

Abstract
Chronic cholestasis is associated with retention of bile acids and profound cytoskeletal alterations in hepatocytes including Mallory body (MB) formation. The mechanisms responsible for MB formation in cholestatic liver diseases are unclear. The aim of our study was to determine the relevance of cholestasis and bile acids for MB formation. For this purpose mice received a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)-supplemented diet for 2.5 months to induce MB formation. After recovery from DDC intoxication for 4 weeks followed by disappearance of MBs, these drug-primed mice were subjected to DDC refeeding, common bile duct ligation (CBDL), and feeding of a cholic acid (CA)-supplemented diet for 7 days, respectively. Cytokeratin (CK) 8 and CK 18 expression was studied by competitive reverse transcriptase-polymerase chain reaction and Western blot analysis. Cytoskeletal alterations of hepatocytes and MB formation were monitored by immunofluorescence microscopy and immunohistochemistry using CK-, ubiquitin-, and MB-specific antibodies. Like DDC refeeding, both CBDL and CA feeding of drug-primed mice significantly increased CK 8 and CK 18 mRNA and protein levels (with excess of CK 8) and resulted in ubiquitination and abnormal phosphorylation of CKs. Furthermore, CBDL and CA feeding resulted in rapid neoformation of MBs in drug-primed mice. It is concluded that MB formation in cholestatic liver diseases may be triggered by the action of potentially toxic bile acids.
Cytokeratin (CK) intermediate filaments (IFs) are major cytoskeletal components and are concentrated in the perinuclear and submembraneous regions of epithelial cells. 1 The CK subfamily has more than 20 members forming heteropolymers of type I and type II CKs. 1 CK 8 and CK 18 are subunits of the IFs of hepatocytes and were also identified as major components of Mallory bodies (MBs) associated with certain human liver diseases and related mouse models. 2 MBs are characteristic cytoplasmic hyaline inclusions in hepatocytes reflecting a peculiar morphological manifestation of chronic liver cell injury. 2,3 Their appearance is related to alterations of the CK-IF cytoskeleton including overexpression and posttranslational modifications of CKs (eg, cross-linking, abnormal phosphorylation, ubiquitination). 2,4-10 In humans, MBs are typically associated with alcoholic and nonalcoholic steatohepatitis, but are also found in chronic cholestatic conditions such as primary biliary cirrhosis and primary sclerosing cholangitis. 2,8,11 A common denominator of these etiologically different liver diseases is their association with cholestasis and elevated serum bile acid levels. In mice, MBs can be induced by chronic griseofulvin (GF) or 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) intoxication. 12-15 Administration of these porphyrinogenic agents also induces cholestasis in mice indicated by elevated serum bile acid levels that may at least partly result from the formation of protoporphyrin plugs and stones obstructing the bile drainage system. 16,17 MB formation requires prolonged intoxication (∼2.5 months) with GF or DDC. 2,4 Interestingly, after recovery from intoxication with disappearance of MBs, which takes ∼4 weeks (primed mouse liver), MBs are reinduced within days by reintoxication with DDC or GF as well as application of colchicine. 4,18-21
We recently demonstrated that obstructive cholestasis or cholic acid (CA) feeding leads to CK overexpression accompanied by abnormal phosphorylation in the mouse liver; 22 nonetheless, the causal relationship between cholestasis with retention of potentially toxic bile acids and MB formation remained unclear. This study was designed to clarify whether cholestasis and bile acids by themselves represent causative factors in MB formation. We therefore assessed the influence of obstructive cholestasis by common bile duct ligation (CBDL) and CA feeding (to mimic retention of a major primary bile acid) on the IF cytoskeleton and MB formation in a well-defined experimental mouse model (ie, the drug-primed mouse liver). 2,4,18-21 Evidence that cholestasis and bile acids play a central role in MB formation is reported.
Materials and Methods
Animals
Male Swiss albino mice (strain Him OF1 SPF) were obtained from the Institute for Laboratory Animal Research, University of Vienna School of Medicine, Himberg, Austria, housed with a 12:12 hour light-dark cycle and permitted ad libitum consumption of water and a standard mouse diet (Marek, Vienna, Austria). Experiments were performed with 2-month-old mice weighing 25 to 30 g. The experiments were approved by the local ethics committee and followed the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the United States National Academy of Sciences, as published by the National Institutes of Health (NIH publication 86-23, revised 1985). CA and DDC were obtained from Aldrich (Steinheim, Germany).
DDC Intoxication
Mice were fed a diet containing 0.1% DDC for 2.5 months to induce MBs. 2,4 After this time period one group of animals was sacrificed to assess DDC-induced cytoskeletal alterations including MB formation, whereas another group was sacrificed 4 weeks after discontinuation of DDC feeding to study the reversibility of these changes as described previously. 4 In addition, recovered primed mice were refed a diet containing 0.1% DDC for 7 days or subjected to CBDL or CA feeding (see Figure 1 ▶ for experimental design).
Figure 1.

Experimental design to study the role of cholestasis and bile acids in MB formation in drug-primed mice. Mice were fed a control diet or 0.1% DDC-supplemented diet for 2.5 months to induce MBs. One group of animals was sacrificed to study DDC-induced cytoskeletal alterations (including MB formation), whereas another group was sacrificed 4 weeks after discontinuation of DDC feeding (recovery) to assess the reversibility of these changes. In addition, recovered (primed) mice received control diet (Co) or were subjected to DDC refeeding (DDC), CBDL, CA feeding, and sham operation for 7 days, respectively. Five animals were studied in each group.
CBDL
All surgical procedures were performed under sterile conditions. To study the effects of obstructive cholestasis on CK expression and MB formation in drug-primed mice, the common bile duct was ligated close to the liver hilum immediately below the bifurcation and dissected between the ligatures as described previously. 23 In addition, cholecystectomy was performed after ligation of the cystic duct. Controls underwent a sham operation with exposure but without ligation of the common bile duct and removal of the gallbladder. The livers were excised under general anesthesia (10 mg Avertin i.p.) 7 days after surgery. Liver tissue samples were frozen in liquid nitrogen for molecular analysis and immunofluorescence microscopy or fixed in 4% neutral buffered formaldehyde solution for light microscopy and immunohistochemistry. Serum samples from each mouse were stored at −70°C for analysis of aspartate aminotransferase/alanine aminotransferase, alkaline phosphatase, and total bile acid levels.
Bile Acid Feeding
To study the effects of bile acids on CK expression and MB formation, drug-primed mice were fed a diet supplemented with CA (1%) for 7 days. 22,24 Livers and sera were processed as described above.
Determination of mRNA Copy Numbers
mRNA copy numbers for CK 8, CK 18, and glyceraldehyde-3-phosphate dehydrogenase were determined by competitive reverse transcriptase-polymerase chain reaction. 10
Western Blotting of CK 8 and CK 18
CK 8 and CK 18 protein levels were determined by Western blot analysis. 4,22
Immunofluorescence Microscopy
Immunofluorescence microscopy was performed on frozen liver sections (3-μm thick, fixed in acetone at −20°C for 10 minutes) using the monoclonal antibody MM120-1 specifically recognizing MBs and the polyclonal rabbit CK antibody 50K160 against CK 8 and CK 18 as described previously. 4,22,25 In addition, phosphorylation of CK 8 was assessed with the antibody 5B3 against a hyperphosphorylated epitope of CK 8. 4,26 Double immunolabeling was performed combining the monoclonal antibodies MM120-1 or 5B3 with the polyclonal antibody 50K160. As secondary antibodies, Cy2-conjugated goat anti-mouse IgG (Amsersham, Buckinghamshire, UK) and tetramethylrhodamine isothiocyanate-conjugated swine anti-rabbit Ig (DAKO, Glostrup, Denmark) were used. For control the primary antibodies were omitted or replaced by isotype-matched immunoglobulins (DAKO). Immunofluorescent specimens were analyzed with a MRC 600 (Bio-Rad, Richmond, CA) laser-scanning confocal device attached to a Zeiss Axiophot microscope. The fluorescent images were collected using the confocal photomultiplier tube at full frame 768 × 512 pixels).
Histology
Mouse livers were fixed in 4% neutral buffered formaldehyde solution and embedded in paraffin. Sections 4 μm thick were stained with hematoxylin and eosin.
Immunohistochemistry
Immunohistochemistry was performed using an antibody against ubiquitin (dilution 1:200; DAKO). Paraffin sections (4 μm thick) of formaldehyde-fixed tissue were deparaffinized, rehydrated, and digested with 0.1% protease (type XXIV; Sigma, St. Louis, MO). Binding of the ubiquitin antibody was detected using the ABC system (DAKO) as previously described. 8
Routine Serum Biochemistry and Bile Acid Measurements
Serum alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase levels were determined by routine testing on a Hitachi 717 analyzer (Boehringer Mannheim, Mannheim, Germany) as measures of liver toxicity of the different treatment regimens and the degree of cholestasis. Total serum bile acid levels were determined by a commercially available 3α-hydroxysteroid dehydrogenase assay (Merck, Darmstadt, Germany). Tests were performed in duplicate.
Statistical Analysis
Five animals from each group were studied at each time point. Data are reported as arithmetic means ± SEM. Statistical analysis was performed using Student’s t-test when appropriate, or analysis of variance with posttesting when three or more groups were compared. A P value <0.05 was considered significant.
Results
Effects of Initial DDC Intoxication, Drug Withdrawal, and DDC Reintoxication
DDC treatment for 2.5 months resulted in disseminated necroses, apoptotic bodies, ballooning of hepatocytes, deposition of brown pigment in macrophages, pigment plugs in the lumina of bile ducts, ductular proliferation, and MB formation as previously described (not shown). 4 After discontinuation of DDC feeding for 4 weeks (drug-primed recovered mice) the hepatocellular changes subsided with the exception of pigment plugs in some interlobular bile ducts and some pigment-containing macrophages. Small residual granular MBs remained at the cell periphery in a few hepatocytes (Figures 2C, 3A, and 4A) ▶ ▶ ▶ . After DDC reintoxication for 7 days, liver morphology closely resembled that after DDC treatment for 2.5 months and was associated with significant elevations of serum transaminase, alkaline phosphatase, and serum bile acid levels in comparison to drug-primed recovered controls fed standard chow (Table 1) ▶ . Moreover, DDC reintoxication of drug-primed mice resulted in significantly increased CK 8 and CK 18 mRNA and protein expression (Figure 2, A and B) ▶ as well as rapid neoformation of MBs in many hepatocytes (Figures 2D, 3B, and 4B) ▶ ▶ ▶ . 4 The relative increase of CK 8 protein expression clearly exceeded that of CK 18, indicating disturbance of the CK 8 to CK 18 ratio with an excess of CK 8 (Figure 2B) ▶ .
Figure 2.
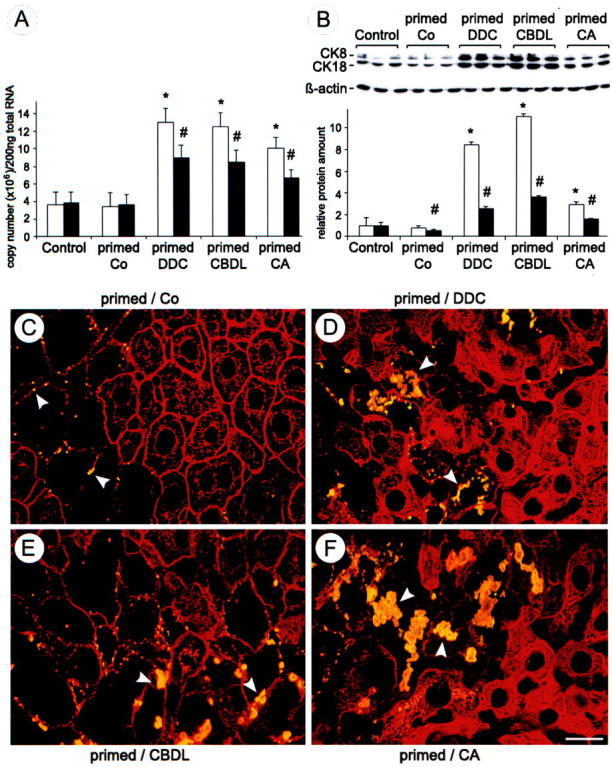
DDC, CBDL, and CA feeding induce overexpression of CK 8 and CK 18 and MB formation in drug-primed mice. A: Competitive reverse transcriptase-polymerase chain reaction revealed a significant increase of CK 8 (open bars) and of CK 18 (filled bars) mRNA in DDC-refed (primed DDC), bile duct-ligated (primed CBDL), and CA-fed (primed CA) drug-primed mouse liver in comparison to drug-primed recovered mice (primed Co) and naive control diet-fed mice (control). Data (means + SEM) are expressed as copy numbers/200 ng of total RNA (n = 5 in each group; * and #, P < 0.05, DDC, CBDL, and CA versus controls). B: Representative Western blots for CK 8, CK 18, and β-actin in naive control diet-fed (control), drug-primed recovered (primed Co), DDC-refed (primed DDC), bile duct-ligated (primed CBDL), and CA-fed (primed CA) drug-primed mouse liver. DDC refeeding, CBDL feeding, and CA feeding significantly increased CK 8 and CK 18 protein levels, whereas β-actin levels remained unchanged. Data (means + SEM) are expressed as relative protein amounts (n = 3 in each group; * and #, P < 0.05, primed Co, DDC, CBDL, and CA versus controls). The increase of CK 8 (open bars) was more pronounced than that of CK 18 (filled bars). The corresponding Western blots are shown at top of B. C to F: Double-immunofluorescence microscopy was performed on frozen liver sections of recovered (primed/Co) (C), DDC-refed (primed/DDC) (D), bile duct-ligated (primed/CBDL) (E), and CA-fed (primed/CA) (F) drug-primed mice combining the monoclonal antibody MM120-1 specifically recognizing MBs (yellow because of co-localization with CKs) with the polyclonal antibody 50K160 against CKs (red). Note the increased density of CK-IF network (particularly in D and F) and the increasing number and size of MBs (arrowheads) in hepatocytes after DDC refeeding, CBDL feeding, and CA feeding, whereas in the primed-recovered mouse liver (primed/Co) only small residual MBs still remain in enlarged hepatocytes with diminished or absent immunostainable IF network (arrowheads). Scale bar, 20 μm.
Figure 3.
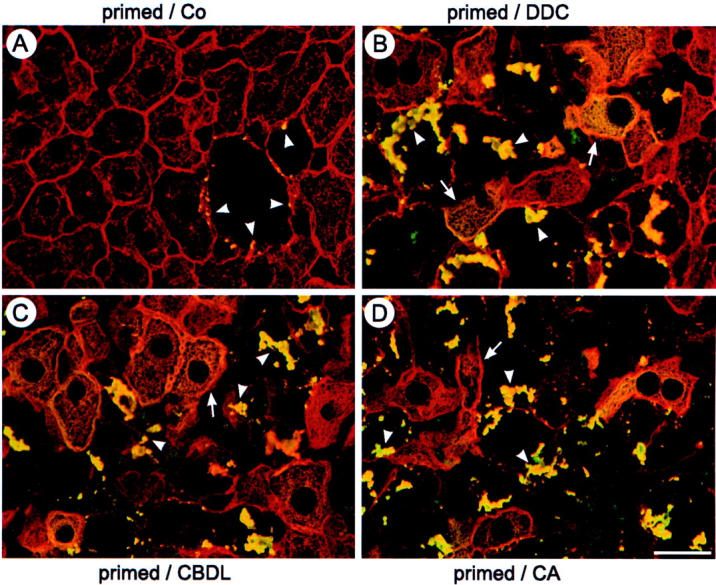
DDC, CBDL, and CA feeding induce abnormal phosphorylation of the CK-IF network and MBs in drug-primed mice. Double-immunofluorescence microscopy was performed on frozen liver sections of recovered (primed/Co) (A), DDC-refed (primed/DDC) (B), bile duct-ligated (primed/CBDL) (C), and CA-fed (primed/CA) (D) drug-primed mice combining the monoclonal antibody 5B3 specifically recognizing an abnormally phosphorylated epitope of CK 8 (yellow because of co-localization with CK) with the polyclonal antibody 50K160 against CKs (red). Note the abnormal phosphorylation of the CK-IF network (arrows) and newly formed MBs (arrowheads) in hepatocytes after DDC refeeding, CBDL feeding, and CA feeding (B–D). Abnormally phosphorylated larger MBs are usually present in enlarged hepatocytes with diminished or missing IF network. In the primed recovered liver (primed/Co) there are only small residual MBs in still enlarged hepatocytes with diminished IF network (arrowheads) (A). Scale bar, 20 μm.
Figure 4.

DDC, CBDL, and CA feeding induce ubiquitination of neoformed MBs in drug-primed mice. Immunohistochemistry was performed on liver sections of recovered (primed/Co) (A), DDC-refed (primed/DDC) (B), bile duct-ligated (primed/CBDL) (C), and CA-fed (primed/CA) (D) drug-primed mice using an antibody against ubiquitin. DDC refeeding, CBDL feeding, and CA feeding resulted in newly formed ubiquitin-positive MBs (arrowheads) (B–D). Arrows designate pigment deposits (A, B). Original magnifications, ×20.
Table 1.
Serum Liver Enzymes in the Different Treatment Groups
| Group | AST | ALT | AP | BA |
|---|---|---|---|---|
| Controls | 180 ± 60 | 60 ± 20 | 40 ± 15 | 7.4 ± 0.8 |
| Primed/recovered | 176 ± 110 | 92 ± 32 | 133 ± 37 | 9.3 ± 8.4 |
| Primed/DDC | 1408 ± 569* | 1340 ± 391* | 329 ± 94* | 202 ± 167* |
| Primed/CBDL | 486 ± 86* | 396 ± 85* | 1349 ± 333* | 468 ± 378* |
| Primed/CA | 599 ± 296* | 366 ± 167* | 475 ± 96* | 109 ± 96* |
Values are means ± SEM. AST, aspartate aminotransferase (U/L); ALT, alanine aminotransferase (U/L); AP, alkaline phosphatase (U/L); BA, bile acids (μmol/L).
*P < 0.05, DDC-refed, bile duct-ligated, and CA-fed drug-primed mice versus recovered drug-primed mice fed standard diet and naive controls.
CBDL Induces CK Overexpression and MB Formation in Drug-Primed Mouse Liver
To determine the effects of obstructive cholestasis on MB formation, drug-primed mice were subjected to biliary obstruction by CBDL for 7 days. This treatment resulted in disseminated and confluent hepatocellular necroses and some apoptotic bodies, vacuolization of hepatocytes, increased number of liver cell mitoses, dilatation of interlobular bile ducts, ductular proliferation, granulocytic infiltration within lobules and portal tracts, and marked induction of MBs (predominantly in acinar zones 1 and 2) (Figures 2E, 3C, and 4C) ▶ ▶ ▶ . Interestingly, the extent of bile infarcts was much less pronounced in drug-primed CBDL mice than in naive CBDL mice (located predominantly in acinar zones 1 and 2; not shown). 22,27 CBDL resulted in a significant increase of CK 8 and CK 18 mRNA levels in comparison to drug-primed recovered mice and naive control diet-fed mice (Figure 2A) ▶ accompanied by an increase in CK protein expression (Figure 2B) ▶ . Similar to DDC refeeding, the relative increase of CK 8 exceeded that of CK 18 in CBDL drug-primed mice (Figure 2B) ▶ . Sham operation had no influence on CK 8 and CK 18 mRNA and protein expression as well as MB formation (not shown).
CA Feeding Induces CK Overexpression and MB Formation in Drug-Primed Mouse Liver
To further discriminate between mechanical (eg, increased canalicular pressure) and toxic effects (eg, bile acid-induced toxicity because of increased bile acid concentrations) of cholestasis with respect to MB formation, drug-primed mice were fed a diet supplemented with potentially toxic CA, which represents a major bile acid in human cholestatic liver disease. 22,24,28,29 In line with our previous studies in naive CA-fed mice, 22,24 CA feeding significantly increased the levels of serum transaminases, alkaline phosphatase, and serum bile acids in drug-primed mice (Table 1) ▶ . CA feeding of drug-primed mice was associated with enlargement of hepatocytes, disseminated hepatocellular necroses, mitoses, dilatation of interlobular bile ducts, and periductal edema (not shown). Moreover, this treatment significantly elevated CK 8 and CK 18 mRNA and protein levels in comparison to drug-primed recovered mice and naive control diet-fed mice (Figure 2, A and B) ▶ . The increase of CK 8 protein levels exceeded that of CK 18; however, the difference was less pronounced than in DDC-refed and CBDL drug-primed mice (Figure 2B) ▶ . CA feeding induced MB formation to a similar extent as observed in drug-primed CBDL mice (Figures 2F, 3D, and 4D) ▶ ▶ ▶ .
CBDL and CA Feeding Induce Abnormal Phosphorylation and Ubiquitination of CKs in Drug-Primed Mouse Liver
Because abnormal phosphorylation and ubiquitination of CKs represent a common event in MB formation in our model system as well as in human alcoholic hepatitis, 4,8,30-33 CK phosphorylation and ubiquitination were studied in bile duct-ligated and CA-fed drug-primed mice in comparison to controls. Drug-primed recovered mouse livers only showed a few residual MBs present in enlarged hepatocytes with missing CK immunostaining that were decorated by an antibody against a hyperphosphorylated CK 8 epitope (Figure 3A) ▶ . CBDL and CA feeding induced abnormal phosphorylation of the CK-IF network in hepatocytes in the vicinity of liver cells containing MBs similar to the situation observed in DDC-refed mice (Figure 3, B, C, and D) ▶ . In addition, newly formed MBs contained abnormally phosphorylated CK 8 (Figure 3, B and C) ▶ . Comparable to DDC refeeding, CBDL and CA feeding of drug-primed mice also resulted in pronounced ubiquitination of newly formed MBs (Figure 4, B, C, and D) ▶ . In contrast, recovered controls only showed ubiquitin-related immunoreactivity of some small residual MBs (Figure 4A) ▶ . Taken together, the findings of abnormal phosphorylation and ubiquitination of MBs in the course of CBDL and CA feeding in drug-primed mouse liver support the importance of cholestasis with retention of potentially toxic bile acids for posttranslational CK modifications and MB formation.
Discussion
MBs are characteristic hepatocellular cytoplasmic inclusions and morphological hallmarks of a variety of chronic liver diseases, including alcoholic and nonalcoholic steatohepatitis as well as chronic cholestasis. 2,3 They may also serve as study objects to obtain insights into the general principles of cellular injury associated with inclusion bodies (ie, diverse protein aggregation diseases). 2,3,30,34,35 The aim of this study was to determine how cholestasis and potentially toxic bile acids trigger MB formation in drug-primed mouse liver. It is demonstrated that both obstructive cholestasis and CA feeding lead within a few days to overexpression, abnormal phosphorylation, and ubiquitination of CKs, and finally MB formation in drug-primed mouse liver. These findings provide evidence for a key role of potentially toxic bile acids for MB formation in cholestatic liver disorders.
The appearance of MBs is related to alterations of the CK-IF cytoskeleton of hepatocytes and can be studied in mice chronically intoxicated with the porphyrinogenic and cholestatic substances GF and DDC. 2,4,18-21 Rapid reinduction of MBs in drug-primed recovered mouse liver is an interesting feature. 2,18,21 Substances known to reinduce MB formation in drug-primed mouse liver (eg, GF, DDC, colchicine, heat shock) also cause cholestasis with elevated serum bile acid levels. 4,16-18 In addition, bile acid-induced CK overexpression was recently demonstrated in our laboratory in common bile duct ligated and CA-fed mice, further underlining the importance of cholestasis with retention of potentially toxic bile acids for CK-IF alterations. 22 Because MB formation seems to involve two steps, namely priming of hepatocytes during prolonged intoxication and triggering in the primed cells, it is attractive to speculate that cholestasis with bile acid retention could represent a major pathogenetic factor. Consequently, retention of toxic bile acids associated with cholestasis could also be causally involved in cytoskeletal alterations with MB formation in primary biliary cirrhosis or primary sclerosing cholangitis. 2,11 It is interesting in this context that MBs in cholestatic liver diseases (eg, primary biliary cirrhosis) arise in acinar zone 1, resembling the zone of cholate stasis, which is exposed to the highest concentration of bile acids.
CK-IFs have long been considered to be rather static structures primarily responsible for the mechanical stability of cells. 1 It is becoming increasingly clear, however, that CK-IF proteins are engaged in additional cellular functions (eg, defense against toxic stress and modulation of apoptotic pathways). 10,26,36-40 The importance of CKs for maintenance of functional integrity of hepatocytes has been demonstrated in several gene knockout mouse models. 2,10,40 Moreover, CK mutations as well as posttranslational modifications of CKs may also play a role in the pathogenesis of human liver diseases because mutations in the CK 8 gene in patients with cryptogenic cirrhosis and pronounced abnormal CK phosphorylation observed in human alcoholic steatohepatitis have been recently demonstrated. 8,41,42 Based on these findings, one may speculate that increased hepatic CK expression with abnormal phosphorylation, ubiquitination, and resulting MB formation represents a hitherto unknown defense response of the liver cell to toxic injury (eg, toxic bile acid-induced oxidative stress).
The development of MBs is a result of a multistep process including increased synthesis and posttranslational modifications of CKs. 2 Increased synthesis of CK 8 and CK 18 monomers with predominance of CK 8 and disturbance of the normal 1:1 ratio necessary for correct IF assembly seems to represent an important initiating step in MB formation as shown here and in previous studies. 2,4 In addition to overexpression of CKs, posttranslational modifications were previously identified as playing a critical role in MB formation. 2,4,30 The importance of CK hyperphosphorylation was illustrated by MB induction in drug-primed mice treated with phosphatase inhibitors (eg, okadaic acid, tautomycin). 19,20 In addition, CKs are polyubiquitinated in MBs as observed not only under experimental conditions but also in human liver disease. 2,8,30 Ubiquitin is a small highly conserved protein that is universally present in eukaryotic cells. It targets proteins for degradation by the 26S proteasome complex. 43-47 Ubiquitin-mediated proteolysis of cellular proteins plays an important role in many basic cellular processes, including the response to stress and various other extracellular stimuli. Substrates usually undergo posttranslational modifications, in most cases phosphorylation, before degradation. 43-47 CK 8 and CK 18 can be ubiquitinated, particularly when each CK is expressed individually, and targeted for degradation by the proteasome. 33 Increased and particularly imbalanced CK synthesis as observed in models for MB formation may, therefore, predispose to CK ubiquitination followed by proteasomal degradation. Consequently aggregation of polyubiquitinated and hyperphosphorylated CKs as MBs could reflect exhausted proteolytic capacity. 48,49
The results of the current study suggest that potentially toxic bile acids lead to MB formation by inducing imbalanced overexpression, abnormal phosphorylation and polyubiquitination, and decreased degradation of CKs in drug-primed mouse liver. It will be interesting to see whether these findings obtained in animal models are also relevant to human cholestatic liver diseases. Oxidative stress may be the final common principle in different types of protein aggregation disorders including cholestatic liver diseases. 49-51
Acknowledgments
We thank Dr. Bishr M. Omary (Department of Medicine, Palo Alto VA Medical Center and Stanford University, Stanford, CA) for providing antibodies against hyperphosphorylated CK epitopes, Dr. W. Erwa (Graz) and colleagues for performing liver function tests, and Dr. R. Aigner (Graz) and colleagues for measurement of serum bile acid levels.
Footnotes
Address reprint requests to Helmut Denk, M.D., FRCPath, Department of Pathology, Karl-Franzens University, Auenbruggerplatz 25, A-8036 Graz, Austria. E-mail: helmut.denk@kfunigraz.ac.at.
Supported by the Austrian Science Foundation (grant S 7401-MOB to K. Z. and P-15502 to M. T.), the Jubilee Fund of the Austrian National Bank (grants 7171 and 8522 to M. T.), and by the Joseph Skoda Prize from the Austrian Society of Internal Medicine (to M. T.).
References
- 1.Moll R, Franke WW, Schiller DL, Geiger B, Krepler R: The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982, 31:11-24 [DOI] [PubMed] [Google Scholar]
- 2.Denk H, Stumptner C, Zatloukal K: Mallory bodies revisited. J Hepatol 2000, 32:689-702 [DOI] [PubMed] [Google Scholar]
- 3.Mallory FB: Cirrhosis of the liver: five different lesions from which it may arise. Bull Johns Hopkins Hosp 1911, 22:69-75 [Google Scholar]
- 4.Stumptner C, Fuchsbichler A, Lehner M, Zatloukal K, Denk H: Sequence of events in the assembly of Mallory body components in mouse liver: clues to the pathogenesis and significance of Mallory body formation. J Hepatol 2001, 34:665-675 [DOI] [PubMed] [Google Scholar]
- 5.Zatloukal K, Böck G, Rainer I, Denk H, Weber K: High molecular weight components are main constituents of Mallory bodies isolated with fluorescence activated cell sorter. Lab Invest 1991, 64:200-206 [PubMed] [Google Scholar]
- 6.Zatloukal K, Denk H, Spurej G, Lackinger E, Preisegger KH, Franke WW: High molecular weight component of Mallory bodies detected by monoclonal antibody. Lab Invest 1990, 62:427-434 [PubMed] [Google Scholar]
- 7.Denk H, Franke WW, Dragosics B, Zeiler I: Pathology of cytoskeleton of liver cells: demonstration of Mallory bodies (alcoholic hyalin) in murine and human hepatocytes by immunofluorescence microscopy using antibodies to cytokeratin polypeptides from hepatocytes. Hepatology 1981, 1:9-20 [DOI] [PubMed] [Google Scholar]
- 8.Stumptner C, Omary MB, Fickert P, Denk H, Zatloukal K: Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in Mallory bodies. Am J Pathol 2000, 156:77-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hazan R, Denk H, Franke WW, Lackinger E, Schiller DL: Change of cytokeratin organization during development of Mallory bodies as revealed by a monoclonal antibody. Lab Invest 1986, 54:543-553 [PubMed] [Google Scholar]
- 10.Zatloukal K, Stumptner C, Lehner M, Denk H, Baribault H, Eshkind LG, Franke WW: Cytokeratin 8 protects from hepatotoxicity, and its ratio to cytokeratin 18 determines the ability of hepatocytes to form Mallory bodies. Am J Pathol 2000, 156:1263-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerber MA, Orr W, Denk H, Schaffner F, Popper H: Hepatocellular hyalin in cholestasis and cirrhosis: its diagnostic significance. Gastroenterology 1973, 64:89-98 [PubMed] [Google Scholar]
- 12.Denk H, Franke WW, Kerjaschki D, Eckersdorfer R: Mallory bodies in experimental animals and man. Int Rev Exp Pathol 1979, 20:1061-1077 [PubMed] [Google Scholar]
- 13.Denk H, Gschnait F, Wolff K: Hepatocellular hyalin (Mallory bodies) in long term griseofulvin-treated mice. A new experimental model for the study of hyalin formation. Lab Invest 1975, 32:773-776 [PubMed] [Google Scholar]
- 14.Denk H, Eckersdorfer R, Gschnait F, Konrad K, Wolff K: Experimental induction of hepatocellular hyalin (Mallory bodies) in mice by griseofulvin treatment. Light microscopic observations. Lab Invest 1976, 35:377-382 [PubMed] [Google Scholar]
- 15.Yokoo H, Harwood TR, Racker D, Arak S: Experimental production of Mallory bodies in mice by diet containing 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Gastroenterology 1982, 83:109-113 [PubMed] [Google Scholar]
- 16.Yokoo H, Craig RM, Harwood TR, Cochrane C: Griseofulvin-induced cholestasis in Swiss albino mice. Gastroenterology 1979, 77:1082-1087 [PubMed] [Google Scholar]
- 17.Meermann L, Koopen NR, Bloks V, Van Goor H, Havinga R, Walthers BG, Kramer W, Stegelin S, Muller M, Kuipers F, Jansen PL: Biliary fibrosis associated with altered bile composition in a mouse model of erythropoietic protoporphyria. Gastroenterology 1999, 117:696-705 [DOI] [PubMed] [Google Scholar]
- 18.Yuan QX, Marceau N, French BA, Fu P, French SW: Mallory body induction in drug-primed mouse liver. Hepatology 1996, 24:603-612 [DOI] [PubMed] [Google Scholar]
- 19.Yuan QX, Nagao Y, Gaal K, Hu B, French SW: Mechanism of Mallory body formation induced by okadaic acid in drug-primed mice. Exp Mol Pathol 1998, 65:87-103 [DOI] [PubMed] [Google Scholar]
- 20.Yuan Q, French BA, French SW: Tautomycin induces extensive Mallory body formation in drug-primed mouse livers. Hepatology 1998, 28:256A [Google Scholar]
- 21.Zatloukal K, Spurej G, Lackinger E, Rainer I, Denk H: Fate of Mallory body containing hepatocytes: disappearance of Mallory bodies and restoration of the hepatocytic intermediate filament cytoskeleton after drug withdrawal in the griseofulvin treated mouse. Hepatology 1990, 11:652-661 [DOI] [PubMed] [Google Scholar]
- 22.Fickert P, Trauner M, Fuchsbichler A, Stumptner C, Zatloukal K, Denk H: Cytokeratins as targets for bile acid induced cytotoxicity. Am J Pathol 2002, 160:491-499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trauner M, Arrese M, Soroka CJ, Anathanarayanan M, Koeppel TA, Schlosser SF, Suchy FJ, Keppler D, Boyer JL: The rat canalicular conjugate export pump (Mrp2) is downregulated in intrahepatic and obstructive cholestasis. Gastroenterology 1997, 113:255-264 [DOI] [PubMed] [Google Scholar]
- 24.Fickert P, Zollner G, Fuchsbichler A, Pojer C, Zenz R, Stumptner C, Pojer C, Zenz R, Lammert F, Stieger B, Meier PJ, Zatloukal K, Denk H, Trauner M: Effects of ursodeoxycholic and cholic acid feeding on hepatocellular transporter expression in mouse liver. Gastroenterology 2001, 120:170-183 [DOI] [PubMed] [Google Scholar]
- 25.Hutter H, Zatloukal K, Winter G, Stumptner C, Denk H: Disturbance of keratin homeostasis in griseofulvin-intoxicated mouse liver. Lab Invest 1993, 69:576-582 [PubMed] [Google Scholar]
- 26.Liao J, Ku N, Omary MB: Stress, apoptosis, and mitosis induce phosphorylation of human keratin 8 at ser73 in tissues and cultured cells. J Biol Chem 1995, 272:17565-17573 [DOI] [PubMed] [Google Scholar]
- 27.Miyoshi H, Rust C, Guicciardi ME, Gores GJ: NF-kappa B is activated in cholestasis and functions to reduce liver injury. Am J Pathol 2001, 158:967-975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trauner M, Graziadei IW: Mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver disease. Aliment Pharm Ther 1999, 13:979-996 [DOI] [PubMed] [Google Scholar]
- 29.Hofmann AF: The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med 1999, 159:2647-2658 [DOI] [PubMed] [Google Scholar]
- 30.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H: p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002, 160:255-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohta M, Marceau N, Perry G, Manetto V, Gambetti P, Autilio-Gambetti L, Metuzals J, Kawahara H, Cadrin M, French SW: Ubiquitin is present on the cytokeratin intermediate filaments and Mallory bodies of hepatocytes. Lab Invest 1988, 59:848-856 [PubMed] [Google Scholar]
- 32.Lowe J, Blanchard K, Morrell K, Lennox L, Reynolds M, Billett M, Landon M, Mayer RJ: Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibers in cerebellar astrocytomas, cytoplasmic bodies in muscle, and Mallory bodies in alcoholic liver disease. J Pathol 1988, 155:9-15 [DOI] [PubMed] [Google Scholar]
- 33.Ku NO, Omary MB: Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J Cell Biol 2000, 149:547-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carrell RW, Lomas DA: Conformational diseases. Lancet 1997, 350:134-138 [DOI] [PubMed] [Google Scholar]
- 35.Haruda M, Skisaka S, Terada K, Kimura K, Krawaguchi T, Koga H, Kim H, Taniguchi E, Harada S, Sugamura T, Furuta K, Sugiyama T, Salon M: A mutation of Wilsons’ disease protein, ATP 7B, is degraded in the proteasomes and forms protein aggregates. Gastroenterology 2001, 120:967-974 [DOI] [PubMed] [Google Scholar]
- 36.Caulin C, Ware CF, Magin TM, Oshima RG: Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J Cell Biol 2000, 149:17-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacFarlan M, Merrison W, Dinskale D, Cohen MG: Active caspases and cleaved cytokeratins are sequestered into cytoplasmic inclusions in TRAIL-induced apoptosis. J Cell Biol 2000, 148:1239-1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ku NO, Michie SA, Soetikno RM, Resurreccion EZ, Broome RL, Oshima RG, Omary MB: Susceptibility to hepatotoxicity in transgenic mice that express a dominant-negative human keratin 18 mutant. J Clin Invest 1996, 98:1034-1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilbert S, Loranger A, Daigle N, Marceau N: Simple epithelium keratins 8 and 18 provide resistance to Fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J Cell Biol 2001, 154:763-773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ku N, Michie SA, Soetikno RM, Resurreccion EZ, Broome RL, Omary MB: Mutation of a major keratin phosphorylation site predisposes to hepatotoxic injury in transgenic mice. J Cell Biol 1998, 143:2023-2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ku NO, Wright TL, Terrault NA, Grish R, Omary MB: Mutation of human keratin 18 is associated with cryptogenic cirrhosis. J Clin Invest 1997, 99:19-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ku NO, Grish R, Wright TL, Omary MB: Keratin 8 mutations in patients with cryptogenic liver disease. N Engl J Med 2001, 344:1580-1587 [DOI] [PubMed] [Google Scholar]
- 43.Cienchanover A, Schwartz AL: Ubiquitin-mediated degradation of cellular proteins in health and disease. Hepatology 2002, 35:3-6 [DOI] [PubMed] [Google Scholar]
- 44.Hershko A, Ciechanover A: The ubiquitin system for protein degradation. Annu Rev Biochem 1992, 67:761-807 [DOI] [PubMed] [Google Scholar]
- 45.Rivett AJ: Proteasomes: multicatalytic proteinase complexes. Biochem J 1993, 291:1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goldberg AL, Rock KL: Proteolysis, proteasomes and antigen presentation. Nature 1992, 357:375-379 [DOI] [PubMed] [Google Scholar]
- 47.Hochstrasser M: Ubiquitin and intracellular protein degradation. Curr Opin Cell Biol 1992, 4:1024-1031 [DOI] [PubMed] [Google Scholar]
- 48.Oiao D, Goritonde SV, Qi W, Martinez JD: Deoxycholic acid suppresses p53 by stimulation proteasome-mediated p53 degradation. Carcinogenesis 2001, 22:957-964 [DOI] [PubMed] [Google Scholar]
- 49.Denk H, Stumptner C, Fuchsbichler A, Zatloukal K: Alcoholic and nonalcoholic steatohepatitis. Histopathologic and pathogenetic considerations. Pathologe 2001, 22:388-398 [DOI] [PubMed] [Google Scholar]
- 50.Sokol RJ, Straka MS, Dahl R, Devereaux MW, Yerushalmi B, Gumricht E, Elkins N, Everson G: Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr Res 2001, 49:519-531 [DOI] [PubMed] [Google Scholar]
- 51.Sokol RJ, McKim AM, Goff MC, Ruyle SZ, Devereaux MW, Han D, Packer L, Everson G: Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology 1998, 114:164-174 [DOI] [PubMed] [Google Scholar]