

Abstract
Ectopic calcification, the abnormal calcification of soft tissues, can have severe clinical consequences especially when localized to vital organs such as heart valves, arteries, and kidneys. Recent observations suggest that ectopic calcification, like bone biomineralization, is an actively regulated process. These observations have led a search for molecular determinants of ectopic calcification. A candidate molecule is osteopontin (OPN), a secreted phosphoprotein invariantly associated with both normal and pathological mineral deposits. In the present study, OPN was found to be a natural inhibitor of ectopic calcification in vivo. Glutaraldehyde-fixed aortic valve leaflets showed accelerated and fourfold to fivefold greater calcification after subcutaneous implantation into OPN-null mice compared to wild-type mice. In vitro and in vivo studies suggest that OPN not only inhibits mineral deposition but also actively promotes its dissolution by physically blocking hydroxyapatite crystal growth and inducing expression of carbonic anhydrase II in monocytic cells and promoting acidification of the extracellular milieu. These findings suggest a novel mechanism of OPN action and potential therapeutic approach to the treatment of ectopic calcification.
Biomineralization is an exquisitely regulated physiological process normally restricted to bones and teeth. Under certain pathological conditions, however, calcium salts may deposit in soft tissues with detrimental effects. Although most soft tissues can undergo ectopic calcification as a result of injury, aging, disease or metabolic imbalance, blood vessels, heart valves, and the kidney seem particularly susceptible. Ectopic calcification of blood vessels is widespread in individuals with atherosclerosis, diabetes, and end-stage renal disease and is correlated with several poor outcomes. In arteries, calcification is positively correlated with atherosclerotic plaque burden and increased risk of myocardial infarction, 1-3 increased ischemic episodes in peripheral vascular disease 4 and increased risk of dissection after angioplasty. 5 In addition, medial arterial calcification is strongly correlated with coronary artery disease in type I diabetics 6 and is a strong independent marker of future cardiovascular events in diabetic patients. 7 In the heart, calcific aortic stenosis, characterized by mineralization of aortic valve leaflets resulting in their deterioration and subsequent mechanical failure, occurs in ∼1 to 2% of the elderly population. 8 Although a common therapy for aortic stenosis is bioprosthetic tissue valve implantation, these replacement valves also suffer from ectopic mineralization, which is the leading cause of implant failure. 9 In the kidney, renal stones affect millions yearly, and can lead to incapacitating pain as well as hydronephrosis and hydroureter. 10
Recent insights into the mechanisms regulating ectopic calcification have predominantly come from studies of cardiovascular calcification. Similarities to bone mineralization were suggested by the finding that bone morphogenetic protein-2 and bone matrix proteins including osteopontin (OPN), osteonectin, osteocalcin, and matrix GLA protein are found in calcified vascular tissues. 11 In addition, smooth muscle cells and other vascular media derived-cells demonstrate osteoblast-like properties and can mineralize their extracellular matrices under appropriate conditions in vitro. 12-14 Moreover, ectopic bone formation has occasionally been found in calcified vascular lesions. 15 Finally, matrix vesicles similar to those proposed to nucleate mineral in bone have been identified in calcified vascular tissues. 16-18
Although these studies suggest cell-mediated regulation of ectopic mineralization, the most compelling data have come from studies of mutant mice. The matrix GLA protein-null mouse displays extensive calcification of the arterial tree, and animals die within 2 months apparently from aortic rupture. 19 In addition, several other mutant mouse strains show enhanced susceptibility to ectopic calcification, including mice deficient in osteoprotegerin; 20 β-glucosidase; carbonic anhydrase II; 21 desmin; 22 fetuin; 23 the progressive ankylosis gene, ANK; 24 Npps, a nucleotide triphosphate pyrophosphohydrolase; 25 and an intracellular mediator of transforming growth factor-β signaling, Smad6. 26 These findings highlight the fundamental importance and potential diversity of mechanisms that are genetically programmed in animals to prevent ectopic calcification.
One molecule that consistently co-localizes with ectopic calcification is OPN. OPN is an acidic phosphoprotein normally found in bone, teeth, kidney, and epithelial lining tissues. OPN′s expression is increased under conditions of injury and disease in many tissues, and it is closely associated with the calcified deposits found in numerous pathologies including atherosclerotic lesions, aortic stenosis, kidney stones, and tumors. 27 OPN is a multifunctional protein containing several structural domains including an integrin-binding (RGD) adhesive domain and aspartic acid-rich calcium-binding regions. In addition, OPN can be highly phosphorylated on serine and threonine residues. The combination of electronegative glutamic and aspartic acid residues, serine/threonine kinase substrate sites, and the putative calcium-binding motifs endow OPN with the ability to bind significant amounts of Ca2+ (50 mol calcium to 1 mol osteopontin). 28 These properties likely contribute to OPN′s ability to bind and regulate apatite crystal growth, the predominant calcium-phosphate mineral phase found in bones and teeth as well as at sites of ectopic calcification. Indeed, in vitro studies support a role for OPN as an inhibitor of apatite growth in both cell-free and cell-dependent in vitro models. 13,29-31 On the other hand, the co-localization of OPN with biomineralization in hard tissues, and its ability to bind and potentially orient calcium suggest that OPN might function to promote calcification in vivo. 32 To determine the role of OPN in ectopic calcification, we developed a model of ectopic calcification in OPN-replete and -deficient mice. Our studies indicate that OPN is a potent inhibitor of ectopic calcification, and suggest a novel function for OPN in controlling mineral-dissolving inflammatory cell function at sites of ectopic calcification.
Materials and Methods
Mouse Ectopic Calcification Model
OPN mutant mice were generated in a 129/SvJ X Black Swiss background and genotyped as previously described by Liaw and colleagues. 33 Animals were maintained in a specific pathogen-free environment, and fed standard chow and water ad libitum. Hybrid mutant mice were backcrossed onto the Black Swiss background for >7 generations. OPN homozygous wild-type (+/+), heterozygote (+/−), and homozygous null mice on the fixed Black Swiss background were used in these studies. Porcine aortic valve leaflets (kindly provided by St. Jude Medical, Inc., Minneapolis, MN) were aseptically dissected from porcine aortic valves obtained from the local abattoir, fixed in 0.6% glutaraldehyde in phosphate-buffered saline (PBS), pH 7.4, and stored in 0.6% glutaraldehyde in PBS, pH 7.4 until use. Four-mm 2 biopsy punches of glutaraldehyde-fixed aortic valve leaflets (GFAVs) were prepared, washed extensively in sterile PBS, and subcutaneously implanted into the dorsal side of anesthetized 5- to 6-week-old, female OPN +/+, +/−, or −/− mice (two GFAVs per mouse). At the indicated times, mice were euthanized, and implants removed for either histological analysis or calcium quantitation. All protocols were approved by the animal use committee, University of Washington, Seattle, WA.
Histological Analysis
Explants were fixed with methyl Carnoy’s solution (3:1, methanol:acetic acid) and embedded in paraffin. Immunostaining was performed in 5-μm sections with goat anti-rat OPN antibody (OP199) 34 at 10 μg/ml, macrophage-specific rat anti-mouse BM-8 (Accurate Chemical & Scientific Corp., Westbury, NY) at 6 μg/ml, or sheep anti-human carbonic anhydrase II (CAII) (Biodesign International, Kennebunk, ME) at 5 μg/ml, and counterstained with methyl green as previously described. 35 Calcium phosphate deposition was visualized by Alizarin Red S staining and von Kossa staining. 36 Transmission electron microscopy was performed as described by Wada and colleagues. 13
Image Analysis
Four to 8 OPN+/+, +/− and −/− mice were subcutaneously implanted with two GFAVs as described above. At each time point, implants and adherent host tissues were retrieved, fixed, and embedded as described above. For each recovered specimen (two per mouse), two histological sections were prepared at least 50 μm apart, for a total of four sections per animal. OPN, BM-8, and CAII staining in sections was performed as described above and quantitated using the ProImage Analysis Program. The area containing the GFAV implant and the associated foreign body capsule was circumscribed and the percentage of this area stained with each antibody determined and averaged to generate the percent area stained per implant per mouse. The percent area stained per implant per mouse for four to eight mice were averaged to obtain the average percent area stained for each genotype ± SE.
Calcium Quantitation
Explants were freeze-dried to constant weight and decalcified with 0.6 N HCl overnight at room temperature. Calcium quantitation was performed by the ο-cresolphthalein complexone as directed in the Sigma Diagnostic kit (Sigma, St. Louis, MO) and normalized to dry weight, as previously described. 13 Accuracy of the kit was confirmed by atomic absorption spectroscopy (kindly performed by Baxter Health Care Corporation, Irvine, CA).
GFAV Explant pH Analysis
GFAV explants were freeze-dried then immersed in Universal Indicator Solution (Fisher Scientific, Pittsburgh, PA). The pH of the GFAV solution was determined by three different, blinded readers by comparing the color of this solution to the manufacturer’s provided graded color scale. Accuracy of the color indicator solution was determined using an MI-408 needle pH microelectrode (Microelectrodes, Inc., Bedford, NH). Control experiments indicated that tissue lyophilization did not significantly alter pH of GFAV using either the dye or microelectrode measurement method (data not shown).
Cell Culture and Western Blot Analysis
HL-60 cells (American Type Culture Collection, Rockville, MD) were maintained in RPMI media (Life Technologies, Inc., Grand Island, NY) containing 5% fetal bovine serum, 100 U/ml penicillin (Life Technologies, Inc.), and 100 mg/ml streptomycin (Life Technologies, Inc.). Primary human monocytes were differentially isolated from human blood, 37,38 and maintained in RPMI containing 5% autologous human serum. Preparation of protein extracts, gel electrophoresis, and Western blotting were performed as previously described 39 using 5 μg/ml of sheep anti-human CA II (Biodesign International, Kennebunk, ME).
Statistical Analysis
Data were analyzed for statistical significance using analysis of variance statistics with Fischer’s protected least significance difference test. Calculations were performed using the computer program StatView version 4.11 (Abacus Concepts, Berkeley, CA).
Results
OPN Inhibits Calcification of GFAV Leaflets in Mice
Four-mm 2 pieces of GFAV were subcutaneously implanted into mice carrying either the OPN homozygous wild-type (OPN+/+), heterozygous (OPN+/−), or homozygous null (OPN−/−) alleles. 33 At 7, 14, 30, and 90 days, implants were removed and assayed for mineral deposition, protein accumulation, and cell recruitment. In all animals, a robust foreign body response was induced by GFAV implantation. This host response was characterized by accumulation of macrophages, formation of giant cells, and fibrous encapsulation by days 14 and 30 (Figure 1; A to C ▶ ). There were no obvious differences in quantity of the foreign body response between the different genotypes as determined by hematoxylin and eosin staining (Figure 1; A to C ▶ and data not shown), and this response was similar to the foreign body response typically observed after GFAV implantation in rats 40 and rabbits. 41 However, differences in the quality of the foreign body response, especially in terms of macrophage number, was noted between genotypes as described below.
Figure 1.

Immunolocalization of OPN on and around GFAV implants. GFAV implants and adherent tissues were removed after 14 days (A, D, G) or 30 days (B, C, E, F, H, and I) from OPN+/+ (A, B, D–F), OPN+/− (G, H), or OPN−/− (C, I) mice. Samples were fixed, embedded in paraffin, and stained with H&E (A–C) or analyzed for OPN accumulation using the anti-OPN antibody, OP199, followed by DAB detection substrate (brown) and counterstained with methyl green (D–I). v, GFAV; f, foreign body response; c, calcium deposits; G, giant cells; M, macrophages. Arrowheads demarcate the interface between the GFAV tissue and the host foreign body response. Scale bars: 250 μm (A–C); 100 μm (D–E); 15 μm (F); 50 μm (G); 30 μm (H–I).
As shown in Figure 1; D to I ▶ , OPN was highly expressed in host cells surrounding the GFAV implant in OPN+/+ mice at days 14 and 30. In contrast, OPN+/− mice showed little OPN staining at day 14 (Figure 1G) ▶ , even when examined at high magnification (data not shown), but showed marked accumulation at day 30 (Figure 1H) ▶ . Staining of adjacent sections with macrophage-specific marker, BM-8 (not shown), indicated that cells expressing OPN were predominantly of macrophage origin, and included a subset of mononuclear macrophages as well as multinucleated giant cells (Figure 1F) ▶ . In addition, punctate OPN staining was also observed within the GFAV implant itself, especially in the 30-day OPN+/+ mice (Figure 1, E and F) ▶ . This staining was co-localized with GFAV mineral deposits as determined by alizarin red staining of an adjacent section (data not shown, as well as the double-staining experiment shown in Figure 3, G and H ▶ , described in detail below). Levels of OPN associated with GFAV increased in OPN+/+ and OPN+/− mice with increasing time of implantation (Figures 1 and 2A) ▶ ▶ . As expected, no OPN was detected in OPN−/− mice at any time point examined (Figures 1I and 2A) ▶ ▶ .
Figure 3.

Histochemical localization of mineral deposition and OPN in GFAV implants. GFAV implants removed from OPN+/+ (A, B), OPN+/− (C, D), or OPN−/− (E, F) mice at 14 (A, C, E) or 30 (B, D, F) days after subcutaneous implantation were fixed, embedded in paraffin, and stained for calcium deposition using Alizarin Red S. G: Sections from 30-day-old OPN +/+ mice stained with antibody directed against OPN (brown) shows cellular staining in host foreign body response as well as punctate staining within the valve. H: Same section shown in G subsequently stained with Alizarin Red S (red) indicates that OPN is co-localized with punctate mineral deposits within the GFAVs. Inset: high-power magnification of boxed region shows OPN-positive giant cell and OPN/alizarin red-positive mineral deposit. v, Valve; f, foreign body response. Arrowheads demarcate interface between GFAV and host foreign body response.
Figure 2.

Quantitation of OPN and mineral deposition associated with GFAV implants. A: OPN accumulation was quantitated using the Pro Image Analysis Program as described in Materials and Methods. Data are represented as percent area. B: Calcium deposition was quantitated from acid-hydrolyzed GFAV explants using Sigma’s Calcium Diagnostic Kit and confirmed by atomic absorption spectroscopy. Numbers represent the average ± SE (n = 4 to 8). *, P < 0.005.
To determine whether OPN expression influenced the propensity of GFAVs to calcify in mice, implants were analyzed for calcium mineral deposition using histochemical and biochemical methods. The OPN null mutation affected both the extent and temporal appearance of calcification as measured by total calcium content (Figure 2B) ▶ , alizarin red S staining (Figure 3) ▶ , and von Kossa staining (not shown). None of the mice showed significant calcification at day 7. This did not appear to be because of a sensitivity problem, as we were unable to detect calcification using the sensitive fluorescent stain, calcein, in these samples (data not shown). Low levels of calcification were observed in GFAV implants at day 14 and day 30 in OPN+/+ mice (Figure 2B ▶ and Figure 3, A and B ▶ ), and these levels were not statistically different. In contrast, significant calcification was observed in both OPN+/− and OPN−/− mice at day 14, at levels fourfold to fivefold greater than that observed at day 14 in OPN+/+ mice (Figure 2B ▶ and Figure 3, C and E ▶ ). Finally, it did not appear that calcification was restricted to the OPN-deficient mice, as GFAVs implanted for 90 days in OPN wild-type mice showed considerable calcification, although still less than OPN null mice (OPN+/+ = 3000 ± 600 mmol Ca/L/g protein versus OPN+/− = 1900 ± 500 mmol Ca/L/g protein versus OPN−/− = 6900 ± 250 mmol Ca/L/g protein). Hence, these studies indicate that OPN plays a role in modulating GFAV calcification in vivo.
As shown in Figure 3 ▶ , calcium deposits visualized by alizarin red staining were deposited along the periphery as well as throughout the interior of the implant. Calcification occurred predominantly in foci within the GFAV leaflet. OPN, in addition to being localized to cells of the foreign body reaction, was found associated with these punctate-mineralized deposits within the GFAV implant as shown by double staining with antibody to OPN and alizarin red S for mineral (Figure 3, G and H) ▶ . By transmission electron microscopy, the foci of mineralization within the GFAV implant were found to be associated predominantly with cell remnants including membranous debris resembling matrix vesicles (Figure 4) ▶ . This localization is consistent with reports describing experimental and patient GFAV mineralization on remnant cell membranes and organelles. 42-44 Taken together, these studies implicate OPN, most likely derived from infiltrating macrophages and giant cells, as an inhibitor of ectopic calcification in this model system. Co-localization of OPN protein with mineralization within the GFAV suggest that OPN binding to nascent sites of mineralization may be one mechanism by which OPN exerts its anti-calcific effect.
Figure 4.
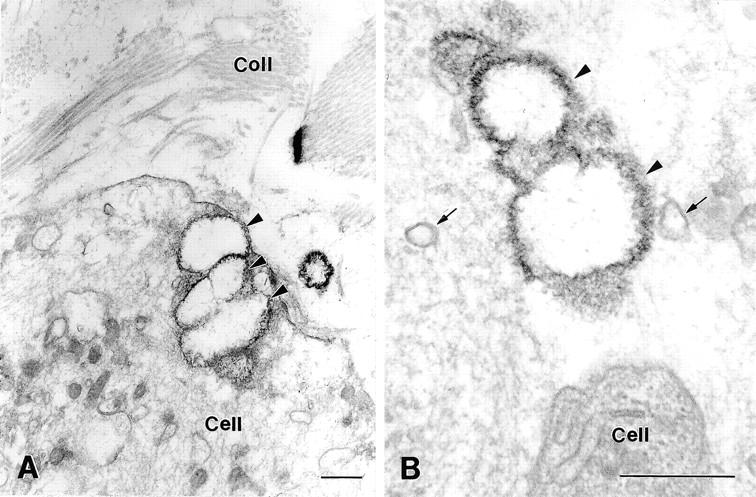
Transmission electron micrographs showing GFAV ultrastructure after implantation into OPN−/− mice for 30 days. Calcification of implanted valve tissue generally occurred in close association with intracellular membranous compartments (arrowheads, A), or with similar membranous debris found within the valve extracellular matrix (arrowheads, B). Calcified deposits typically were spherical in appearance with a peripheral rim of particularly dense organic material that associated with the mineralized nodules. Membranous structures resembling matrix vesicles (arrows, B) were frequently observed in the vicinity of calcification sites. Coll, collagen fibrils. Scale bars, 0.5 μm.
OPN Promotes Regression of Ectopic Calcification
Comparison of calcification with OPN levels in GFAV at day 14 after implantation indicated that OPN was an inhibitor of ectopic calcification. These findings are consistent with previous in vitro studies showing that OPN can bind to and block hydroxyapatite crystal growth. 29 However, examination of the OPN mutants at day 30 after implantation suggested that simple physical inhibition alone could not explain the inhibitory effect of OPN on ectopic calcification in this model system. By 30 days, significant levels of OPN accumulated in OPN+/− mice throughout the GFAV implant and was similar to levels observed in OPN+/+ mice (Figure 2A ▶ , and immunostaining data not shown). Strikingly, OPN accumulation in OPN+/− mice at day 30 was concurrent with a significant reduction in GFAV mineralization from the levels observed at day 14 (Figure 2B ▶ and Figure 3, C and D ▶ ). If OPN acted simply as a crystal poison, one would expect little difference in the day 14 and day 30 calcification values after implant in the OPN+/− mice. Thus, these findings suggested that OPN additionally acted to promote regression of ectopic calcification.
Macrophage Number and Phagocytosis Do Not Correlate with Extent of Mineralization in GFAV Implants in OPN Mutant Mice
Because OPN binding alone cannot mediate the dissolution of calcium phosphate, we hypothesized that the observed mineral regression was the result of an OPN-regulated cell-mediated dissolution of mineral. The only known mechanisms capable of removing calcium phosphate crystals are phagocytosis and acidification. 45 To address the former possibility, we examined macrophage accumulation in our model. In all genotypes and time points examined, BM-8-positive cells accumulated in the foreign body reaction, preferentially along the edge of the GFAV implant (Figure 5; A to C ▶ , and data not shown). However, few, if any, macrophages were able to penetrate the GFAV at any time point examined (Figure 5; D to F ▶ , and data not shown). This is consistent with previous studies demonstrating the effect of glutaraldehyde fixation in blocking tissue degradation, and is one of the main reason that glutaraldehyde crosslinked valves are used clinically. 46 Because macrophages were unable to enter the GFAV, phagocytosis of mineral deposits within the implant is unlikely to account for the observed mineral loss in heterozygous mice.
Figure 5.

Analysis of macrophage recruitment to site of GFAV implantation by BM-8 immunohistochemistry. GFAVs and adherent tissues were removed 30 days after subcutaneous implantation from OPN+/+ (A, D), OPN+/− (B, E), or OPN−/− (C, F) mice and analyzed for macrophage accumulation using an antibody to the macrophage surface marker, BM-8 (brown staining). v, GFAV. Arrowheads demarcate the interface between the GFAV tissue and the host foreign body response. Scale bars: 250 μm (A–C); 50 μm (D–F).
However, consistent with previous studies, 47,48 OPN−/− mice did display an overall defect in macrophage recruitment to the GFAV implant. As shown in Figure 6 ▶ , OPN−/− mice demonstrated BM-8-positive cell staining, but at levels 50 to 25% lower than OPN+/+ at 14 and 30 days of implantation, respectively. Although OPN+/− mice also had lower BM-8-positive cell accumulation at day 30, levels were not significantly different from those observed in OPN +/+ mice at day 14. Thus, total macrophage levels did not correlate with the mineral loss pattern observed in the OPN+/− mice between day 14 and at day 30 (compare Figure 2B ▶ and Figure 6 ▶ ). Therefore, it seemed unlikely that the loss of mineral was strictly regulated by macrophage number alone.
Figure 6.

Quantitation of macrophage levels associated with GFAV implants and associated foreign body response. BM-8-positive cells in the foreign body response surrounding GFAV implants at 14 and 30 days after subcutaneous implantation were quantitated using the ProImage Analysis System and presented as percent BM-8-positive area. Numbers represent the average ± SE (n = 4 to 8). *, P < 0.005.
OPN Controls GFAV Implant Acidification
These findings led us to investigate whether OPN promotes implant acidification. The ability of cells to dissolve calcium phosphate mineral by promoting local decreases in pH has been described for both macrophages and osteoclasts. 41 Osteoclasts and macrophages are derived from a similar hematopoietic origin, and previous reports suggest multinucleated osteoclasts can be produced from the fusion of monocytes or macrophages. To test the acidification hypothesis, GFAVs were explanted from OPN+/+, OPN+/−, and OPN−/− mice after 14 days of implantation, and pH determined using Fisher’s universal indicator solution (a mixture of pH sensitive dyes including phenolphthalein, bromothymol blue, methyl red, and thymol). As shown in Figure 7 ▶ , GFAV explants from OPN+/+ mice at 2 weeks had an acidic pH (pH 6.0), whereas explants from OPN−/− mice were more neutral (pH 6.7). These differences in pH were highly statistically significant (P = 0.0002), and confirmed using a needle microelectrode in a second set of GFAV valves (data not shown). GFAV explants from OPN+/− mice were intermediate with respect to those from OPN+/+ and OPN−/− mice with pH 6.2. Because apatite stability is low at pH less than 7 at 37°C, 49 the observed pH of the explants would be physiologically relevant with respect to ability to dissolve apatitic mineral and could explain the observed loss in GFAV mineralization. These findings strongly suggest that OPN regulates the ability of host cells to acidify the GFAV microenvironment, thereby controlling mineral dissolution.
Figure 7.

GFAV implant pH analysis. Implanted GFAVs and adherent tissues were removed from OPN +/+, +/−, and −/− mice at 2 weeks, freeze-dried, then immersed in Universal Indicator Solution (Fisher Scientific). The pH of each implant was independently determined by three different observers comparing the color of the GFAV solution to the manufacturer’s provided color scale. Numbers represent the average pH ± SE (n = 4 to 9). *, P < 0.005.
OPN Regulates Expression of CAII in Macrophages and Multinucleated Giant Cells
To shed light on the mechanism by which OPN might mediate GFAV acidification, we examined levels of CAII in the cells surrounding the implants. CAII is an enzyme that promotes the conversion of carbon dioxide to carbonic acid. In osteoclasts, CAII-driven carbonic acid production serves as an intracellular source of protons that are exported from the cell via a vacuolar H+-ATPase thereby reducing local pH, and promoting mineral dissolution. 50 To evaluate the acid-producing potential of cells recruited to the GFAV implant, we examined the presence of CAII immunohistochemically. CAII was strongly expressed by a subset of BM-8-positive mononuclear cells (arrowheads) as well as all multinucleated giant cells (arrows) located within the foreign body response adjacent to the mineralized implant (Figure 8, A and B) ▶ . Although the giant cells resembled osteoclasts in being multinucleated and expressing high levels of OPN, αvβ3, and cathepsin K, no characteristic clear zones or ruffled borders were observed by electron microscopy (Figure 8B ▶ and data not shown).
Figure 8.
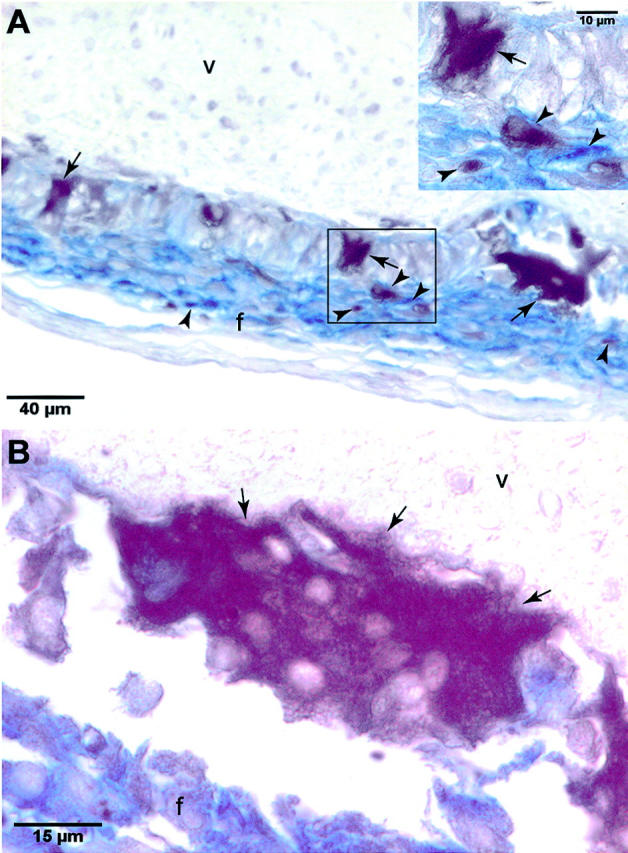
Carbonic anhydrase II expression in macrophages and giant cells associated with GFAV implantation. A and B: Carbonic anhydrase II (CAII) expressing multinucleated and mononuclear cells were co-localized in the foreign body response surrounding GFAV explants using an anti-human CAII antibody (brown stain) and anti-BM-8 antibody (blue stain). A, Inset: Higher magnification of boxed region showing that CAII-positive cells make up a subset of the responding BM-8-positive macrophages, as well as BM-8-negative giant cells. f, foreign body response; v, GFAV. Arrows indicate giant cells and arrowheads indicate macrophages.
Quantitation of CAII immunostaining revealed a mineral- and OPN-dependent regulation of CAII expression. At 14 days, OPN was present and GFAV implants showed little mineralization in OPN+/+ mice (Figure 2) ▶ . At this time point, CAII levels were low in OPN +/+ mice (Figure 9) ▶ . At 30 days, when GFAV mineralization increased and OPN levels accumulated in OPN +/+ mice (Figure 3) ▶ , CAII-expressing cells were dramatically increased (Figure 7B) ▶ . Conversely, the OPN−/− mice expressed very low CAII levels despite the elevated mineral levels at all observed time points. The OPN-mediated CAII response appeared accelerated in the OPN+/− mice. At 14 days, OPN+/− mice demonstrated substantial GFAV mineralization and low levels of OPN (Figure 2) ▶ , but appeared to be able to respond to the mineralization by producing CAII-positive cells (Figure 9) ▶ .
Figure 9.

Quantitation of CAII expression in GFAV implants and associated foreign body response. CAII expression surrounding GFAV implants was examined immunohistochemically as in Figure 8 ▶ at 14 and 30 days after subcutaneous implantation into OPN +/+, +/−, and −/− mice. Quantitation was performed using the ProImage Analysis Program. Data are presented as percent CAII-positive area. Numbers represent the average ± SE (n = 4 to 8). *, P < 0.005.
To further examine the possibility that OPN directly regulates CAII levels in monocytic cells, HL-60 cells, a promyelocytic leukemia line demonstrating monocyte to macrophage differentiation, 51 and primary human monocyte-derived macrophages were treated with either soluble or immobilized recombinant OPN, respectively. As shown in Figure 10 ▶ , OPN induced CAII protein expression in both cell types. This activity was mimicked by the addition of the αvβ3-crosslinking antibody, LM609, to HL60 cells. 52 Furthermore, RGD peptides inhibited the ability of OPN to induce CAII expression in primary human monocyte-derived macrophages (data not shown). These findings support the hypothesis that OPN promotes acidification by αvβ3-mediated CAII induction and subsequent proton production in monocytic cells.
Figure 10.

Effect of OPN on CAII expression in cultured macrophages. HL-60 cells were treated with either 40 nmol/L of soluble recombinant OPN, equal volume vehicle control, 15 μg/ml of LM609 mouse IgG (αvβ3 agonist), or mouse IgG (mIgG) control for 24 hours. Primary human monocytes (hMo) were plated on tissue culture plates immobilized with 100 nmol/L of recombinant OPN or poly-d-lysine (Sigma) for 48 hours. Cell lysates were prepared, quantitated, and analyzed for CAII expression by immunoblotting.
Discussion
Ectopic calcification contributes to clinically significant sequelae in patients with valve disease, bioprosthetic valves, diabetes, end-stage renal disease, and nephrolithiasis. Until recently, the study of ectopic calcification has been restricted to immunochemical description of molecules co-localized with the abnormally deposited mineral. In the present studies, we have demonstrated that one of these molecules, OPN, is a natural regulator of ectopic calcification. Mice deficient in OPN through targeted gene deletion showed much greater propensity to mineralize subcutaneously implanted GFAVs than wild-type mice. These studies are the first to demonstrate an inhibitory role for OPN in ectopic mineralization in vivo. Our studies suggest that OPN not only acts by physical inhibition of mineral crystal growth, but also mediates an anti-calcific cellular response to ectopic mineralization that leads to active dissolution. Consistent with this hypothesis, mice deficient in OPN were characterized by an inability to acidify GFAV implants, and had decreased CAII expression by macrophages and giant cells. These findings suggest a novel mechanism of action for effects of OPN on ectopic mineralization, and open the door for new therapeutic approaches in treating and perhaps reversing ectopic calcification.
To determine the function of OPN in vivo, we developed a subcutaneous GFAV implantation model in the mouse. In this model, bioapatites were deposited throughout the GFAV implant with time. By electron microscopy, crystals appeared to nucleate and grow early within residual cell bodies and membrane-bound vesicular cell fragments resembling matrix vesicles. OPN was expressed by monocyte-derived macrophages and giant cells in the surrounding foreign body response, and accumulated at foci of mineralization within the implants. These findings are consistent with observations made in GFAV implantation models in rats and rabbits. 40,41,53 Although accelerated, all of these models mimic the human pathology observed during native and bioprosthetic aortic valve failure both in levels of mineral deposition (∼100 μg calcium/mg tissue), presence of inflammation, and OPN accumulation. 54,55 In fact, the subcutaneous implantation model in rats and rabbits has become a standard preclinical animal model used to analyze the effectiveness of potential bioprosthetic valve anti-calcific agents. 53
Using the GFAV subcutaneous model in mice bearing targeted deletion of the OPN gene, we show that OPN is a potent, endogenous inhibitor of ectopic calcification. OPN levels in mutant mice were inversely proportional to calcification levels. OPN−/− mice showed not only more GFAV calcification at all time points studied, but they also became calcified at earlier time points than OPN+/+ or OPN+/− mice. These findings are consistent with previous in vitro studies showing that OPN inhibits hydroxyapatite growth in solution and in calcifying smooth muscle cell cultures. 13,29,30 In those studies, OPN was found to bind tightly to hydroxyapatite, suggesting that it acted as a physical inhibitor of crystal growth by binding to growing crystal faces. Evidence for this mechanism was seen in our studies as OPN was found to co-localize with mineral deposits within the calcifying GFAVs. However, our studies also provide new mechanistic insights into the role of OPN in controlling ectopic calcification that could not be predicted based on the in vitro findings. Although significant calcification was observed in GFAVs in OPN+/− mice at 2 weeks, by 4 weeks calcification had regressed to levels observed in wild-type mice. This decrease in calcification correlated with an increase in OPN accumulation between 2 and 4 weeks in GFAVs of OPN+/− mice. These data indicate that OPN does not merely physically inhibit ectopic calcification, but promotes dissolution as well.
How does OPN promote regression of GFAV calcification? Our studies indicate that OPN contributes to the regulation of pH in the implant environment. We found that OPN−/− mice, and to a lesser extent, OPN+/− mice, were defective in their ability to acidify the GFAV implants when compared to OPN+/+ mice. Because bioapatites are unstable below pH 7 at body temperature, 45 this failure to acidify likely contributes to the increased mineralization observed in OPN−/− mice. Furthermore, OPN regulation of implant pH could explain the reversal of calcification observed between 2 and 4 weeks in OPN+/− mice, because OPN levels accumulate almost to wild-type levels in OPN+/− mice by 4 weeks.
We were able to measure pH differences within the microenvironment of the GFAVs because of the encapsulation caused by the foreign body reaction that follows GFAV implantation. Avascular foreign body capsules such as those observed in our studies are well known to essentially wall off the foreign body, thereby isolating the implants physically and chemically from the surrounding host tissues. 56 Such encapsulation of foreign bodies is thus a major problem in the development of blood chemistry-sensing devices such as glucose sensors. 57 Thus, what is typically considered a detrimental foreign body response enabled us to monitor pH within the microenvironment of the GFAV implant in these studies. Finally, the finding that macrophage levels as well as the ability of macrophages and giant cells to express CAII were dependent on OPN genotype indicates that OPN may be involved in regulating the foreign body reaction in general.
To determine the mechanism by which OPN might regulate implant acidity, we examined CAII levels in cells surrounding the GFAV implant. CAII is found in most cells, but is highly expressed in osteoclasts, the major mineral resorbing cell in the body. CAII enzymatically converts carbon dioxide to carbonic acid. In osteoclasts, CAII-driven carbonic acid production serves as an intracellular source of protons that are exported from the cell via a vacuolar H+-ATPase thereby reducing local pH, and facilitating mineral dissolution. 50 Furthermore, CAII deficiency in humans and mice leads to soft tissue calcifications, in addition to osteopetrosis. 21 We found that CAII was expressed in a subset of macrophages, as well as most giant cells, associated with the foreign body response to GFAV implants. Although the giant cells resembled osteoclasts by being multinucleated and expressing high levels of OPN, CAII, αvβ3, and cathepsin K, no structures resembling clear zones or ruffled borders characteristic of actively resorbing osteoclasts were observed. Thus, these cells represent a previously uncharacterized cell type that may be particularly important for resorption of ectopic mineralization.
We found that CAII expression in macrophages and multinucleated giant cells surrounding the GFAV implants correlated with two parameters: OPN levels and presence of mineral in the implant. These data suggest that OPN localization to bioapatite within the implant may be required for localized up-regulation of CAII levels in adjacent macrophages/giant cells. In support of a direct effect of OPN on CAII expression, we found that OPN induced CAII protein levels in monocytic HL60 as well as primary human peripheral blood-derived macrophages. Furthermore, the effect of OPN on CAII expression in HL-60 cells was mimicked by an αvβ3 crosslinking antibody (LM609), and blocked by RGD peptides (data not shown), suggesting that OPN may act through αvβ3 to control CAII expression in monocytic cells.
These findings have led us to propose a new, dual model for OPN function in regulating ectopic calcification (Figure 11) ▶ . According to this model, OPN, elaborated by stromal or inflammatory cells at sites of ectopic mineralization, binds to bioapatites and initially physically inhibits crystal growth. Binding of OPN to bioapatite simultaneously provides a recognition site and/or concentration gradient for macrophages and giant cells thereby leading to localized accumulation and up-regulation of CAII, increased proton efflux, and acidification of the local microenvironment. Local acidification would then lead to dissolution of residual bioapatite. Although our studies support this model and indicate that OPN may directly interact with monocytic cell to induce CAII expression and proton release via a receptor mediated pathway, further studies are needed to definitively show that this mechanism is operative during regression of GFAV implant calcification, or other types of ectopic mineralization, in vivo. Finally, OPN promotes receptor-mediated recruitment and migration of additional macrophages, thereby further enhancing mineral regressive mechanisms.
Figure 11.

Model for mechanisms of OPN action in regulating ectopic calcification. It is proposed that OPN made by stromal or inflammatory cells at sites of ectopic mineralization binds to mineral and initially physically inhibits crystal growth. Binding of OPN to bioapatite then provides a recognition site or concentration gradient for macrophage and giant cells leading to localized accumulation as well as up-regulation of CAII via cell surface receptors. This leads to increased proton efflux, acidification of the local microenvironment, and dissolution of the bioapatite.
OPN has also been implicated in osteoclastic resorption of bone. Osteoclasts are monocyte-derived, multinucleated giant cells that resorb bone matrix and mineral by elaborating proteases and protons, respectively, into a sealed microenvironment known as the resorption lacunae. 58 OPN is concentrated at cement lines and lamina limitantes in bone, 59 is a major ligand for αvβ3 receptors on osteoclasts, 60 and is thought to play a role in initial osteoclast recognition and attachment to bone. 61 Indeed, OPN promotes cytoskeletal rearrangements 62 as well as osteoclast differentiation 63 necessary for osteoclastic resorption in vitro, and OPN−/− mice display a deficit in bone resorption after ovariectomy, 64 mechanical unloading, 65 and PTH treatment. 66 Although αvβ3 is known to regulate osteoclastic resorption, 67 the mechanisms by which αvβ3 facilitates this process are still under investigation. Given these similarities, it is interesting to speculate that control of CAII may also be involved in OPN/αvβ3-mediated osteoclastic bone resorption.
Finally, our findings are among the first to provide evidence that regression of ectopic calcification can occur in vivo. Consistent with this idea, osteoclast-like cells have been detected in mineralized atherosclerotic lesions, 68 native cardiac valves, 69 and bioprosthetic valves. 70 Of particular interest, a recent study of 102 symptomatic patients with coronary vascular calcification found that 15% of patients showed evidence of regression of calcification based on electron beam computed tomography (EBCT) calcium scores measured at baseline and ∼6 months later. 71 Along with our findings, these studies suggest provocative new physiological mechanisms controlling ectopic mineralization and suggest novel therapeutic approaches in treating and perhaps reversing ectopic calcification.
Acknowledgments
We thank Dr. Wenda Carlyle and St. Jude Medical Inc. (Minneapolis, MN) for GFAV reagents and Dr. Crystal Cunanan for atomic absorption spectroscopy analysis.
Footnotes
Address reprint requests to Cecilia Giachelli, Ph.D., Department of Bioengineering, University of Washington, Seattle, WA 98195. E-mail: ceci@u.washington.edu.
Supported by the National Institutes of Health (training grant GM07037 to S. A. S. and grant IRO1 HL62329-01 to C. M. G.), the University of Washington Engineered Biomaterials National Science Foundation (grant EEC9529161 to C. M. G.), and the Medical Research Council (grant MT11360 to M. D. M.).
M. D. M. is a member of the Medical Research Council of Canada Group in Periodontal Physiology, and a scholar of the Fellow of the Royal Society of Quebec.
References
- 1.Beadenkopf WG, Daoud AS, Love BM: Calcification in the coronary arteries and its relationship to arteriosclerosis and myocardial infarction. Am J Roentgenol 1964, 92:865-871 [PubMed] [Google Scholar]
- 2.Locker TH, Schwartz RS, Cotta CW, Hickman JR: Fluoroscopic coronary artery calcification and associated coronary disease in asymptomatic young men. J Am Coll Cardiol 1992, 19:1167-1172 [DOI] [PubMed] [Google Scholar]
- 3.Puentes G, Detrano R, Tang W, Wong N, French W, Narahara K, Brundage B, Baksheshi H: Estimation of coronary calcium mass using electron beam computed tomography: a promising approach for predicting coronary events? Circulation 1995, 92:I313 [Google Scholar]
- 4.Niskanen LK, Suhonen M, Siitonen O, Lehtinen JM, Uusitupa MI: Aortic and lower limb artery calcification in type II (non-insulin-dependent) diabetic patients and non-diabetic control subjects: a five year follow-up study. Atherosclerosis 1990, 84:61-71 [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald PJ, Ports TA, Yock PG: Contribution of localized calcium deposits to dissection after angioplasty: an observational study using intravascular ultrasound. Circulation 1992, 86:64-70 [DOI] [PubMed] [Google Scholar]
- 6.Olson JC, Edmundowicz D, Becker DJ, Kuller LH, Orchard TJ: Coronary calcium in adults with type 1 diabetes: a stronger correlate of clinical coronary artery disease in men than in women. Diabetes 2000, 49:1571-1578 [DOI] [PubMed] [Google Scholar]
- 7.Lehto S, Niskanen L, Suhonen L, Ronnemaa T, Laakso M: Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 1996, 16:978-983 [DOI] [PubMed] [Google Scholar]
- 8.O’Keefe JH, Lavie CJ, Nishimura RA, Edwards WD: Degenerative aortic stenosis. One effect of the graying of America. Postgrad Med 1991, 89:143-154 [DOI] [PubMed] [Google Scholar]
- 9.Schoen FJ, Levy RJ, Piehler HR: Pathological considerations in replacement cardiac valves. Cardiovasc Pathol 1992, 1:29-52 [DOI] [PubMed] [Google Scholar]
- 10.Cotran RS, Kumar V, Robbins SL: Cellular injury and cellular death. Robbins SL eds. Pathological Basis of Disease ed 6 1999:pp 1-35 WB Saunders Philadelphia
- 11.Giachelli CM: Ectopic calcification: gathering hard facts about soft tissue mineralization. Am J Pathol 1999, 154:671-675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL: Bone morphogenic protein expression in human atherosclerotic lesions. J Clin Invest 1993, 91:1800-1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wada T, McKee MD, Stietz S, Giachelli CM: Calcification of vascular smooth muscle cell cultures: inhibition by osteopontin. Circ Res 1999, 84:1-6 [DOI] [PubMed] [Google Scholar]
- 14.Shioi A, Nishizawa Y, Jono S, Koyama H, Hosoi M, Morii H: Beta-glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1995, 15:2003-2009 [DOI] [PubMed] [Google Scholar]
- 15.Virchow R: Cellular Pathology: As Based upon Physiological and Pathological Histology. 1863:pp 404-408 Dover New York [DOI] [PubMed]
- 16.Kim KM: Calcification of matrix vesicles in human aortic valve and aortic media. Fed Proc 1976, 35:156-162 [PubMed] [Google Scholar]
- 17.Tanimura A, McGregor DH, Anderson HC: Calcification in atherosclerosis. I Human studies. J Exp Path 1986, 2:261-297 [PubMed] [Google Scholar]
- 18.Hsu HHT, Camacho N: Isolation of calcifiable vesicles from human atherosclerotic aortas. Atherosclerosis 1999, 143:353-362 [DOI] [PubMed] [Google Scholar]
- 19.Luo GDP, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G: Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386:78-81 [DOI] [PubMed] [Google Scholar]
- 20.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS: Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998, 12:1260-1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spicer SS, Lewis SE, Tashian RE, Schulte BA: Mice carrying a CAR-2 null allele lack carbonic anhydrase II immunohistochemically and show vascular calcification. Am J Pathol 1989, 134:947-954 [PMC free article] [PubMed] [Google Scholar]
- 22.Thornell LE, Carlsson L, Li Z, Mericskay M, Paulin D: Null mutation in the desmin gene gives rise to cardiomyopathy. J Mol Cell Cardiol 1997, 29:2107-2124 [DOI] [PubMed] [Google Scholar]
- 23.Jahnen-Dechent W, Schinke T, Trindl A, Muller-Esterl W, Sablitzky F, Kaiser S, Blessing M: Cloning and targeted deletion of the mouse fetuin gene. J Biol Chem 1997, 272:31496-31503 [DOI] [PubMed] [Google Scholar]
- 24.Ho AM, Johnson MD, Kingsley DM: Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289:265-270 [DOI] [PubMed] [Google Scholar]
- 25.Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S: Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament. Nat Genet 1998, 19:271-273 [DOI] [PubMed] [Google Scholar]
- 26.Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone Jr MA, Falb D, Huszar D: A role for Smad6 in development and homeostasis of the cardiovascular system. Nat Genet 2000, 24:171-174 [DOI] [PubMed] [Google Scholar]
- 27.Giachelli CM, Schwartz SM, Liaw L: Molecular and cellular biology of osteopontin. Trends Cardiovasc Med 1995, 5:88-95 [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Bal BS, Gorski JP: Calcium and collagen binding properties of osteopontin, bone sialoprotein, and bone acidic glycoprotein-75 from bone. J Biol Chem 1992, 276:24871-24878 [PubMed] [Google Scholar]
- 29.Hunter GK, Kyle CL, Goldberg HA: Modulation of crystal formation by bone phosphoproteins: structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem J 1994, 300:723-728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boskey AL, Maresca M, Ullrich W, Doty SB, Butler WT, Prince CW: Osteopontin-hydroxyapatite interactions in vitro: inhibition of hydroxyapatite formation and growth in a gelatin-gel. Bone Miner 1993, 22:147-159 [DOI] [PubMed] [Google Scholar]
- 31.Jono S, Peinado C, Giachelli CM: Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J Biol Chem 2000, 275:20197-20203 [DOI] [PubMed] [Google Scholar]
- 32.Gorski JP: Acidic phosphoproteins from bone matrix: a structural rationalization of their role in biomineralization. Calcif Tissue Int 1992, 50:391-396 [DOI] [PubMed] [Google Scholar]
- 33.Liaw L, Birk DE, Ballas CB, Whitsitt JS, Davidson JM, Hogan BL: Altered wound healing in mice lacking a functional osteopontin gene (spp1). J Clin Invest 1998, 101:1468-1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liaw L, Almeida M, Hart CE, Schwartz SM, Giachelli CM: Osteopontin promotes vascular cell adhesion and spreading and is chemotactic for smooth muscle cells in vitro. Circ Res 1994, 74:214-224 [DOI] [PubMed] [Google Scholar]
- 35.Murry CE, Giachelli CM, Schwartz SM, Vracko R: Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol 1994, 145:1450-1462 [PMC free article] [PubMed] [Google Scholar]
- 36.Meloan SN, Puchtler H, Valentine LS: Alkaline and acid alizarin red S stains for alkali-soluble and alkali-insoluble calcium deposits. Arch Pathol 1972, 93:190-197 [PubMed] [Google Scholar]
- 37.Beezhold H, Personius C: Fibronectin fragments stimulate tumor necrosis factor secretion by human monocytes. J Leukoc Biol 1992, 51:59-64 [DOI] [PubMed] [Google Scholar]
- 38.Defife KM, Yun JK, Azeez A, Stack S, Ishihara K, Nakabayashi N, Colton E, Anderson JM: Adhesion and cytokine production by monocytes on poly(2-methacryloyloxyethyl phosphorylcholine-co-alkyl methacrylate)-coated polymers. J Biomed Mater Res 1995, 29:431-439 [DOI] [PubMed] [Google Scholar]
- 39.Malyankar UM, Almeida M, Johnson RJ, Pichler RH, Giachelli CM: Osteopontin regulation in cultured rat renal epithelial cells. Kidney Int 1997, 51:1766-1773 [DOI] [PubMed] [Google Scholar]
- 40.Levy RJ, Schoen FJ, Levy JT, Nelson AC, Howard SL, Oshry LJ: Biologic determinants of dystrophic calcification and osteocalcin deposition in glutaraldehyde-preserved porcine aortic valve leaflets implanted subcutaneously in rats. Am J Pathol 1983, 133:143-155 [PMC free article] [PubMed] [Google Scholar]
- 41.Fishbein MC, Levy RJ, Ferrans VJ, Dearden LC, Nashef A, Goodman AP, Carpentier A: Calcification of cardiac valve bioprostheses. Biochemical, histological and ultrastructural observations in a subcutaneous implantation model system. J Thorac Cardiovasc Surg 1982, 83:602-609 [PubMed] [Google Scholar]
- 42.Schoen FJ, Tsao JW, Levy RJ: Calcification of bovine pericardium used in cardiac valve bioprostheses. Implications for the mechanisms of bioprosthetic tissue mineralization. Am J Pathol 1986, 123:134-145 [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrans VJ, Boyce SW, Billingham ME, Jones M, Ishihara T, Roberts WC: Calcific deposits in porcine bioprostheses: structure and pathogenesis. Am J Cardiol 1980, 46:721-734 [DOI] [PubMed] [Google Scholar]
- 44.Valente M, Bortolotti U, Thiene G: Ultrastructural substrates of dystrophic calcification in porcine bioprosthetic failure. Am J Pathol 1985, 119:12-21 [PMC free article] [PubMed] [Google Scholar]
- 45.Fallon MD, Teitelbaum SL, Kahn AJ: Multinucleation enhances macrophage-mediated bone resorption. Lab Invest 1983, 49:159-164 [PubMed] [Google Scholar]
- 46.Levy RJ: Glutaraldehyde and the calcification mechanism of bioprosthetic heart valves. J Heart Valve Dis 1994, 3:101-104 [PubMed] [Google Scholar]
- 47.Ophascharoensuk V, Giachelli CM, Gordon K, Hughes J, Pichler R, Brown P, Liaw L, Schmidt R, Shankland SJ, Alpers CE, Couser WG, Johnson RJ: Obstructive uropathy in the mouse: role of osteopontin in interstitial fibrosis and apoptosis. Kidney Int 1999, 56:571-580 [DOI] [PubMed] [Google Scholar]
- 48.Crawford HC, Matrisian LM, Liaw L: Distinct roles of osteopontin in host defense activity and tumor survival during squamous cell carcinoma progression in vivo. Cancer Res 1998, 58:5206-5215 [PubMed] [Google Scholar]
- 49.LeGeros RZ: Calcium phosphates in oral biology and medicine. Myers HM eds. Calcium Phosphates in Oral Biology and Medicine. 1991:pp 1-200 Karger New York [PubMed]
- 50.Maren TH: Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev 1967, 47:595-781 [DOI] [PubMed] [Google Scholar]
- 51.Collins SJ: The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood 1987, 70:1233-1244 [PubMed] [Google Scholar]
- 52.Cheresh DA, Spiro RC: Biosynthetic and functional properties of an Arg-Gly-Asp-directed receptor involved in human melanoma cell attachment to vitronectin, fibrinogen, and von Willebrand factor. J Biol Chem 1987, 262:17703-17711 [PubMed] [Google Scholar]
- 53.Schoen FJ, Golomb G, Levy RJ: Calcification of bioprosthetic heart valves: a perspective on models. J Heart Valve Dis 1992, 1:110-114 [PubMed] [Google Scholar]
- 54.Schoen FJ, Kujovich JL, Webb CL, Levy RJ: Chemically determined mineral content of explanted porcine aortic valve bioprosthesis: correlation with radiographic assessment of calcification and clinical data. Circulation 1987, 76:1061-1066 [DOI] [PubMed] [Google Scholar]
- 55.Shen M, Marie P, Farge D, Carpentier S, De Pollack C, Hott M, Chen L, Martinet B, Carpentier A: Osteopontin is associated with bioprosthetic heart valve calcification in humans. C R Acad Sci Paris, Sciences de la Vie 1997, 320:49-57 [DOI] [PubMed] [Google Scholar]
- 56.Tang L, Eaton JW: Inflammatory responses to biomaterials. Am J Clin Pathol 1995, 103:466-471 [DOI] [PubMed] [Google Scholar]
- 57.Pfeiffer EF: The glucose sensor: the missing link in diabetes therapy. Horm Metab Res Suppl 1990, 24:S154-S164 [PubMed] [Google Scholar]
- 58.Blair HC: How the osteoclast degrades bone. BioEssays 1998, 20:837-846 [DOI] [PubMed] [Google Scholar]
- 59.McKee D, Nanci A: Ostepontin at mineralized interfaces in bone, teeth, and osseointegrated implants: ultrastructural distribution and implications for mineralized tissue formation, turnover and repair. Microsc Res Tech 1996, 33:141-164 [DOI] [PubMed] [Google Scholar]
- 60.Helfrich MH, Nesbitt SA, Dorey EL, Horton MA: Rat osteoclasts adhere to a wide range of RGD (Arg-0Gly-Asp) peptide-containing proteins, including the bone sialoproteins and fibronectin, via a b3 integrin. J Bone Miner Res 1992, 7:335-343 [DOI] [PubMed] [Google Scholar]
- 61.Reinholt FP, Hultenby K, Oldberg C, Heinegard D: Osteopontin—a possible anchor of osteoclasts to bone. Proc Natl Acad Sci USA 1990, 8:4473-4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chellaiah MA, Soga N, Swanson S, McAllister S, Alvarez U, Wang D, Dowdy SF, Hruska KA: Rho-A is critical for osteoclast podosome organization, motility, and bone resorption. J Biol Chem 2000, 275:11993-12002 [DOI] [PubMed] [Google Scholar]
- 63.Rittling SR, Matsumo HN, McKee MD, Nanci A, An XR, Novick KE, Kowalski AJ, Noda M, Denhardt DT: Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res 1998, 13:1101-1111 [DOI] [PubMed] [Google Scholar]
- 64.Yoshitake H, Rittling SR, Denhardt DT, Noda M: Osteopontin-deficient mice are resistant to ovariectomy-induced bone resorption. Proc Natl Acad Sci USA 1999, 96:8156-8160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishijima M, Rittling SR, Yamashita T, Tsuji K, Kurosawa H, Nifuji A, Denhardt DT, Noda M: Enhancement of osteoclastic bone resorption and suppression of osteoblastic bone formation in response to reduced mechanical stress do not occur in the absence of osteopontin. J Exp Med 2001, 193:399-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ihara H, Denhardt DT, Furuya K, Yamashita T, Muguruma Y, Tsuji K, Hruska KA, Higashio K, Enomot S, Nifuji A, Rittling SR, Noda M: Parathyroid hormone-induced bone resorption does not occur in the absence of osteopontin. J Biol Chem 2001, 276:13065-13071 [DOI] [PubMed] [Google Scholar]
- 67.McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, Feng X, Ross FP, Hynes RO, Teitelbaum SL: Mice lacking b3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest 2000, 105:433-440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deneke T, Langner K, Gewe PH, Hrrer E, Muller KM: Ossification in atherosclerotic carotid arteries. Z Kardiol 2001, 90(suppl 3):III/106-III/115 [DOI] [PubMed] [Google Scholar]
- 69.Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS: Bone formation and inflammation in cardiac valves. Circulation 2001, 103:1522-1528 [DOI] [PubMed] [Google Scholar]
- 70.Srivatsa SS, Harrity PJ, Maercklein PB, Kleppe L, Veinot J, Edwards WD, Johnson CM, Fitzpatrick LA: Increased cellular expression of matrix proteins that regulate mineralization is associated with calcification of native human and porcine xenograft bioprosthetic heart valves. J Clin Invest 1997, 99:996-1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmermund A, Baumgart D, Mohlenkamp S, Kriner P, Pump H, Gronemeyer D, Seibel R, Erbel R: Natural history and topographic pattern of progression of coronary calcification in symptomatic patients: an electron beam CT study. Arterioscler Thromb Vasc Biol 2001, 21:421-426 [DOI] [PubMed] [Google Scholar]