

Abstract
A better understanding of cellular mechanisms that occur in Parkinson’s disease and related Lewy body diseases is essential for development of new therapies. We previously found that 6-hydroxydopamine (6-OHDA) elicits sustained extracellular signal-regulated kinase (ERK) activation that contributes to neuronal cell death in vitro. As subcellular localization of activated kinases affect accessibility to downstream targets, we examined spatial patterns of ERK phosphorylation in 6-OHDA-treated cells and in human postmortem tissues representing the full spectrum of Lewy body diseases. All diseased human cases exhibited striking granular cytoplasmic aggregates of phospho-ERK (P-ERK) in the substantia nigra (involving 28 ± 2% of neurons), which were largely absent in control cases (0.3 ± 0.3%). Double-labeling studies and examination of preclinical cases suggested that these P-ERK alterations could occur relatively early in the disease process. Development of granular cytoplasmic P-ERK staining in 6-OHDA-treated cells was blocked by neuroprotective doses of catalase, supporting a role for oxidants in eliciting neurotoxic patterns of ERK activation. Evidence of nuclear translocation was not observed in degenerating neurons. Moreover, granular cytoplasmic P-ERK was associated with alterations in the distribution of downstream targets such as P-RSK1, but not of P-Elk-1, suggesting functional diversion of ERK-signaling pathways in Lewy body diseases.
Parkinson’s disease (PD) is a debilitating movement disorder characterized by degenerating neurons containing cytoplasmic inclusions called Lewy bodies. 1-3 Although classic cases of PD are characterized by neuron loss in the substantia nigra, prominent cortical involvement by Lewy bodies is seen in patients exhibiting concurrent symptoms of dementia (dementia with Lewy bodies, DLB). The etiology of sporadic PD is unknown, although mutations in α-synuclein, a major component of Lewy bodies, and in enzymes involved in ubiquitination have been identified in familial cases. 3,4 Regardless of etiology, oxidative stress has been widely implicated in the pathogenesis of PD. 5-7 One mechanism by which oxidative species could contribute to neurodegeneration is through modulation of key intracellular signaling pathways that regulate neuronal survival.
The mitogen-activated protein kinases constitute a major family of signaling proteins 8 that regulate neuronal survival, differentiation, and plasticity. In particular, the extracellular signal-regulated protein kinase (ERK) branch is involved in neuronal development, hippocampal learning, and survival. 9,10 However, recent studies indicate that ERK signaling may also play a detrimental role in neuronal responses to stress. 11-18 In animal models of cerebral ischemia-reperfusion, inhibitors of MEK, the upstream kinase that activates ERK, confers significant protection. 12,15 Moreover, suppression of ERK phosphorylation protects neuronal cell lines and primary neuronal cultures subjected directly to oxidative stressors. 13,14,17,18 These studies highlight a potentially detrimental role for ERK signaling in oxidative neurotoxicity.
6-Hydroxydopamine (6-OHDA) is an oxidative neurotoxin commonly used in animal models to lesion the nigrostriatal system that degenerates in PD. We previously found that 6-OHDA elicits ERK activation in a central nervous system-derived, tyrosine-hydroxylase-expressing neuronal cell line. 14 Although transient activation and nuclear translocation characterize classic ERK-signaling responses to trophic factors, 6-OHDA elicits ERK activation that is sustained >20 hours after removal of the toxin. Inhibition of ERK phosphorylation confers significant protection, implicating ERK signaling in a causal role. The phospho-ERK (P-ERK) is catalytically active (SMK and CTC, unpublished observation), suggesting that alterations in the spectrum of downstream targets may account for differences between detrimental and protective patterns of ERK activation.
Subcellular localization of activated signaling complexes is central to regulating accessibility to downstream targets and regulatory phosphatases. 8 Thus, we examined spatial patterns of P-ERK localization in our cell culture model and in postmortem human tissues representing the full spectrum of Lewy body diseases. We present evidence for altered spatial patterns of ERK signaling in PD and other human Lewy body diseases, including early presymptomatic cases. Moreover, we show that development of similar cytoplasmic P-ERK staining in cultured cells is inhibited by catalase, and that catalase protects against 6-OHDA toxicity. Our results indicate that both spatial and temporal alterations in ERK signaling may contribute to parkinsonian neurodegeneration.
Materials and Methods
Human Tissues
Paraffin-embedded midbrain sections were obtained from the Joseph and Kathleen Price Bryan Brain Bank 19 and the University of Pittsburgh Brain Bank. All banked patients have undergone extensive standardized premortem neurological and postmortem neuropathological assessments. All available PD and incidental Lewy body disease cases were requested, along with a set of DLB cases with low Braak scores (pure DLB), and a set of normal control cases matched for age and postmortem intervals. Incidental Lewy body disease cases were defined as patients who had been followed as normal Alzheimer’s Disease Research Center control patients, having neither symptoms of dementia nor of parkinsonism, but who had Lewy bodies identified on pathological examination of their midbrains. A total of seven PD cases, five incidental Lewy body disease cases, eight DLB cases, and seven control cases were initially identified. Of these cases, two cases (one incidental Lewy body and one DLB) had no substantia nigra left in the paraffin block. One incidental case was reclassified as a PD case because clinical correlation obtained by the Bryan Brain Bank revealed that the subject had developed PD symptoms before death. This resulted in a set of eight PD cases, three incidental Lewy body disease cases, seven DLB cases, and seven control cases. Only one of the seven DLB cases had an advanced Braak stage of V/VI, representing the common type of DLB (Lewy body variant of Alzheimer’s disease). Clinical and pathological data are summarized in Table 1 ▶ . When available, frozen substantia nigra was also obtained. The study design was approved by the University of Pittsburgh Institutional Review Board.
Table 1.
Summary of Cases Examined
| Age, years | PMI, hours | Symptoms | Braak stage | n | |
|---|---|---|---|---|---|
| Control | 82.3 (3.1) | 9.5 (2.8) | N/N | 1 (1–2) | 7 |
| Preclinical | 78.3 (3.2) | 3.7 (1.4) | N/N | 1 (1–2) | 3 |
| PD | 83.1 (1.6) | 7.0 (2.2) | N/Y | 1 (1–3) | 8 |
| DLB | 81.9 (2.2) | 4.8 (1.8) | Y/Y | 3 (2–5)* | 7 |
Values are expressed as Mean (SEM) for age and post-mortem interval (PMI), and as mode (range) for Braak stage. Symptoms reflect presence or absence of dementia/parkinsonism.
*P < 0.05 compared to the other three groups using the Kruskal-Wallis test followed by multiple comparison using the Mann-Whitney U test.
Immunohistochemistry
Details concerning source and dilution of antibodies used in this study are shown in Table 2 ▶ . Dewaxed sections were treated with 3% H2O2 for 30 minutes to quench endogenous peroxidases, heated in target retrieval solution at 95°C for 1 hour, treated with Immunon protein blocking agent (Shandon, Pittsburgh, PA), and incubated at 4°C overnight with one of three different rabbit anti-P-ERK1/2 antisera, anti-phospho-p90RSK, or anti-ERK1/2, followed by biotinylated anti-rabbit IgG (1:500; Jackson ImmunoResearch, West Grove, PA) at 25°C for 1 hour and streptavidin-horseradish peroxidase (1:500). For P-ERK1/2, biotinyl tyramide (1:100; TSA; Perkin-Elmer, Emeryville, CA) was applied for 30 minutes at 25°C, followed by streptavidin-horseradish peroxidase. The peroxidase reaction was visualized with 3-amino-9-ethylcarbazole (AEC) substrate (BioGenex, San Ramon, CA) or NovaRED (Vector Laboratories, Burlingame, CA), and sections counterstained with Mayer’s hematoxylin.
Table 2.
Summary of Antibodies Used
| Antibody name | Source | Company | Catalog no. | Dilution |
|---|---|---|---|---|
| Phospho-MAP kinase (P-ERK 1/2) | Rabbit | Calbiochem | 442705 | 1:2500 (IH) |
| Phospho-MAPK (P-ERK 1/2) | Rabbit | Sigma | E 7028 | 1:2000 (IH), 1:500 (IC) |
| Active MAPK (P-ERK 1/2) | Rabbit | Promega | V803A | 1:2000 (IH) |
| Phospho-p44/42 MAP kinase (T202/Y204) (E10 monoclonal) | Mouse | Cell Signaling Technol | 9106 | 1:2000 (W), 1:4000 (IC) |
| MAP kinase (ERK 1/2) | Rabbit | Oncogene | PC54 | 1:100 (IH) |
| MAP kinase 1/2 (ERK 1/2) | Rabbit | Upstate Biotechnology | 06-182 | 1:20000 (W) |
| alpha-Synuclein | Mouse | Zymed Laboratories | 18-0215 | 1:1500 (IH) |
| Ubiquitin | Rabbit | DAKO | Z 0458 | 1:50 (IH) |
| Phospho-p90RSK (T359/S363) | Rabbit | Cell Signaling Technol | 9344 | 1:100 (IH), 1:10000 (W) |
| RSK-1 | Rabbit | Santa Cruz | sc-231 | 1:10000 (W) |
| Phospho-ELK-1 | Mouse | Cell Signaling Technol | 9186 | 1:100 (IH) |
| Actin | Rabbit | Sigma | A2066 | 1:4000 (W) |
| Lamin A | Rabbit | Cell Signaling Technol | 2032 | 1:2000 (W) |
Dilutions used for paraffin immunohistochemistry (IH), immunocytochemistry (IC), and Western blots (W) are shown.
P-ERK immunofluorescence was performed as above except sections were incubated with fluorophor-tyramide (1:100; Perkin-Elmer), and nuclei counterstained with propidium iodide (Molecular Probes, Eugene, OR). For double-labeling studies, anti-α-synuclein or anti-ubiquitin antibodies were applied to P-ERK1/2 (Calbiochem, La Jolla, CA, or Sigma Chemical Co., St. Louis, MO) prestained sections for 2 hours at 25°C, followed by Cy3-conjugated secondary antibodies (Jackson ImmunoResearch) for 1 hour at 25°C. The slides were observed using a Molecular Dynamics laser-scanning confocal imaging system, and co-localization was confirmed using Z-sectioning and orthogonal image analysis on a Zeiss Axioplan 2 confocal imaging system. For negative controls, primary antibody was replaced with nonimmune rabbit IgG. Specificity of antisera was verified by immunoblot analysis.
Cell Culture and Toxicity Assays
B65 cells (ECACC 85042305) were the gift of David Schubert of the Salk Institute, La Jolla, CA. B65 cells were cultivated and toxicity assays performed as described previously. 14 Active catalase (Roche Molecular Biochemicals, Indianapolis, IN) or catalase inactivated by boiling for 5 minutes, was added with fresh culture media 30 minutes before addition of 6-OHDA or vehicle.
Immunocytochemistry
B65 cells were plated on glass coverslips at a density of 300 cells/mm2, treated with 6-OHDA or vehicle, washed with phosphate-buffered saline (PBS) containing NaVO4, fixed in 3% paraformaldehyde, permeabilized with 0.1% Triton X-100/PBS, and blocked sequentially with 1% bovine serum albumin/PBS and 10% normal goat serum. To visualize activated ERK, E10 monoclonal antibody (overnight incubation at 4°C) or rabbit polyclonal anti-P-ERK1/2 (1 hour at 25°C; Sigma) were used at concentrations indicated in Table 2 ▶ . Coverslips were washed with PBS and incubated with Alexa 488 goat anti-mouse IgG (1:300, 30 minutes at 37°C; Molecular Probes) or Cy3-conjugated secondary antibodies (1:200, 1 hour at 25°C; Jackson ImmunoResearch). Nuclei were counterstained with propidium iodide after treatment with RNase A.
Negative controls included either E10 antibody preincubated for 2 hours at room temperature with a fivefold excess of immunizing peptide, or substituting an equivalent concentration of mouse or rabbit IgG for primary antibody. Double labeling was performed using the E10 monoclonal antibody (or E10 preincubated with immunizing peptide) and P-RSK rabbit antibody (or nonimmune rabbit IgG) followed by the appropriate combinations of secondary antibodies. Coverslips were mounted in gelvatol, and cells visualized using a Nikon Eclipse II microscope or a Zeiss Axioplan 2 confocal imaging system. Co-localization was confirmed using Z-sectioning and orthogonal image analysis.
Cell Fractionation, Tissue Homogenization, and Immunoblotting
Cytosolic and nuclear subcellular fractionation of B65 cells was performed using NE-PUR kit (Pierce, Rockford, IL). B65 cell lysis and immunoblot analysis were performed as described previously. 14,20 Brain tissue was homogenized with protease and phosphatase inhibitors in 4 vol of ice-cold buffer A2 (25 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid) for 1 minute on high speed, then centrifuged at 3000 rpm for 30 minutes at 4°C (SH29000 rotor). The pellet was then homogenized in buffer B (25 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, 0.5% Triton X-100, 10% glycerol) and the supernatants were used for immunoblot analysis. Antibody dilutions used are shown in Table 2 ▶ . In vitro kinase activity was assessed by addition of recombinant Elk-1 substrate using the p44/42 MAP kinase assay kit (Cell Signaling Technology, Beverly, MA).
Data Analysis and Statistics
Substantia nigra sections were analyzed independently by two to three pathologists who were blinded with respect to the diagnoses. Neuronal profiles of substantia nigra neurons, as defined by location with respect to the cerebral peduncles, large size (>35 μm), pyramidal shape, and pigmentation were counted and scored with respect to the presence or absence of discrete P-ERK granules, presence or absence of diffuse P-ERK immunoreactivity, involvement of nuclear profiles, Lewy bodies or pale bodies, and extent of cell loss. One of the eight PD cases was excluded from quantitative analysis because of acute ischemic injury to portions of the ventral midbrain. All three preclinical cases and nine of the PD/DLB cases displayed well-oriented sections at the level of the third cranial nerve that could be analyzed by region. Cell groups were defined using the nomenclature of Gibb and Lees, 21 with the paranigral nucleus and intermediate group comprising the medial (M) region and the ventrolateral group (VL), dorsal group (D), and pars lateralis comprising the lateral (L) region. Data were expressed as either raw number of neurons containing granular P-ERK within each region or normalized as percentage of neurons within each region with P-ERK positivity. Data are expressed as mean percentages ± SEM. Differences between groups were analyzed by analysis of variance followed by two-tailed t-tests.
Results
Abnormal Cytoplasmic Accumulations of P-ERK Are Found in PD and DLB Cases
To determine whether P-ERK alterations could contribute to PD pathogenesis, we examined the substantia nigra of eight PD patients, seven DLB patients, and seven age-matched control persons by immunohistochemistry using three different rabbit polyclonal antisera for P-ERK (Table 2) ▶ . We found that pigmented substantia nigra neurons from PD and DLB brains showed coarse, granular cytoplasmic accumulations of P-ERK (Figure 1; A, C, E, F, and G) ▶ . (Aggregates and granules will be used interchangeably as brief morphological descriptors without implying additional biochemical or ultrastructural information.) The P-ERK granules were observed surrounding Lewy bodies in the region of the halo (Figure 1; A, C, and E) ▶ , in pale bodies (Figure 1, F and G) ▶ , and elsewhere in the cytoplasm, but were excluded from the Lewy body dense core and the nucleus (Figure 1; A, C, and G) ▶ . These observations were confirmed by confocal microscopy of sections labeled with P-ERK and ubiquitin (Figure 1; K, L, and M) ▶ , α-synuclein (Figure 1; N, O, and P) ▶ , and propidium iodide (not illustrated). P-ERK aggregates were observed in neurons of the substantia nigra and to a lesser degree, in the ventral tegmental area (see below). Pigmented neurons of the locus ceruleus, which was present for analysis in one case, also displayed Lewy bodies and P-ERK granules. This pattern of staining was distinct from the diffuse cytoplasmic and nuclear pattern sometimes observed in widely scattered cortical neurons in both diseased and control cases (not illustrated), or near acute infarcts, and was not observed in glial cells, neurons of cranial nerve or pontine nuclei, or vascular elements.
Figure 1.

Abnormal ERK distribution in human Lewy body diseases. A–G: Midbrain sections from PD (A), age-matched control (B), or DLB (C, E–G) patients were stained for P-ERK using antibodies from Calbiochem (A–C), Sigma (E, F), and Promega (G). Note abnormal cytoplasmic distribution of P-ERK (coarse red granules of varying sizes) in endogenously pigmented substantia nigra neurons (fine brown uniform granules). D: Nonimmune rabbit serum was used as a negative staining control. H–J: Representative images from PD (H), DLB (I), and control (J) patients stained for T-ERK. Note discrete T-ERK granules in the diseased cases (H and I, arrows) against a background of mottled cytoplasmic staining. K–M: Double-label confocal immunofluorescence study of a PD case showing association (yellow) of P-ERK granules (green) at the periphery of ubiquitinated (red) Lewy bodies. N–P: Double-label confocal immunofluorescence study of a DLB case showing association and partial co-localization (yellow) of P-ERK granules (green) with abnormal α-synuclein aggregates (red). Scale bars, 50 μm.
All of the eight PD and seven DLB cases showed numerous coarse P-ERK granules in pigmented substantia nigra neurons. P-ERK immunoreactivity was completely absent in five of seven age-matched control cases (Figure 1B) ▶ . In one control case, a single P-ERK granule was identified in a single neuron, and in a second case, rare P-ERK-positive neurons could be identified. Quantitative analysis of substantia nigra neuronal profiles revealed that PD and DLB cases showed significantly more P-ERK-positive neurons than control cases (Figure 2A) ▶ . There was no significant difference between PD (26 ± 3%) and DLB (29 ± 2%) groups. When averaged together, the diseased PD/DLB groups showed coarse P-ERK granules in 28% (±2%) of substantia nigra neurons. Although all of the PD cases showed discrete cytoplasmic P-ERK granules in the substantia nigra, cytoplasmic granules were not always observed in the ventral tegmental area and, when present, the percentage of affected ventral tegmental pigmented neurons was significantly lower (2.9 ± 3%, P < 0.05 compared to all PD cases).
Figure 2.
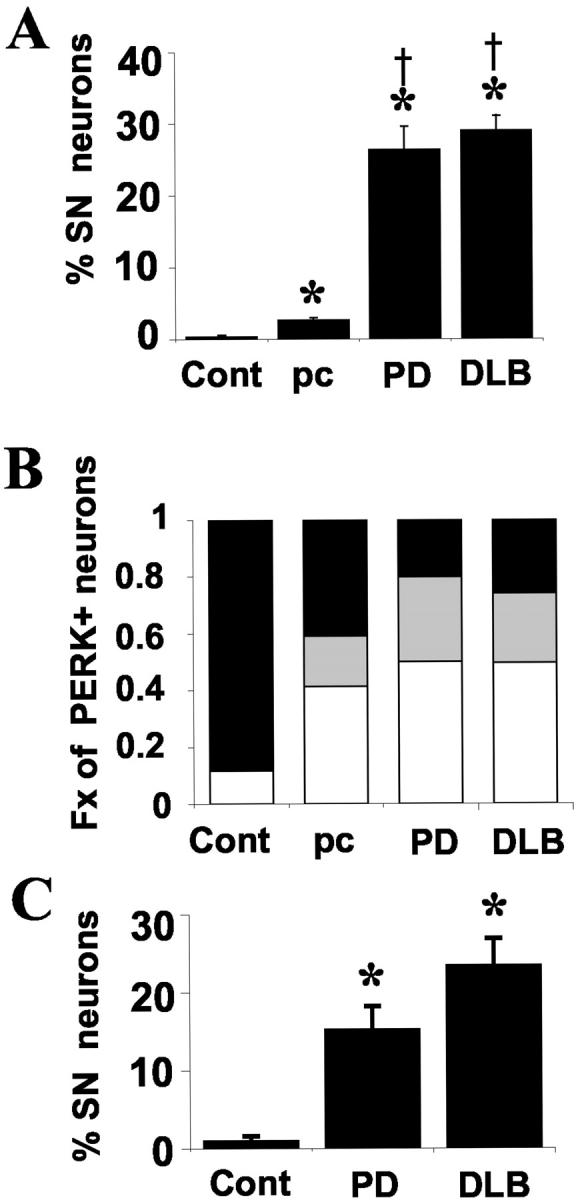
Distribution of P-ERK and P-RSK abnormalities in control, preclinical, and diseased cases. A: Percentage of pigmented substantia nigra neurons with granular cytoplasmic P-ERK immunoreactivity is shown for control (Cont), preclinical (pc), PD, and DLB groups. *, P < 0.05 compared to control; †, P < 0.05 compared to preclinical. B: Bar graph showing proportion of P-ERK-positive neurons that are overtly abnormal by routine histology (white bar), histologically unremarkable with early α-synucleinopathy (gray bar), or lacking either histological or α-synuclein pathology (black bar) was assessed as described in the text. Note that 20 to 25% of P-ERK-positive neurons do not manifest other pathology in PD and DLB cases (black bar), and this fraction is increased to 41% in preclinical cases. None of the rare P-ERK-positive neurons observed in the control cases showed abnormal α-synuclein positivity, but ∼10% did show pigment loss. C: The percentage of pigmented substantia nigra neurons with cytoplasmic P-RSK immunoreactivity was determined and the mean ± SEM determined for each group. *, P < 0.05 compared to control.
Discrete Granular T-ERK Staining Could Be Detected in PD/DLB Cases against a Background of More Diffuse Staining
Immunohistochemistry for total ERK (T-ERK) showed diffuse cytoplasmic staining in glial and neuronal elements, with mottled regions of intense accentuation in the paranuclear and submembranous regions. Aside from staining of Lewy bodies, the overall appearance of T-ERK staining did not differ significantly between control and diseased cases (Figure 1, I and J) ▶ . However, in more lightly stained substantia nigra neurons, sharply defined, punctate T-ERK granules were occasionally observed in PD/DLB cases (Figure 1, H and I ▶ , arrows), but not in control cases (Figure 1J) ▶ . These were on average smaller than the granules observed using the three P-ERK antisera, but were within the range of sizes observed with these antisera (Figure 1, F and G ▶ , arrows). The T-ERK granules were detected in a smaller number of neurons than recognized by the P-ERK antibodies. The granules may be more poorly recognized by the T-ERK antibody for several reasons, including differences in amplification procedures, differences in epitope accessibility after assembly into regulatory or pathological complexes, or differential patterns of phosphorylation at other sites remote from the activation motif. 22
Discrete Granular Accumulations Are Present in Incidental Lewy Body Disease
Incidentally discovered Lewy bodies are encountered in 4 to 10% of routine autopsies involving older individuals (>60 years), and have been proposed to represent an early preclinical form of PD. 3,23,24 We were able to examine the substantia nigra from three such patients, enrolled as normal age-matched controls without evidence of dementia or parkinsonism. The presence of Lewy bodies in neuronal soma was confirmed by α-synuclein immunohistochemistry. All three of these cases displayed multiple discrete P-ERK granules (Figure 2A) ▶ , suggesting that these changes can occur relatively early in the disease process.
P-ERK-Positive Neurons Were Concentrated in the Ventrolateral Substantia Nigra
Because cell loss is generally greater in the ventrolateral group of substantia nigra neurons, 21 the numbers of P-ERK-positive neurons was analyzed by region. In preclinical cases, there were significantly more P-ERK-positive neurons in the lateral versus medial substantia nigra, and in the ventrolateral versus dorsal groups (Figure 3, A and B) ▶ . In PD/DLB cases, although the overall pattern was retained, the raw number of P-ERK-positive neurons in ventrolateral group was no longer significantly different from the dorsal group (Figure 3C) ▶ . This most likely reflects greater cell loss in the ventrolateral group, as supported by the fact that a significantly higher percentage (∼60%) of the remaining ventrolateral neurons displayed P-ERK granules compared to the dorsal neurons (Figure 3D) ▶ .
Figure 3.

Distribution of P-ERK-positive neurons by substantia nigra region. Preclinical (A, B) and PD/DLB (C, D) cases were analyzed for neuronal P-ERK positivity in medial (M) versus lateral (L) regions of the substantia nigra as described in Materials and Methods. The lateral region was further subdivided into ventrolateral (VL) and dorsal (D) groups. 21 The data are expressed as either the total number of P-ERK-positive neurons in each region (A, C), or as the percentage of neurons in each region with P-ERK granules (B, D). *, P < 0.05 compared to medial region; †, P < 0.05 compared to dorsal group.
Immunoblot Analysis of Substantia Nigra Tissues
Immunoblot analysis of homogenized substantia nigra highlights increased P-ERK band intensity within all three groups of Lewy body diseases compared to control tissues (Figure 4A) ▶ . The same pattern was observed using both polyclonal and monoclonal antibodies, and the intensity of the P-ERK bands corresponded with level of in vitro kinase activity (Figure 4B) ▶ . It is interesting that the PD case showed a fainter P-ERK band than the DLB cases’ with a level of kinase activity comparable to the preclinical case. This may be related to the particularly severe neuronal loss seen in this PD case. Alternatively, it is possible that the longer 8-hour postmortem interval of this case may have contributed to the fainter signal, as postmortem intervals were 3 to 5 hours for the control, DLB, and preclinical cases.
Figure 4.
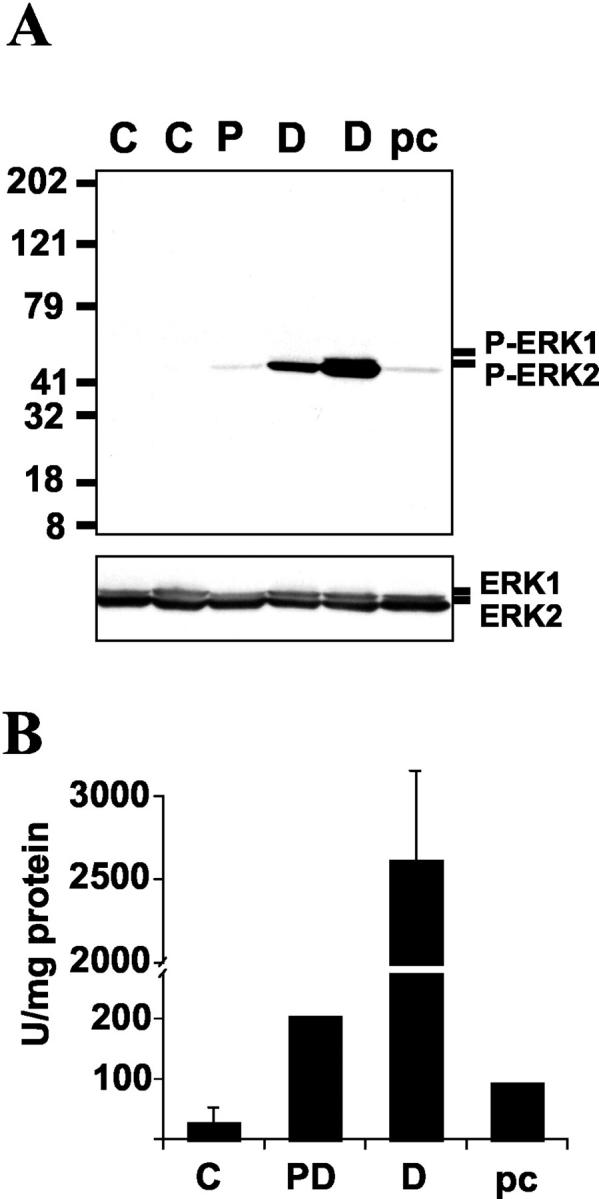
Frozen substantia nigra from two control cases (C), one PD case (P), two DLB cases (D), and one preclinical case (pc) were homogenized and the soluble fraction was subjected to immunoblot analysis (A) and in vitro kinase activity (B) as described in Materials and Methods. Shown is a representative immunoblot using the Calbiochem antibody for P-ERK (top), with reprobing for T-ERK (bottom). The same pattern of P-ERK staining was observed using the E10 monoclonal antibody. Kinase activity is expressed in arbitrary units based on phosphorylation of a recombinant ERK substrate.
Relationship of P-ERK Immunoreactivity to Abnormal α-Synuclein or Ubiquitin Immunoreactivity
To determine whether cytoplasmic P-ERK granules tended to occur in obviously abnormal neurons or in neurons in which abnormalities are more subtle (because the latter may reflect an earlier phase of degeneration), P-ERK-positive neuronal profiles were classified as overtly abnormal, if they contained Lewy bodies or pale bodies or significant regions of depigmentation, or as otherwise histologically normal. In Lewy body disease cases, P-ERK accumulations were present in both histologically normal neuronal profiles (Figure 5A) ▶ and in neurons showing clear pathological changes (Figure 5B) ▶ , at approximately equal ratios (Figure 2B ▶ , black/gray versus white bars). In preclinical cases, the accumulations were more likely to involve histologically normal neurons than overtly abnormal neurons by a ratio of 3:2 (Figure 2B) ▶ .
Figure 5.

Relationship of P-ERK granules to α-synuclein and P-RSK staining. A–C: Double-label immunohistochemical stains for P-ERK and P-RSK revealed abnormal P-ERK (red) in otherwise morphologically normal neurons without evidence of abnormal α-synuclein immunoreactivity (A); neurons with both P-ERK (red) and abnormal synuclein inclusions (brown) (B); and neurons with advanced synucleinopathy (brown), but no P-ERK immunoreactivity (C). Cresyl violet was used to convert endogenous pigment to a gray-green color. D–F: Abnormal cytoplasmic P-RSK1 staining in PD/DLB cases. Substantia nigra from normal control (D) and PD (E, F) cases were stained for P-RSK1 (red). The substantia nigra neurons of PD/DLB cases showed significantly increased levels of coarse, granular cytoplasmic staining. Note involvement of pale bodies (E), and the halo region of Lewy bodies (F), a distribution similar to that of P-ERK. G–I: Confocal immunofluorescent microscopy showing co-localization (yellow) of a subset of P-ERK granules (green) with P-RSK1 (red). Scale bars, 50 μm.
α-synuclein is a synaptic vesicle-associated protein whose physiological function is unknown. It is normally not observed immunohistochemically within neuronal soma. Because α-synuclein is a sensitive marker for early neuronal abnormalities in PD and DLB, 25,26 we performed double-labeling studies of preclinical, PD, and DLB cases for quantitative analysis. As expected, double labeling for P-ERK and α-synuclein detected involvement of an additional population of histologically normal neurons with early α-synucleinopathy in the diseased cases (Figure 2B ▶ , gray bars). Although the majority of P-ERK-positive neurons also showed abnormal synucleinopathy (R 2 = 0.985), ∼20 to 25% of these neurons did not show detectable abnormalities. Again, in preclinical cases, abnormal P-ERK staining was more often observed in otherwise normal-appearing, α-synuclein-negative neurons (41%), compared to in PD and DLB cases (Figure 2B ▶ , black bars). Moreover, neurons that were severely affected by abnormal α-synuclein aggregation sometimes did not show P-ERK granules (Figure 5C) ▶ , supporting the hypothesis that P-ERK abnormalities may occur relatively early in the disease process.
Specific Increases in P-RSK1 But Not P-Elk Immunoreactivity in PD and DLB Cases
One major downstream target for P-ERK is the transcription factor Elk-1, which participates in enhancing serum response element-gene transcription. There was no disease-related increase in P-Elk immunoreactivity in substantia nigra neurons (not illustrated).
Among other downstream targets that are phosphorylated by P-ERK are the ribosomal S6 kinases (RSK), which link the ERK signaling cascade to cyclicAMP response element (CRE)-mediated transcription through phosphorylation of CREB. As CRE gene transcription is generally believed to mediate neuroprotection, 27 we examined the distribution of P-RSK1 in control and diseased substantia nigra sections. Control cases showed variable degrees of nuclear P-RSK staining in substantia nigra neurons, other nonpigmented neurons of the midbrain/pons, endothelial cells, and white matter glial cells (Figure 5D) ▶ . Although one control case showed diffuse cytoplasmic P-RSK in 2% of neurons, granular cytoplasmic P-RSK was virtually never observed. In PD and DLB cases, there were significant increases in cytoplasmic P-RSK1 (Figure 2C) ▶ , with coarse, granular staining in either a random cytoplasmic distribution (Figure 5E) ▶ or surrounding Lewy bodies (Figure 5F) ▶ , a pattern similar to that observed for P-ERK. In the diseased cases, granular cytoplasmic immunoreactivity for P-RSK1 was also observed in neurons of the ventral tegmental area and the locus ceruleus, regions affected in PD/DLB, but not in other neurons or nonneuronal cells. When analyzed by region, cytoplasmic P-RSK distribution paralleled that of P-ERK, with the highest burden in the ventrolateral group (Table 3) ▶ . Double-label confocal immunofluorescent studies showed that a subset of cytoplasmic P-RSK1 aggregates co-localized with P-ERK (Figure 5; G, H, and I) ▶ .
Table 3.
Number and Percent of Pigmented Neurons within Subregions of the Substantia Nigra Showing Granular Cytoplasmic P-RSK Staining
| Preclinical | PD/DLB | |
|---|---|---|
| No. medial region | 1 | 4.7 (1.2) |
| No. lateral region | 6 | 36.1 (11.5)* |
| No. ventrolateral | 5 | 33.2 (10) |
| No. dorsal | 1 | 11.8 (3.9) |
| Percent medial region | 0.53 | 11.4 (3.0) |
| Percent lateral region | 1.14 | 20.7 (2.4)* |
| Percent ventrolateral | 1.22 | 25.5 (3.5)* |
| Percent dorsal | 0.85 | 17.0 (2.1) |
| n | 1 | 7 |
The substantia nigra was divided into medial and lateral regions. The lateral region was further subdivided into ventrolateral and dorsal groups as described in the text. The data for PD/DLB cases are expressed as mean (SEM).
*P < 0.05 when compared to medial group.
6-OHDA Elicits a Coarsely Granular Cytoplasmic P-ERK-Staining Pattern in the B65 Neuronal Cell Line
We have previously found that inhibition of ERK phosphorylation using an inhibitor of its upstream kinase MEK resulted in significant protection of the B65 cell line from 6-OHDA. 14 This is a rat central nervous system-derived cell line that expresses the dopamine transporter (Kulich SM and Chu CT, unpublished observation) tyrosine hydroxylase and other neuronal features including the ability to form regenerative action potentials. 28,29 Given the striking cytoplasmic distribution of P-ERK in the diseased human tissues, we performed immunocytochemical and biochemical analyses to examine the subcellular distribution of P-ERK elicited by 6-OHDA treatment. We found that, although minimal diffuse cytoplasmic P-ERK could be observed in ascorbate (vehicle)-treated cells (Figure 6A) ▶ , 6-OHDA treatment resulted in a robust, coarsely granular cytoplasmic-staining pattern (Figure 6, B and C) ▶ . The P-ERK immunoreactivity could be abolished by competition with the immunizing peptide (Figure 6F) ▶ . As observed in the human system, the immunofluorescent signal did not co-localize with propidium iodide-stained nuclei (Figure 6B) ▶ . These immunocytochemical results are supported by immunoblot analysis of nuclear and cytoplasmic extracts from 6-OHDA-treated cells, which likewise show little nuclear localization at 15 minutes, 3 hours, 6 hours, or 12 hours (representative data from a 6-hour experiment is shown in Figure 7 ▶ ).
Figure 6.

P-ERK- and P-RSK-staining patterns in B65 cells. A–F: Immunofluorescent labeling of P-ERK (green) in B65 cells treated for 6 hours with ascorbate vehicle (A), 500 μmol/L of 6-OHDA (B, C), 6-OHDA plus 25 U/ml of catalase (D), or 6-OHDA plus heat-inactivated catalase (E). F: Negative control: 6-OHDA-treated cells stained with E10 antibody absorbed with immunizing peptide. A, B, and F show merged image with red propidium iodide-stained nuclei. G–J: Immunofluorescent labeling of P-RSK (red) in B65 cells treated for 6 hours with ascorbate vehicle (G) or 500 μmol/L of 6-OHDA (H). I: Double-label confocal immunofluorescence study of P-ERK (green) and P-RSK (red) in 6-OHDA-treated cells demonstrating scattered co-localization (yellow). J: Orthogonal image analysis of Z-sectioned series confirming co-localization of P-ERK and P-RSK signals in a subset of granules. Scale bars: 30 μm (C, E); 20 μm (H).
Figure 7.

Subcellular fractionation of 6-OHDA elicited P-ERK and P-RSK. B65 cells were treated with 500 μmol/L of 6-OHDA for 6 hours. Cell lysates obtained using 0.1% Triton X-100 (L) were compared to cytoplasmic (C) and nuclear (N) extracts. Blots were analyzed for P-ERK (E10 monoclonal) and P-RSK, and stripped and reprobed for T-ERK, RSK1, nuclear marker Lamin A, and cytoplasmic marker actin.
6-OHDA Elicits Coarsely Granular Cytoplasmic P-RSK Staining
Treatment of B65 cells with 6-OHDA also elicited an increase in RSK phosphorylation. Again, the majority of phosphorylated RSK remained in the cytoplasm (Figure 7) ▶ in a granular distribution (Figure 6H) ▶ . Similar to our observations in human tissues, double-label confocal immunofluorescent studies indicated that a subset of P-ERK and P-RSK granules were co-localized (Figure 6, I and J) ▶ .
Neuroprotective Treatment with Catalase Inhibits Granular P-ERK Staining
As 6-OHDA spontaneously generates hydrogen peroxide and other reactive oxygen species, we assessed the effects of catalase on 6-OHDA toxicity. Because hydrogen peroxide diffuses across cell membranes, catalase treatment reduces both extracellular and intracellular levels of hydrogen peroxide as assessed using 2′,7′-dichlorofluorescein as an indicator. 30 Not only did catalase protect the cells from toxicity (Figure 8, A and D) ▶ , but it also attenuated the P-ERK response to 6-OHDA treatment (Figure 8F) ▶ . Heat-inactivated catalase had no protective effect (Figure 8, A and E) ▶ , nor did it decrease ERK phosphorylation (not illustrated). Immunocytochemical studies show that granular cytoplasmic P-ERK staining was inhibited by catalase, but not heat-inactivated catalase (Figure 6, D and E) ▶ . As direct inhibition of ERK phosphorylation using PD98059 also conferred significant protection to these cells, 14 these results support the hypothesis that altered patterns of ERK signaling contribute to detrimental neuronal responses to oxidative stress.
Figure 8.

Catalase confers protection from 6-OHDA and inhibits ERK phosphorylation. A: B65 cells were treated with 6-OHDA (filled squares), 6-OHDA with 5 U/ml of active catalase (open circles), and 6-OHDA with heat-inactivated catalase (open triangles). Viability was assessed using the MTS metabolic assay, normalized to vehicle-treated cells, and expressed as mean ± SEM. Phase microscopic images of B65 cells treated with ascorbate (the vehicle for 6-OHDA) (B), 1 mmol/L 6-OHDA (C), 6-OHDA plus 25 U/ml catalase (D), or 6-OHDA plus heat-inactivated catalase (E). F: Cells were treated with ascorbate or 500 μmol/L of 6-OHDA for 22 hours in the absence or presence of 5 U/ml of catalase and analyzed for ERK phosphorylation. Blots were stripped and reprobed for T-ERK.
Discussion
Regardless of etiology, oxidative stress has been widely implicated in PD pathogenesis. Although ERK signaling pathways are generally thought to promote neuronal survival, ERK activation can also contribute to neuronal injury in several in vivo and in vitro experimental models. 11-18,31 Sustained, rather than transient, ERK activation characterizes many of the oxidative models in which ERK activation is detrimental. We now present evidence of distinctly altered subcellular distribution of P-ERK in both 6-OHDA-treated cells and in human Lewy body diseases, including early preclinical cases. In the cell line, development of this granular P-ERK-staining pattern could be blocked by active catalase, which also conferred significant protection, supporting a causal role for oxidants in the development of neurotoxic patterns of ERK activation. These findings support the hypothesis that oxidative stress elicits distinct spatial-temporal patterns of ERK activation that contribute to parkinsonian neuronal injury.
In contrast to diffuse cytoplasmic and nuclear P-ERK immunoreactivity typical of acute ischemic injury, 15 diseased human nigral neurons and 6-OHDA-treated cells both demonstrated discrete, coarsely granular accumulations of P-ERK limited to the cytoplasm. Moreover, granular cytoplasmic ERK was associated with granular alterations in the distribution of a downstream kinase RSK1 (which typically trafficks to the nucleus after phosphorylation by ERK in the cytoplasm), but not of another ERK target, the transcription factor Elk-1. Although classic pathways of ERK-mediated neuronal survival involve transcriptional activation, the association of P-ERK with P-RSK1 (the major CREB kinase activated by ERK) in cytoplasmic granules suggests a functional diversion of ERK-signaling pathways under conditions of oxidative stress. It is interesting to note that sequestration of CREB-binding protein into aggregates away from its normal nuclear distribution has been shown to play an important causal role in models of Huntington’s disease. 32 Given the reported ability of α-synuclein to associate with several members of the Ras-ERK-signaling cascade, 33,34 it is possible that a similar sequestration mechanism plays a role in PD and DLB. It is also possible that abnormal phospho-protein profiles elicited by altered patterns of ERK activation contribute to synuclein aggregatability. Alternatively, it has been proposed that α-synuclein may play a compensatory role in regulating detrimental kinase responses to oxidative stress. 35
It is unclear whether the coarse, well-demarcated P-ERK granules observed by immunohistochemistry indicate association of P-ERK with cytoplasmic organelles or formation of small discrete complexes or inclusions. Although our cell culture model indicates that ERK activity is important to 6-OHDA toxicity, and the P-ERK granules in human PD/DLB cases are recognized by several activation-specific antibodies, it is unknown whether P-ERK distributed in these granules are functionally active. We do observe increased in vitro kinase activity in homogenates of diseased PD/DLB substantia nigra compared to control cases, but, in the absence of an in situ activity assay, cannot directly verify that the P-ERK granules contributed to this increased activity. Activated ERK is known to assemble onto the scaffolded surfaces of cellular vesicles. 36 Thus, these structures may reflect dysregulated trafficking or regulation of organelles. Further work to more clearly define the nature of these P-ERK accumulations are in progress.
Given the fact that altered proteasome function has been implicated in PD, 4,37 it is also possible that cytoplasmic accumulation of proteins occurs passively as a consequence of deficient proteolytic degradation. We believe this explanation cannot fully account for our observations. Given the micromolar abundance of T-ERK in cells, 8 phosphatase dysregulation would be expected to play a much more significant role than degradation in regulating ERK activity. In support of this, double-labeling studies indicate that, although P-ERK and P-RSK1 granules are sometimes associated peripherally with ubiquitinated inclusions, they are not themselves heavily ubiquitinated. Moreover, nonspecific accumulation of other downstream ERK targets such as Elk-1 is not observed. The presence of P-ERK in otherwise normal appearing neurons and its absence in a subset of neurons showing extensive α-synuclein burdens also argues against a strictly passive end-stage accumulation. Finally, in our experimental culture system, significant neuroprotection is observed with treatments that inhibit ERK phosphorylation, including MEK inhibition 14 and antioxidants (Figures 6D and 8F) ▶ ▶ , implicating ERK activation in a causal role. These experiments do not, however, distinguish between activity-dependent sequestration of P-ERK into granules with subsequent loss of downstream function versus direct toxic consequences of sustained cytoplasmic ERK activity.
There is growing consensus that PD patients and at least a subset of Alzheimer’s disease patients may reflect two ends of a disease continuum, with DLB, and the Lewy body variant of Alzheimer’s disease in the middle. 2,38 Indeed, sensitive new techniques indicate that up to half of Alzheimer’s disease brains show co-morbid abnormalities in α-synuclein immunoreactivity. 39 It is interesting to note that abnormal P-ERK immunoreactivity has also been observed in cases of Alzheimer’s disease. 40,41 Given the prevalence of synuclein abnormalities in Alzheimer’s disease patients, it is possible that abnormal P-ERK immunoreactivity may reflect common pathogenic mechanisms in both Parkinson’s and Alzheimer’s disease patients.
Acknowledgments
We thank Dr. Christine Hulette of the Joseph and Kathleen Bryan Alzheimer’s Disease Research Center Brain Bank, Duke University, Durham, NC, and Dr. Ronald Hamilton of the University of Pittsburgh Alzheimer’s Disease Research Center Brain Bank, Pittsburgh, PA, for providing human tissues and clinical information.
Footnotes
Address reprint requests to Charleen T. Chu, M,D., Ph.D., Room A-516, 200 Lothrop St., Pittsburgh, PA 15213. E-mail: chu@np.awing.upmc.edu.
Supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (R01 NS40817), the National Parkinson Foundation, and the Rockefeller Brothers Fund (to C. T. C., who is a Charles E. Culpeper Scholar in Medical Science), and the University of Pittsburgh Pathology Post-doctoral Research Training Program (to S. M. K.).
J. H. Z. and S. M. K. contributed equally to this study.
References
- 1.Dickson DW: Alpha-synuclein and the Lewy body disorders. Curr Opin Neurol 2001, 14:423-432 [DOI] [PubMed] [Google Scholar]
- 2.Galvin JE, Lee VM, Trojanowski JQ: Synucleinopathies: clinical and pathological implications. Arch Neurol 2001, 58:186-190 [DOI] [PubMed] [Google Scholar]
- 3.Goedert M: Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2001, 2:492-501 [DOI] [PubMed] [Google Scholar]
- 4.McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P: Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat Rev Neurosci 2001, 2:589-594 [DOI] [PubMed] [Google Scholar]
- 5.Hirsch EC, Faucheux BA: Iron metabolism and Parkinson’s disease. Mov Disord 1998, 13(Suppl 1):S39-S45 [PubMed] [Google Scholar]
- 6.Jenner P, Olanow CW: Understanding cell death in Parkinson’s disease. Ann Neurol 1998, 44(Suppl 1):S72-S84 [DOI] [PubMed] [Google Scholar]
- 7.Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J, Montine T, Sayre LM, Smith MA: Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett 2002, 319:25-28 [DOI] [PubMed] [Google Scholar]
- 8.Pearson G, Robinson F, Gibson TB, Xu B-E, Karandikar M, Berman K, Cobb MH: Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001, 22:153-183 [DOI] [PubMed] [Google Scholar]
- 9.Segal RA, Greenberg ME: Intracellular signaling pathways activated by neurotrophic factors. Ann Rev Neurosci 1996, 19:463-489 [DOI] [PubMed] [Google Scholar]
- 10.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270:1326-1331 [DOI] [PubMed] [Google Scholar]
- 11.Runden E, Seglen PO, Haug FM, Ottersen OP, Wieloch T, Shamloo M, Laake JH: Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci 1998, 18:7296-7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV: MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA 1999, 96:12866-12869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri M, Johnson J, DeFranco DB: Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem 2000, 275:12200-12206 [DOI] [PubMed] [Google Scholar]
- 14.Kulich SM, Chu CT: Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson’s disease. J Neurochem 2001, 77:1058-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A: Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci USA 2001, 98:11569-11574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cha YK, Kim YH, Ahn YH, Koh JY: Epidermal growth factor induces oxidative neuronal injury in cortical culture. J Neurochem 2000, 75:298-303 [DOI] [PubMed] [Google Scholar]
- 17.Oh-hashi K, Maruyama W, Yi H, Takahashi T, Naoi M, Isobe K: Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun 1999, 263:504-509 [DOI] [PubMed] [Google Scholar]
- 18.Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S: Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci Lett 2000, 288:163-166 [DOI] [PubMed] [Google Scholar]
- 19.Hulette CM, Welsh-Bonner KA, Crain B, Szymanski MH, Sinclaire NO, Roses AD: The Joseph and Kathleen Bryan Alzheimer’s Disease Research Center Experience. Arch Pathol Lab Med 1997, 121:615-618 [PubMed] [Google Scholar]
- 20.Chu CT, Everiss KD, Batra S, Wikstrand CJ, Kung H-J, Bigner DD: Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem J 1997, 324:855-861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibb WR, Lees AJ: Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1991, 54:388-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norman ED, Thiels E, Barrionuevo G, Klann E: Long-term depression in the hippocampus in vivo is associated with protein phosphatase-dependent alterations in extracellular signal-regulated kinase. J Neurochem 2000, 74:192-198 [DOI] [PubMed] [Google Scholar]
- 23.Gibb WR, Lees AJ: The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 1988, 51:745-752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forno LS: Concentric hyalin intraneuronal inclusions of Lewy type in the brains of elderly persons (50 incidental cases): relationship to parkinsonism. J Am Geriatr Soc 1969, 17:557-575 [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT: alpha-synuclein immunoreactivity in dementia with Lewy bodies: morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol (Berl) 2000, 99:352-357 [DOI] [PubMed] [Google Scholar]
- 26.Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM, Clark CM, Glosser G, Stern MB, Gollomp SM, Arnold SE: Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 2000, 54:1916-1921 [DOI] [PubMed] [Google Scholar]
- 27.Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M: CREB phosphorylation promotes nerve cell survival. J Neurochem 1999, 73:1836-1842 [PubMed] [Google Scholar]
- 28.Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinback JH: Clonal cell lines from the rat central nervous system. Nature 1974, 249:224-227 [DOI] [PubMed] [Google Scholar]
- 29.Schubert D, Carlisle W, Look C: Putative neurotransmitters in clonal cell lines. Nature 1975, 254:341-343 [DOI] [PubMed] [Google Scholar]
- 30.Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M: Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol 1983, 130:1910-1917 [PubMed] [Google Scholar]
- 31.Park JA, Koh JY: Induction of an immediate early gene egr-1 by zinc through extracellular signal-regulated kinase activation in cortical culture: its role in zinc-induced neuronal death. J Neurochem 1999, 73:450-456 [DOI] [PubMed] [Google Scholar]
- 32.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA: Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001, 291:2423-2428 [DOI] [PubMed] [Google Scholar]
- 33.Iwata A, Miura S, Kanazawa I, Sawada M, Nukina N: Alpha-synuclein forms a complex with transcription factor Elk-1. J Neurochem 2001, 77:239-252 [DOI] [PubMed] [Google Scholar]
- 34.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, Wolozin B: Alpha-synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci 1999, 19:5782-5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hashimoto M, Hsu LJ, Rockenstein E, Takenouchi T, Mallory M, Masliah E: A-synuclein protects against oxidative stress via inactivation of the C-jun N-terminal kinase stress signaling pathway in neuronal cells. J Biol Chem 2002, 277:11465-11472 [DOI] [PubMed] [Google Scholar]
- 36.Rizzo MA, Shome K, Watkins SC, Romero G: The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J Biol Chem 2000, 275:23911-23918 [DOI] [PubMed] [Google Scholar]
- 37.Rideout HJ, Larsen KE, Sulzer D, Stefanis L: Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem 2001, 78:899-908 [DOI] [PubMed] [Google Scholar]
- 38.Chu CT, Caruso JL, Cummings TJ, Ervin J, Rosenberg C, Hulette CM: Ubiquitin immunochemistry as a diagnostic aid for community pathologists evaluating patients who have dementia. Mod Pathol 2000, 13:420-426 [DOI] [PubMed] [Google Scholar]
- 39.Hamilton RL: Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 2000, 10:378-384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AK, Holbrook N, Siedlak SL, Harris PL, Smith MA: Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 1999, 10:2411-2415 [DOI] [PubMed] [Google Scholar]
- 41.Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Vinais F, Ribalta T: Phosphorylated Map kinase (ERK1, ERK2) expression is associated with early Tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol 2001, 11:144-158 [DOI] [PMC free article] [PubMed] [Google Scholar]