropathy. Kidney Int 1991, 39:1267-1273 [DOI] [PubMed] [Google Scholar]
- 28.Remuzzi A, Ruggenenti P, Mosconi L, Pata V, Viberti G, Remuzzi G: Effect of low-dose enalapril on glomerular size-selectivity in human diabetic nephropathy. J Nephrol 1993, 6:36-43 [Google Scholar]
- 29.Remuzzi A, Malanchini B, Battaglia C, Bertani T, Remuzzi G: Comparison of the effects of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade on the evolution of spontaneous glomerular injury in male MWF/Ztm rats. Exp Nephrol 1996, 4:19-25 [PubMed] [Google Scholar]
- 30.Remuzzi G, Zoja C, Gagliardini E, Corna D, Abbate M, Benigni A: Combining an antiproteinuric approach with mycophenolate mofetil fully suppresses progressive nephropathy of experimental animals. J Am Soc Nephrol 1999, 10:1542-1549 [DOI] [PubMed] [Google Scholar]
- 31.Abbate M, Zoja C, Rottoli D, Corna D, Perico N, Bertani T, Remuzzi G: Antiproteinuric therapy while preventing the abnormal protein traffic in proximal tubule abrogates protein- and complement-dependent interstitial inflammation in experimental renal disease. J Am Soc Nephrol 1999, 10:804-813 [DOI] [PubMed] [Google Scholar]
- 32.Pfeffer JM, Pfeffer MA, Frohlich ED: Validity of an indirect tail-cuff method for determining systolic arterial pressure in unanesthetized normotensive and spontaneously hypertensive rats. J Lab Clin Med 1971, 78:957-962 [PubMed] [Google Scholar]
- 33.Read SM, Northcote DH: Minimization of variation in the response to different proteins of the Coomassie blue G dye-binding assay for protein. Anal Biochem 1981, 116:53-64 [DOI] [PubMed] [Google Scholar]
- 34.Bonsnes RW, Tausski HA: The colorimetric determination of creatinine of the Jaffé reaction. J Biol Chem 1945, 158:581 [Google Scholar]
- 35.Abbate M, Bachinsky D, Zheng G, Stamenkovic I, McLaughlin M, Niles JL, McCluskey RT, Brown D: Location of gp330/α2-m receptor-associated protein (α2-MRAP) and its binding sites in kidney: distribution of endogenous α2-MRAP is modified by tissue processing. Eur J Cell Biol 1993, 61:139-149 [PubMed] [Google Scholar]
- 36.McLean IW, Nakane PF: Periodate-lysine paraformaldehyde fixative: a new fixative for immunoelectron microscopy. J Histochem Cytochem 1974, 22:1077-1083 [DOI] [PubMed] [Google Scholar]
- 37.Floege J, Burns MW, Alpers CE, Yoshimura A, Pritzl P, Gordon K, Seifert RA, Bowen-Pope DF, Couser WG, Johnson RJ: Glomerular cell proliferation and PDGF expression precede glomerulosclerosis in the remnant kidney model. Kidney Int 1992, 41:297-309 [DOI] [PubMed] [Google Scholar]
- 38.Zoja C, Liu XH, Donadelli R, Abbate M, Testa D, Corna D, Taraboletti G, Vecchi A, Dong QG, Rollins BJ, Bertani T, Remuzzi G: Renal expression of monocyte chemoattractant protein-1 in lupus autoimmune mice. J Am Soc Nephrol 1997, 8:720-729 [DOI] [PubMed] [Google Scholar]
- 39.Mundel P, Reiser J, Borja AZM, Pavenstadt H, Davidson GR, Kriz W, Zeller R: Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res 1997, 236:248-258 [DOI] [PubMed] [Google Scholar]
- 40.MacKay K, Striker LJ, Elliot S, Pinkert CA, Brinster RL, Striker GE: Glomerular epithelial, mesangial, and endothelial cell lines from transgenic mice. Kidney Int 1988, 33:677-684 [DOI] [PubMed] [Google Scholar]
- 41.Johnson RJ, Ida H, Alpers CE, Majesky MW, Schwartz SM, Pritzl P, Gordon K, Gown AM: Expression of smooth muscle cell phenotype by rat mesangial cells in immune complex nephritis: α-smooth muscle actin is a marker of mesangial cell proliferation. J Clin Invest 1991, 87:847-858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Floege J, Johnson RJ, Gordon K, Iida H, Pritzl P, Yoshimura A, Campbell C, Alpers CE, Couser WG: Increased synthesis of extracellular matrix in mesangial proliferative nephritis. Kidney Int 1991, 40:477-488 [DOI] [PubMed] [Google Scholar]
- 43.Zhang G, Moorhead PJ, El Nahas AM: Myofibroblasts and the progression of experimental glomerulonephritis. Exp Nephrol 1995, 3:308-318 [PubMed] [Google Scholar]
- 44.Muchaneta-Kubara EC, El Nahas AM: Myofibroblast phenotypes expression in experimental renal scarring. Nephrol Dial Transplant 1997, 12:904-915 [DOI] [PubMed] [Google Scholar]
- 45.Taal MW, Zandi-Nejad K, Weening B, Shahsafaei A, Kato S, Lee K-W, Ziai F, Jiang T, Brenner BM, Mackenzie HS: Proinflammatory gene expression and macrophage recruitment in the rat remnant kidney. Kidney Int 2000, 58:1664-1676 [DOI] [PubMed] [Google Scholar]
- 46.Floege J, Alpers CE, Sage EH, Pritzl P, Gordon K, Johnson RJ, Couser WG: Markers of complement-dependent and complement-independent glomerular visceral epithelial cell injury in vivo. Expression of antiadhesive proteins and cytoskeletal changes. Lab Invest 1992, 67:486-497 [PubMed] [Google Scholar]
- 47.Shimojo H: Adaptation and distortion of podocytes in rat remnant kidney. Pathol Int 1998, 48:368-383 [DOI] [PubMed] [Google Scholar]
- 48.Mundel P, Heid HW, Mundel TM, Kruger M, Reiser J, Kriz W: Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol 1997, 139:193-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mundel P, Reiser J, Kriz W: Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol 1997, 8:697-705 [DOI] [PubMed] [Google Scholar]
- 50.Taal MW, Brenner BM: Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int 2000, 57:1803-1817 [DOI] [PubMed] [Google Scholar]
- 51.Ruggenenti P, Schieppati A, Remuzzi G: Progression, remission, regression of chronic renal diseases. Lancet 2001, 357:1601-1608 [DOI] [PubMed] [Google Scholar]
- 52.Nenov VD, Taal MW, Sakharova OV, Brenner BM: Multi-hit nature of chronic renal disease. Curr Opin Nephrol Hyperten 2000, 9:85-97 [DOI] [PubMed] [Google Scholar]
- 53.Yoshioka T, Mitarai T, Kon V, Deen WM, Rennke HG, Ichikawa I: Role for angiotensin II in an overt functional proteinuria. Kidney Int 1986, 30:538-545 [DOI] [PubMed] [Google Scholar]
- 54.Yoshioka T, Rennke HG, Salant DJ, Deen WM, Ichikawa I: Role of abnormally high transmural pressure in the permselectivity defect of glomerular capillary wall: a study in early passive Heymann nephritis. Circ Res 1987, 61:531-538 [DOI] [PubMed] [Google Scholar]
- 55.Elema JD, Hoyer JR, Vernier RL: The glomerular mesangium: uptake and transport of intravenously injected colloidal carbon in rats. Kidney Int 1976, 9:395-406 [DOI] [PubMed] [Google Scholar]
- 56.Stein HD, Feddergreen W, Kashgarian M, Sterzel RB: Role of angiotensin-II induced renal functional changes in mesangial deposition of exogenous ferritin in rats. Lab Invest 1983, 49:270-280 [PubMed] [Google Scholar]
- 57.Barisoni L, Kriz W, Mundel P, D’Agati VD: The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 1999, 10:51-61 [DOI] [PubMed] [Google Scholar]
- 58.Barisoni L, Bruggeman LA, Mundel P, D’Agati VD, Klotman PE: HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int 2000, 58:173-181 [DOI] [PubMed] [Google Scholar]
- 59.Srivastava T, Garola RE, Whiting JM, Alon US: Synaptopodin expression in idiopathic nephrotic syndrome of childhood. Kidney Int 2001, 59:118-125 [DOI] [PubMed] [Google Scholar]
- 60.Weigert C, Sauer U, Brodbeck K, Pfeiffer A, Haring HU, Schleicher ED: AP-1 proteins mediate hyperglycemia-induced activation of the human TGF-β1 promoter in mesangial cells. J Am Soc Nephrol 2000, 11:2007-2016 [DOI] [PubMed] [Google Scholar]
- 61.Inoki K, Haneda M, Ishida T, Mori H, Maeda S, Koya D, Sugimoto T, Kikkawa R: Role of mitogen-activated protein kinases as downstream effectors of transforming growth factor-β in mesangial cells. Kidney Int 2000, 58:S76-S80 [DOI] [PubMed] [Google Scholar]
- 62.Schiffer M, von Gersdorff G, Bitzer M, Susztak K, Bottinger EP: Smad proteins and transforming growth factor-β signaling. Kidney Int 2000, 58:S45-S52 [DOI] [PubMed] [Google Scholar]
- 63.Morigi M, Zoja C, Angioletti S, Zanchi C, Longaretti L, Donadelli R, Mundel P, Benigni A, Remuzzi G: Podocytes respond to protein overload by cytoskeleton rearrangement and upregulation of AP-1 dependent endothelin-1 (ET-1) gene expression. J Am Soc Nephrol 2001, 12:686A [Google Scholar]
- 64.Purkerson ML, Hoffsten PE, Klahr S: Pathogenesis of the glomerulopathy associated with renal infarction in rats. Kidney Int 1976, 9:407-417 [DOI] [PubMed] [Google Scholar]
- 65.Zatz R, Dunn BR, Meyer TW, Anderson S, Rennke HG, Brenner BM: Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest 1986, 77:1925-1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson S, Rennke HG, Brenner BM: Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 1986, 77:1993-2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benigni A, Tomasoni S, Gagliardini E, Zoja C, Grunkemeyer JA, Kalluri R, Remuzzi G: Blocking angiotensin II synthesis/activity preserves glomerular nephrin in rats with severe nephrosis. J Am Soc Nephrol 2001, 12:941-948 [DOI] [PubMed] [Google Scholar]
- 68.Macconi D, Ghilardi M, Bonassi ME, Mohamed EI, Abbate M, Colombi F, Remuzzi G, Remuzzi A: Effect of angiotensin-converting enzyme inhibition on glomerular basement membrane permeability and distribution of zonula occludens-1 in MWF rats. J Am Soc Nephrol 2000, 11:477-489 [DOI] [PubMed] [Google Scholar]
- 69.Kagami S, Border WA, Miller DE, Noble NA: Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-β expression in rat glomerular mesangial cells. J Clin Invest 1994, 93:2431-2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Riser BL, Ladson-Wofford S, Sharba A, Cortes P, Drake K, Guerin CJ, Yee J, Choi ME, Segarini PR, Narins RG: TGF-β receptor expression and binding in rat mesangial cells: modulation by glucose and cyclic mechanical strain. Kidney Int 1999, 56:428-439 [DOI] [PubMed] [Google Scholar]
- 71.van Det NF, Verhagen NAM, Tamsma JT, Berden JHM, Bruijn JA, Daha MR, van der Woude FJ: Regulation of glomerular epithelial cell production of fibronectin and transforming growth factor-β by high glucose, not by angiotensin II. Diabetes 1997, 46:834-840 [DOI] [PubMed] [Google Scholar]