

Abstract
We previously showed that a mammary-specific dominant-negative p53 transgene (WAP-p53172H) could accelerate ErbB2-induced mammary tumorigenesis in mice, but was not tumorigenic on its own. To identify other genes that cooperate with WAP-p53172H in tumorigenesis, we performed mouse mammary tumor virus (MMTV) proviral mutagenesis. We derived F1, N2, and N4/N5 mice from p53172H transgenic FVB mice backcrossed onto MMTV+ C3H/He mice. Results show the latency of MMTV tumorigenesis is correlated with FVB contribution. F1 tumors had the shortest latency (217 days), had a higher rate of metastasis, and were less differentiated than the N2 and N4/N5 tumors. The latency was 269 days in N2 mice, and lengthened to 346 days in N4/N5 mice. p53172H significantly accelerated MMTV tumorigenesis only in N2 mice, indicating cooperativity between p53172H and MMTV in this cohort. To identify genes that may be causally involved in MMTV-induced mammary tumorigenesis, we identified 60 sites of proviral insertion in the N2 tumors. Among the insertions in p53172H transgenic tumors were 10 genes not previously found as sites of MMTV insertion including genes involved in signaling (Pdgfra, Pde1b, Cnk1), cell adhesion (Cd44), angiogenesis (Galgt1), and transcriptional regulation (Olig1, Olig2, and Uncx4.1). These may represent cellular functions that are likely not deregulated by mutation in p53.
Human breast cancer is the result of deregulation or loss of function of multiple genes, a process that typically takes years to complete. Support for this comes from demographics of cancer occurrence in humans 1 showing the relationship of patient age to tumor incidence, from studies of oncogene cooperativity in transgenic mice that reveal the necessity of two oncogenes for rapid tumor development, 2 and from transformation studies in primary cells. 3 It is evident from oncogene cooperation studies that specific combinations of genes can shorten the latency to cancer development whereas other combinations cannot.
We are using the mouse as a model system to identify genetic combinations that lead to mammary tumor development. We showed previously that ErbB2-induced mammary tumorigenesis in transgenic mice, which had a latency of 230 days, could be accelerated by combining this transgene with a mutant p53 transgene, 4 a result that indicates cooperativity between ErbB2 and p53172H. The latter transgene, murine p53172H driven by the whey acidic protein (WAP) promoter, is analogous to human p53175H, and is a dominant-negative allele that constitutes 8% of the p53 mutations in human breast cancer. By itself, the p53172H transgene is not able to induce mammary cancer in mice. It acts as a dominant-negative tumor suppressor allele rather than a dominant oncogenic allele in vivo. 5 It is evident that additional factors, either hormonal, mutational, viral, or epigenetic, are required for mammary tumorigenesis in p53172H transgenic mice. One way to identify such cooperating events is to use mouse mammary tumor virus (MMTV) proviral tagging.
MMTV is transmitted through germline transmission or through the milk in several mouse strains with high incidence of mammary tumors. 6,7 Tumors are induced by MMTV by proviral integration into the genomic DNA of the mammary epithelial cell; proviral insertion causes alteration of structure and expression of cellular genes that are normally tightly regulated. This can lead to the overproduction of a natural protein product(s), but can also lead to overexpression of truncated proteins. 8 Previous analysis of the sites of MMTV integration in mammary tumors has led to the identification of eight genes that appear to play important roles in murine mammary tumorigenesis: Wnt1, Fgf3/int2, Fgf4/hst, Fgf8/AIGF, Notch4/int3, Wnt3/int4, Wnt10b, and Tpl2/cot. 7 Several other sites have been found, and either the gene product is not known, or has been minimally characterized: IntH/Int5, 9,10 Int6, 11 and Int41. 12 The genes located at known insertion sites can be grouped into three functional categories: Wnt genes, Fgf family genes, and Notch family genes.
MMTV is a potentially useful tool for identifying genes that can cooperate with p53172H in mammary tumorigenesis. However, this requires cooperativity between p53172H and MMTV, ie, a shortening of latency to tumor formation or change in tumor histopathology in MMTV-infected mice that express the p53172H transgene relative to mice with one or the other agent but not both. In this study, we evaluate the tumor susceptibility in such groups of mice. We find that there is cooperativity in tumorigenesis between p53172H and MMTV in N2 and N4/N5 mice derived from backcrosses to C3H/He mice, but not in FVBxC3H/He F1 mice. In addition, we find that susceptibility to MMTV-induced tumors in the absence of p53172H transgene depends on the background strain, with the FVB genotype causing a significant shortening of latency, while the C3H/He genotype lengthened the latency. We further investigate the sites of proviral insertion to find cooperating oncogenes and were surprised to find a spectrum of insertion sites far broader than that previously described. This group includes a wide assortment of genes that play roles in diverse aspects of cellular biology including mitotic stimulation, cell adhesion, and angiogenesis.
Materials and Methods
Creation and Screening of Mice
FVB mice (Taconic, Germantown, NY) bearing the p53172H transgene 4 were crossed with either C3H/HeNCr-MMTV+ (Division of Cancer Treatment, NCI, Frederick, MD) (termed C3H/HeN herein) or C3H/HeOuJ mice as depicted in Figure 1 ▶ . In experiment 1, we created F1 hybrid mice from C3H/HeN and transgenic FVB mice harboring WAP-p53172H to create four groups of mice with or without transgene, and either infected or not infected with MMTV (Figure 1A) ▶ . Because MMTV is transmitted through the C3H/He dam’s milk, virus-negative mice were made using a C3H/HeN sire. In experiment 2, we mated p53172H FVB mice to C3H/HeN MMTV-negative female mice to create five male F1 offspring carrying p53172H as a hemizygous transgene. These five mice were mated with female C3H/HeN MMTV+ mice to generate 15 nontransgenic and 25 transgenic offspring, all MMTV+ because of their having suckled from an MMTV+ dam (Figure 1B) ▶ . In experiment 3 we backcrossed the p53172H transgene onto the C3H/HeOuJ (Jackson Labs, Bar Harbor, ME) background to the N3 or N4 generation, and in the final production cross, used C3H/HeN mice to create virus-positive and -negative and transgene-positive and -negative offspring for study (Figure 1C) ▶ . During the initial phases of this mating scheme, we used only male C3H/HeOuJ mice, to maintain MMTV-negative mice. C3H/HeOuJ was used in the initial crosses because of its reported high incidence of mammary cancer and short latency. 13 Subsequent to our initiating this cross, the Jackson Laboratories (Bar Harbor, ME) reported a shift in the phenotype of C3H/HeOuJ mice, such that the latency of mammary tumors was considerably longer and the incidence lower. This long latency tumor phenotype is apparently not because of lack of infectious MMTV in the milk of C3H/HeOuJ dams, but rather other, genetic factors. Thus, to minimize the inhibitory genetic factors of the C3H/HeOuJ strain, we crossed MMTV-negative generation N3 and N4 C3H/HeOuJx(C3H/HeOuJ × FVB) p53172H transgenic mice to C3H/HeN mice that have a high incidence of mammary tumors (see Figure 1C ▶ for mating scheme).
Figure 1.

Mating schemes for generation of F1 (A), N2 (B), and N4/N5 (C) mice. The boxed areas within A, B, and C designate the cohorts tested for susceptibility to tumors.
The presence of the transgene was determined by transgene-specific polymerase chain reaction (PCR) of DNA from tail biopsies performed as described. 4 Cohorts of mice were placed into mating at 4 to 5 weeks, and maintained in a pregnant and/or lactating state thereafter, to ensure that the transgene remained active. The health of the mice was monitored with biweekly examination and palpation of mammary glands, and the time of the first palpable lesion was noted as the onset date of the mammary tumor. Moribund mice were subjected to a complete necropsy. Tissues were fixed overnight in 3.7% neutral buffered formalin and submitted for embedding and histopathological analysis, or were frozen for DNA or RNA extraction.
Pathology
Five-μm paraffin sections of tumors, mammary gland, and other organs were stained with hematoxylin and eosin. Mammary tumors were classified according to the Sass and Dunn classification for MMTV-induced tumors. 14 Images of representative areas were captured with ×10, ×20 ,and ×40 objectives using a Carl Zeiss (Thornwood, NY) Axiocam camera and were processed using Adobe Photoshop (Adobe Systems Incorporated, San Jose, CA) software.
Southern and Northern Blotting
High-molecular weight DNA was extracted and Southern blotted as described. 15 RNA samples were fractionated on 1.2% formaldehyde-agarose gels and transferred to nylon membrane as described. 16 Northern and Southern blots were hybridized in the same manner: Agarose gel-purified plasmid inserts were radiolabeled by random priming, 17 and were hybridized in 7% sodium dodecyl sulfate/0.25 mol/L phosphate buffer (pH 7.2)/1 mmol/L ethylenediaminetetraacetic acid. 18 Filters were washed in 5% sodium dodecyl sulfate/40 mmol/L phosphate buffer (pH 7.2)/1mmol/L ethylenediaminetetraacetic acid, 15 minutes at 65°C and then 1% sodium dodecyl sulfate/40 mmol/L phosphate buffer (pH 7.2)/1mmol/L ethylenediaminetetraacetic acid, 15 minutes at 65°C. Filters were then autoradiographed. Probes for hybridization were generated as follows: a 1.8-kb hybridization probe for the MMTV env gene was derived from genomic tumor DNA by PCR using the following primers: 5′ GGC ACA GGG AAA TGC CTA TGC GG 3′ and 5′ TCT GTA TGG GGA ACT AGA GCAG 3′. Hybridization probe DNAs for known insertion site genes were obtained from G. Shackleford, (University of Southern California) and were as follows: Wnt1, 2.2-kb EcoR1 fragment from pWNT1; 19 Wnt3A, 0.4-kb BamHI-XhoI fragment of pJ70; 20 Wnt10b, 1.8-kb EcoR1-XhoI fragment of pJ54; 20 Hst/Fgf4, 1-kb HindIII fragment of pHH1; 21 Fgf8, 0.9 kb KpnI-XhoI fragment of pCMV-fgf8c. 22 A probe for the region of Cd44 encompassing the variable exons (exons 6 to 10, corresponding to variable exon V1 to variable exon V5 23 ) was prepared by performing reverse transcriptase (RT)-PCR with tumor 6505 RNA, using primers with sequence 5′ TGG AAG ACT TGA ACA GGA CAG G 3′ and 5′ GAG AGA TGC CAA GAT GAT G 3′.
Cloning of Insertion Sites by Inverse PCR
Inverse PCR was performed essentially as described by Li and colleagues 24 using either SacII or BamHI as restriction enzymes. The sequence of the PCR primers within MMTV was chosen based on the GenBank entry AF228552 for MMTV. Sequences of the primers used are shown in Table 1 ▶ . PCR products from the secondary PCR reaction were fractionated on ethidium bromide-agarose gels, and individual bands were excised and annealed to the pAMP1-cloning vector (Invitrogen, Inc., Carlsbad, CA). Plasmids were sequenced using M13 forward primers by automated sequencing at the Keck Biotechnology Resource Laboratory at Yale University. The resultant sequences were first edited to remove vector and retroviral sequences and were then compared to the sequences deposited in GenBank using BLAST algorithm available at the NCBI and/or DDBJ (http://www.ddbj.nig.ac.jp/E-mail/homology.html) websites searching the nr, est, and htgs databases.
Table 1.
Restriction Enzymes and Primers Used in iPCR
| Enzyme | Direction of amplification | 1° PCR | 2° PCR | Base pairs in MMTV | Primer sequence |
|---|---|---|---|---|---|
| Sac II | 5′ | Sac II 5′F | 5749–5762 | 5′ GGACACACTGGACTTCCCGGTC 3′ | |
| Sac II | 5′ | Sac II 5′E | 1829–1808 | 5′ GTCACAGACTTGGCCTCAGCGG 3′ | |
| Sac II | 5′ | Sac II 5′G | 5767–5789 | 5′ CAUCAUCAUCAUGGCACAGGGAAATGCCTATGCGG 3′ | |
| Sac II | 5′ | SAC II 5′H | 72–50 | 5′ CUACUACUACUACCCTTGGCTGCTTCTCCCCTAG 3′ | |
| Sac II | 3′ | Sac II 3′F | 8605–8629 | 5′ CGAGAGTGTCCTACACCTAGGGGAG 3′ | |
| Sac II | 3′ | Sac II 3′E | 5961–5938 | 5′ CCTAGTTGTGGCGCATGTCCCCAG 3′ | |
| Sac II | 3′ | Sac II 3′A | 5884–5863 | 5′ CAUCAUCAUCAUGTGAAACTGAAACCTAAGCGCC 3′ | |
| Sac II | 3′ | 3′C | 9833–9854 | 5′ CUACUACUACUAATCCTTCACTTTCCAGAGGGTC 3′ | |
| Bam H I | 3′ | 3′A | 8514–8535 | 5′ CTTGACCAAGTGCAGTCAGATC 3′ | |
| Bam H I | 3′ | 3′D | 7644–7624 | 5′ TCATGTTCTTGAAATCCTGGCC 3′ | |
| Bam H I | 3′ | 3′C | 9833–9854 | 5′ CUACUACUACUAATCCTTCACTTTCCAGAGGGTC 3′ | |
| Bam H I | 3′ | 3′B | 7579–7558 | 5′ CAUCAUCAUCAUTCTGTATGGGGAACTAGAGCAG 3′ |
RNA Isolation and Reverse Transcriptase-PCR Reactions
Total RNA was isolated from frozen mammary tumors (numbers 6509, 6536, 6098), normal mammary gland, brain, kidney, and embryo using Trizol (Invitrogen, Inc.) followed by RNeasy kit (Qiagen, Valencia, CA), as per the manufacturer’s protocol. RNA (1 μg) was annealed to oligo dT primer (33 ng) in 12-μl total volume at 65°C for 10 minutes. Reverse transcription was performed in a 20-μl reaction with 1× first strand buffer (Invitrogen, Inc.), dithiothreitol (10 mmol/L), dNTPs (250 μmol/L of each), placenta-derived RNase inhibitor (0.5 μl; Promega, Madison, WI) and Superscript II (200 units; Invitrogen, Inc.) as per the manufacturer’s protocol. In parallel, another 1 μg of RNA was treated exactly like the experimental reverse transcription reaction except that the Superscript enzyme was omitted. All of the samples were incubated at 42°C for 1 hour and then incubated 70°C for 10 minutes to inactivate the RT. The samples were diluted by adding 25 μl of H2O.
PCR amplification of the cDNA samples was performed with gene-specific primers (Table 2) ▶ . β-actin was used to normalize the PCR reactions. A typical 50-μl PCR reaction in 1× Promega buffer with 2.5 mmol/L of MgCl2 containing 1.5 μl of the RT product as template, dNTPs (400 μmol/L of each), the gene-specific primers (0.5 μmol/L of each), and 2.5 U of Taq DNA Polymerase (Promega).
Table 2.
Primers for PCR of cDNAs
| Genes | Direction | Primer sequence |
|---|---|---|
| Tfap2b | Forward | 5′ CAGGCGCAAAGCAGCAAATGTCACG 3′ |
| Reverse | 5′ ATCTTGTCCATGCCTTTGAGCGCCTCGG 3′ | |
| Fgfr2 | Ex. 7 forward | 5′ AAGCTGGACTGCCTGCAAATGC 3′ |
| Ex. 10 reverse | 5′ TTGGTCAGCTTGTGCACAGCTGG 3′ | |
| Ex. 10 forward | 5′ TGAGAGAGAAGGAGATCACGGC 3′ | |
| Ex. 11 reverse | 5′ TTCCCACTTTGGATCCTCTGGC 3′ | |
| Ex. 11 forward | 5′ AGTCCAGCTCCTCCATGAACTCC 3′ | |
| Ex. 15 reverse | 5′ TGGCCAGGCCAAAGTCTGC 3′ | |
| Plzf | Forward | 5′ AGAGGGAGCTGTTCAGCAAGC 3′ |
| Reverse | 5′ TTTCCGCAGAGTTCACACCCG 3′ | |
| Olig 1 | Forward | 5′ TTCCGAGCTGGATGTTACGC 3′ |
| Reverse | 5′ AGGGAAGTGGAGACTAAGTAAGG 3′ |
Results
Effect of Mouse Strain Background on Susceptibility to MMTV-Induced Mammary Tumors
To test for cooperativity between p53172H and MMTV, we generated test mice by crossing FVB (the source of the p53172H transgene) and C3H/He (the source of MMTV, Figure 1 ▶ ). At the outset, no information was available on the susceptibility of FVB to MMTV-induced mammary tumorigenesis. Because it is known that certain strains of mice carry genes that confer resistance to MMTV-induced mammary tumorigenesis (eg, II-TES 25 ), we decided to test FVBxC3H/He F1 hybrids, as well as N2 and N4/N5 mice generated by backcrossing the F1 to C3H/He (Figure 1) ▶ . Thus we created transgenic and nontransgenic F1, N2, and N4/N5, with successively less contribution from FVB.
In Tables 3 and 4 ▶ ▶ we present the nine groups of mice used in this study and their tumor incidence and latency to tumor formation. In comparing MMTV-infected, nontransgenic mice of the F1, N2, and N4/N5 generations, it is evident that the FVB strain has a significant effect on tumor latency: F1 mice (group F1.V) had a median latency to mammary tumor formation of 217 days, whereas for N2 (group N2.V) and N4/N5 (group N4/5.V) mice, that figure is 269 days and 447 days, respectively (Tables 3 and 4 ▶ ▶ and Figure 3 ▶ ). In addition, the percentage of mice developing tumors is also affected by strain background, being 100% in the F1 mice, 86% in the N2 mice, and 50% in the N4/N5 mice (Figure 3) ▶ . Log-rank analysis of the tumor latency data of the F1.V and N2.V cohorts revealed a significant difference between the groups (P < 0.01), as was the case for N2.V versus N4/5.V (P < 0.00001). These data are to be compared with an incidence of 62.5 to 98% and a median latency of 300 to 320 days that is observed in C3H/HeN mice (Table 4) ▶ . 26,27
Table 3.
Tumor Latency and Incidence in FVBxC3H/He F1 and F1xC3H/He Backcross Mice
| Group | Generation | p53172H | MMTV | Total number of mice | Percent with mammary tumor | Median latency (range) |
|---|---|---|---|---|---|---|
| F1 | F1 | − | − | 12 | 17 | 336 |
| F1.V | F1 | − | + | 17 | 100 | 217 (113–314) |
| F1.V.TG | F1 | + | + | 8 | 100 | 234 |
| N2.V | N2 | − | + | 15 | 87 | 269 (213–322) |
| N2.V.TG | N2 | + | + | 25 | 100 | 242 |
| N4/5 | N4/N5 | − | − | 8 | 75 | 453 |
| N4/5.V | N4/N5 | − | + | 14 | 50 | 447 (204–487) |
| N4/5.TG | N4/N5 | + | − | 5 | 20 | 471 |
| N4/5.V.TG | N4/N5 | + | + | 10 | 70 | 346 (243–461) |
Table 4.
Tumor Latency and Incidence in C3H/He and FVB Backcross Mice
Figure 3.
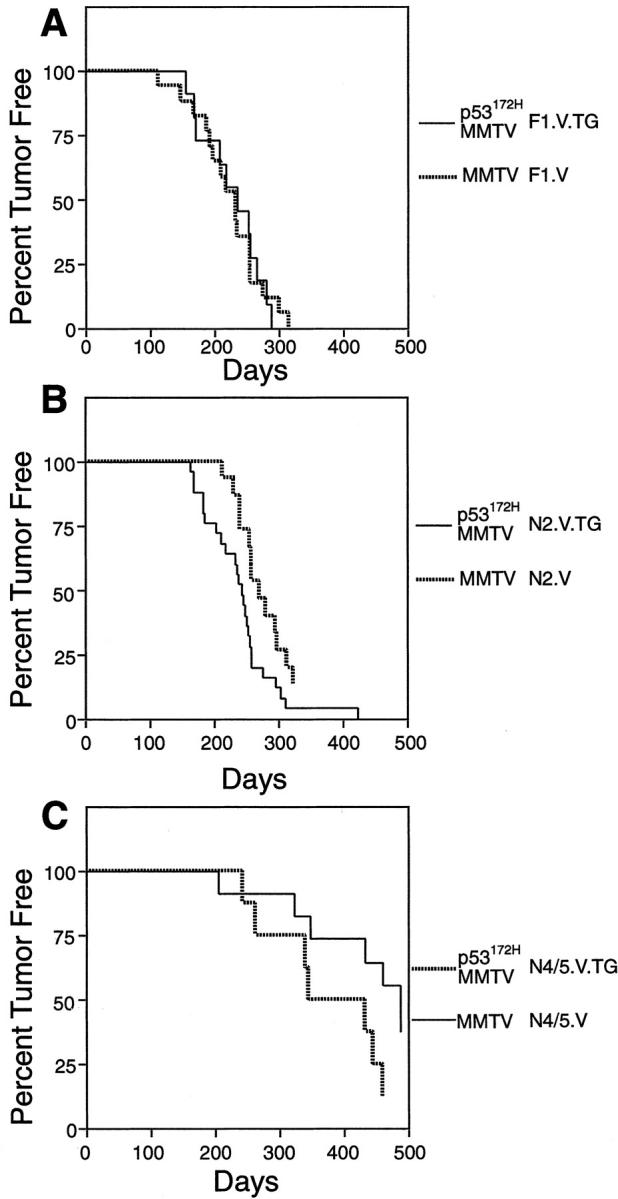
Kaplan-Meier plot of the tumor-free survival of FVB × C3H/N F1 mice (A), N2 mice (B), and N4/N5 mice (C). The groups plotted are indicated to the right.
Six of 17 (35%) MMTV-infected nontransgenic F1 mice had pulmonary metastases (Figure 2, E and F) ▶ but no metastases were observed in the nontransgenic MMTV-infected N2 or N4/N5 mice. In the transgenic MMTV-infected mice, pulmonary metastases were seen not only in the F1 cohort (one in eight tumor-bearing mice), but also in two of eight tumor-bearing N4/N5 mice. Thus, either 50% FVB background or p53 mutation were sufficient to promote metastasis.
Figure 2.
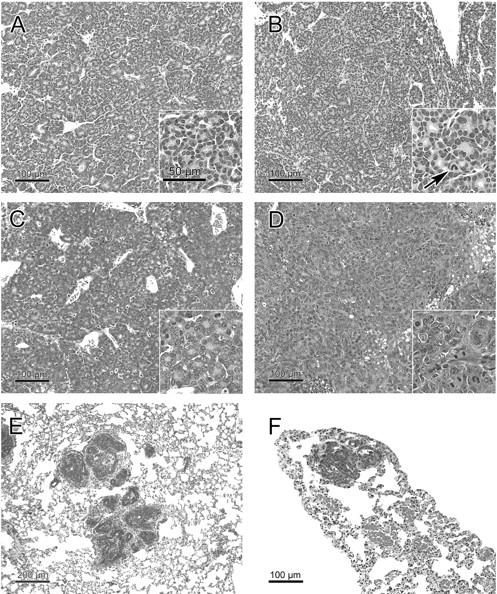
Histopathology of mammary tumors and their metastases from nontransgenic (A, C, E) and transgenic (B, D, F) mice with 6% (A, B), 25% (D), or 50% (C, E, F) FVB contribution to the background. A: Typical acinar tumor (adenocarcinoma type A 30 ) of an MMTV-infected nontransgenic mouse with high C3H background (group N4/5.V). As shown here, acini may vary in size, but must cover >50% of the tumor section for this diagnosis. There is scanty stroma with numerous dilated vessels. Inset shows a rather homogeneous population of small tumor cells with abundant eosinophilic cytoplasm. The small nuclei have a dense chromatin structure. Mitotic and apoptotic rates are low. The inset scale bar is valid for all insets. B: Acinar tumor (adenocarcinoma type A 30 ) of an MMTV-infected p53172H transgenic mouse with high C3H background (group N4/5.V.TG). Using a ×20 objective, no significant histological differences are seen as compared to the nontransgenic tumor in A. Inset shows a mild cytological effect of the transgene with slightly increased tumor cell size, nuclear pleomorphism, and mitotic and apoptotic rates. The nucleocytoplasmic ratio is unchanged. Arrow points to an abnormal mitotic figure with a chromosome leg. C: Acinar tumor (adenocarcinoma type A 30 ) of an MMTV-infected nontransgenic mouse with low C3H background (group F1.V). Irregular acini because of high proliferation and focal transitions to the solid pattern are present. Less stroma than in tumors with less FVB background (A and B). Inset shows increased tumor cell size, nuclear size, nuclear pleomorphism, and mitotic and apoptotic rates. The cytoplasm is basophilic. The nucleocytoplasmic ratio is inversed. The medium-sized nuclei have an open chromatin structure. D: Solid tumor with minor acinar components (adenocarcinoma type B 30 ) of an MMTV-infected transgenic mouse with intermediate C3H background (group N2.V.TG). This is tumor 6115 with an insertional activation of Uncx4. As in all studied solid tumors, there are less stroma and vessels, and more mitoses and apoptoses than in acinar tumors of the same mouse cohort (not shown). Inset shows increased tumor cell size, nuclear size, nuclear pleomorphism, and mitotic and apoptotic rates as compared to A, B, and C. The cytoplasm is scanty and basophilic. The nucleocytoplasmic ratio is inversed. The chromatin structure varies with a predominance of the vesicular pattern. E and F: Lung metastases were observed in nontransgenic (E) and transgenic (F) mice with 50% FVB background (E, group F1.V; F, group F1.V.TG).
Histomorphological Effects of Mouse Background Strain and p53172H
Histopathological analysis of the MMTV-induced tumors revealed a similar spectrum of tumor types in the F1, N2, and N4/N5 mice (Table 5) ▶ . However, the mammary tumors that arose in the F1 mice were less differentiated than those arising in the N2 or N4/N5 mice. Clear cytological differences were found between tumors of the F1 and N2 and N4/5 cohorts: the more C3H contribution to the background genotype, the smaller and less heterogenic the nuclei, the denser the chromatin, and the more cytoplasm (Figure 2, A to D ▶ ). The tumors from the N4/5 mice best resembled the type A and B tumors described by Dunn. 30 However, taking into account the background strain effect described here, all MMTV-induced mammary tumors fit histologically into the Dunn classification for MMTV-induced mouse mammary tumors.
Table 5.
Tumor Type by Generation
| Group | p53172H | MMTV | Number mice | Number tumors* | Type A† | Type B† | Type C† | Type Y† | Others† | Adeno- squamous | Molluscoid | Carcino- sarcoma |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | − | − | 12 | 2 | 0 | 1 (50) | 0 | 0 | 1 (50) | 0 | 0 | 0 |
| F1.V | − | + | 17 | 23* | 8 (35) | 14 (61) | 0 | 0 | 0 | 0 | 1 | 0 |
| F1.V.TG | + | + | 8 | 8 | 1 (13) | 6 (75) | 1 (13) | 0 | 0 | 0 | 0 | 0 |
| N2.V | − | + | 15 | 16 | 7 (44) | 7 (44) | 1 (6) | 1 (6) | 0 | 0 | 0 | 0 |
| N2.V.TG | + | + | 22 | 24* | 8 (33) | 15 (63) | 0 | 0 | 1 (4) | 0 | 0 | 0 |
| N4/5 | − | − | 8 | 6 | 1 (17) | 4 (75) | 0 | 1 (17) | 0 | 0 | 0 | 0 |
| N4/5.V | − | + | 14 | 9 | 2 (22) | 1 (11) | 2 (22) | 1 (11) | 0 | 1 (11) | 1 (11) | 1 (11) |
| N4/5.TG | + | − | 5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 |
| N4/5.V.TG | + | + | 10 | 8 | 1 (13) | 2 (25) | 0 | 0 | 1 (12) | 1 (13) | 1 (13) | 2 (25) |
| Sum | 97 | 28 (29) | 50 (52) | 4 (4) | 3 (3) | 3 (3) | 2 (2) | 4 (4) | 3 (3) |
*Represents more than the total number of mice, since some mice had more than one tumor. Percentages are given in parentheses.
Histopathological comparison of mammary tumors from transgenic and nontransgenic mice showed a mild cytological effect of the p53172H transgene in infected N2 and N4/5 mice that consisted of more pleomorphism, larger nuclei, more open chromatin structure, and less cytoplasm than in wild-type mice (Figure 2, A and B) ▶ . In the F1 transgenic mice, this cytological effect was not observed. Both transgenic and nontransgenic tumors in F1 mice were poorly differentiated because of the background effect (Figure 2C) ▶ . The mutation of p53 had a less profound histopathological effect than the background strain.
Cooperativity between p53172H and MMTV: Effect of Mouse Strain Background
We saw no significant acceleration of tumorigenesis in the F1 cohort when the p53172H transgene was added to MMTV infection: median latencies to mammary tumor were 234 days for MMTV+p53172H (group F1.V.TG) versus 217 days for MMTV alone (group F1.V; Figure 3A ▶ , Tables 3 and 4 ▶ ▶ ). However, in the N2 cohort, mice with both MMTV and p53172H (group N2.V.TG; 25 mice) all developed mammary adenocarcinomas, with latency of tumor development ranging from 161 days to 421 days, and a median latency of 242 days (Figure 3B ▶ ; Tables 3 and 4 ▶ ▶ ). In comparison 86% of nontransgenic MMTV-positive (group N2.V) mice developed mammary adenocarcinomas, with a median latency of 269 days (range, 213 to 322 days; Figure 3B ▶ , Tables 3 and 4 ▶ ▶ ). Results of a log-rank test on these data indicate a statistically significant difference between the p53172H-transgenic and nontransgenic groups (P < 0.02), thus showing that in this genetic background, p53172H can accelerate tumorigenesis.
Among the N4/N5 cohorts, the highest tumor incidence and the shortest tumor latency were observed in mice with both MMTV and p53172H (N4/5.V.TG mice; Tables 3 and 4 ▶ ▶ , Figure 3C ▶ ): 70% of mice with both virus and transgene developed tumors with a median latency to tumorigenesis of 346 days. Mice with virus alone (N4/5.V mice) had a lower incidence and longer latency relative to the N4/5.V.TG mice (Tables 3 and 4) ▶ ▶ ; however, by log-rank analysis this difference was not significant (P = 0.24). Some of the virus-negative N4/5 mice, both with and without p53172H (N4/5 and N4/5.TG cohorts), did develop mammary tumors at long latency (Tables 3 and 4) ▶ ▶ . In contrast to MMTV-induced tumors, the acinar pattern was rare in non-MMTV-induced tumors (Table 5) ▶ . Metastases from non-MMTV-induced tumors were not observed. In the N4/5 cohorts, non-MMTV-induced tumors occurred less frequently in the transgenic (one tumor in five mice) than in the nontransgenic (six tumors in eight mice) cohort. Also in the nontransgenic F1 mice, the non-MMTV-induced tumor incidence was lower than in the N4/5 cohort (Table 4) ▶ .
Analysis of Proviral Insertions in Tumor DNAs
To confirm the presence of somatically acquired MMTV proviruses in the MMTV-induced tumors, we performed Southern blot analysis of tumor DNAs. We were interested in determining the sites of proviral insertion in our tumors with the goal of identifying cooperating oncogenes for p53172H in the N2 mice because in this cohort, the combination of MMTV and p53 transgene resulted in an acceleration of tumorigenesis. Consequently, DNA was prepared from 23 N2.V.TG and 10 N2.V tumors. Southern blot was performed using the envelope probe of MMTV to see the presence of somatically acquired virus in these tumor DNAs. They were further analyzed by Southern blot to detect MMTV-induced restriction fragment length polymorphisms at some of the known sites of proviral insertion. They were also subjected to inverse PCR to clone out heretofore unknown sites of insertion.
Figure 4 ▶ shows representative hybridizations with Wnt1, Wnt3, Wnt10b, Fgf8, and Fgf4. No MMTV-induced polymorphisms were detected for Wnt1, Wnt3, or Fgf4. One N2.V tumor (tumor 6500; Figure 4 ▶ , lane 1) has evidence of a Wnt10b insertion, and in three N2.V.TG tumors, there is an insertion at Fgf8 (tumors 6506 and 6560, Figure 4 ▶ , lanes 4 and 9, respectively; and tumor 6101, not shown).
Figure 4.
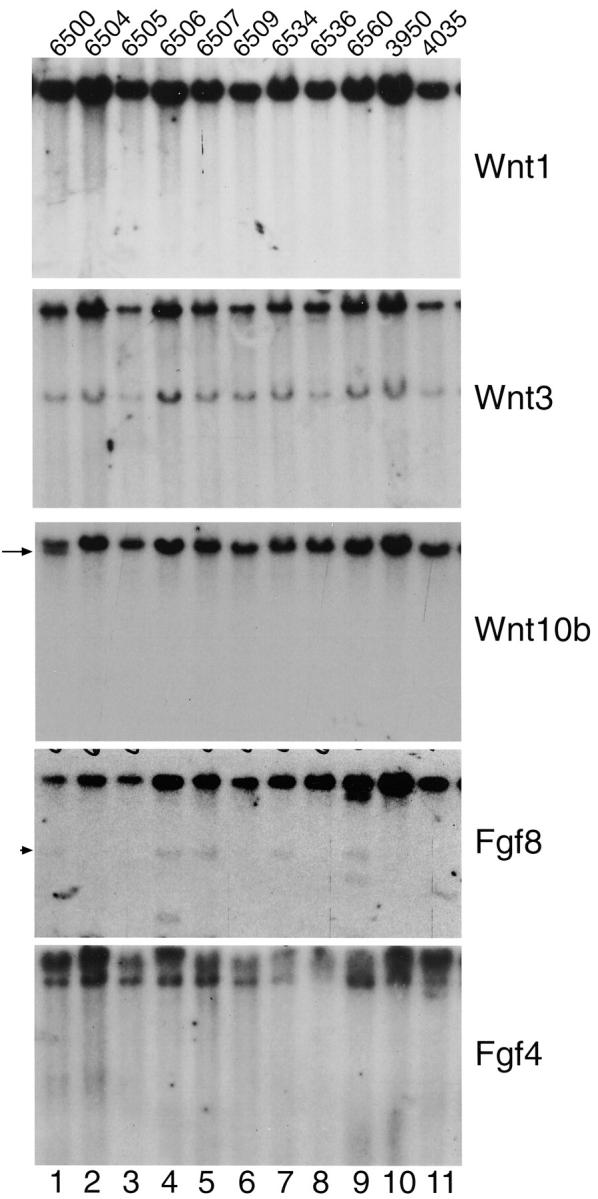
Representative Southern blot analyses of tumor DNAs from the N2.V.TG (6500, 6506, 6507, 6534, and 6560), and N2.V (6504, 6505, 6509, and 6536) groups. ErbB2-induced tumors, 4 3950 and 4035, serve as MMTV-negative controls. Note the presence of Fgf8 polymorphisms in tumors 6506 and 6560 and the presence of Wnt10b polymorphism in tumor 6500 (arrow). No polymorphisms were identified in the Wnt1, Wnt3A, or Fgf4. The band at arrowhead in Fgf8 hybridization represents residual signal from a previous hybridization with a 32P-labeled probe for p53. Not shown is a proviral insertion at Fgf8 in p53172H transgenic tumor 6101.
To identify potentially novel sites of proviral insertion in the N2 tumors, we used inverse PCR cloning. We were able to obtain PCR products on 15 of 24 N2.V.TG tumors and 10 of 16 N2.V tumors. Within the 25 tumors that arose in the N2 backcross mice for which we obtained iPCR product, MMTV insertions were found at 22 different known genes (Table 6) ▶ and 8 uncharacterized genes (Table 7) ▶ . The known genes include four known sites of MMTV proviral insertion: Wnt1, Wnt3, Wnt10b, and Fgf8 as well as 18 genes that heretofore had not been known to be sites of insertion (Table 6) ▶ . These genes play roles in mitotic stimulation, cell adhesion, and angiogenesis. Histopathological analysis of these tumors did not reveal specific tumor phenotypes for the given insertion sites.
Table 6.
Sites of Proviral Insertion in Known Genes
| Tumor number | Gene | Location | Function of gene |
|---|---|---|---|
| p53 transgenic mice | |||
| 6505 | CD 44 | Exon 8 | Cell surface glycoprotein |
| 6099 | Pde1b | Intron 1,11 kb from putative TSS | Cytoplasmic signaling |
| 6116 | Galgt1 | 2 kb 5′ of TSS | Enzyme |
| 6276 | GalNAc4ST2 | 5′ UTR | Enzyme |
| 6536 | Col5a1 | Intron within coding region | Extracellular matrix |
| 6500 | Wnt10b | Not known | Ligand |
| 6112 | Wnt 1 | 3′ UTR | Ligand |
| 6506 | Fgf8 | Not known | Ligand |
| 6560 | Fgf8 | Not known | Ligand |
| 6101 | Fgf8 | Not known | Ligand |
| 6499 | Fgf8 | 18 kb 3′ of gene | Ligand |
| 6499 | Pdgfra | Promoter | Receptor tyrosine kinase |
| 6091 | Cnk1 | 2.2 kb 5′ of TSS | Signaling molecule |
| 6536 | Olig 1 | 3′ UTR | Transcription factor |
| 6536 | Olig 2 | 6 kb 5′ of TSS | Transcription factor |
| 6115 | Uncx4.1 | 2 kb 5′ of TSS | Transcription factor |
| Nontransgenic mice | |||
| 6118 | Bart1 | 3′ UTR | Binds ras-related GTPase |
| 6098 | Cdh23 | Intron 5 | Cell adhesion molecule |
| 6108 | Nod 1 | Intron 11 | Cytoplasmic signaling |
| 6111 | Irs4 | 1.3 kb 5′ of TSS | Cytoplasmic signaling |
| 6108 | Wnt 3A | 4.8 kb 3′ of gene | Extracellular ligand |
| 6090 | Twik 2 | Intron 1 | K+-rectifying channel |
| 6096 | Wnt 1 | Enhancer region | ligand |
| 6509 | Fgfr2 | Intron 10 | Receptor tyrosine kinase |
| 6118 | Pdgfra | 3 kb 5′ of TSS | Receptor tyrosine kinase |
| 6509 | Tfap2B | Intron 1 | Transcription factor |
| 6098 | Plzf | Promoter | Transcription factor |
TSS, Transcription start site.
Table 7.
Uncharacterized Genes at Sites of MMTV Insertions
| Int number | Tumor number | Transgene | EST | Unigene | Locus link | Protein | Motifs |
|---|---|---|---|---|---|---|---|
| Int 9 | 6178 | None | NM_028003 | 12255 | 71919 | NP_082279 | Protein phosphatase? |
| Int 10 | 6090 | None | AK012169 | 23090 | 74207 | BAB28075 | Homology to D. melanogaster slit protein |
| Int 10 | 6090 | None | AK018739 | 41792 | 78885 | BAB31380 | 70 kd WD-repeat tumor-sp.antigen homolog |
| Int 10 | 6090 | None | BB267392 | 145416 | None | None | None |
| Int 11 | 6099 | p53 | KIAA0952 | None | None | None | BTB/POZ domain, nuc matx, p53 ind’d |
| Int 12 | 6115 | p53 | 3167888 | 151390 | None | None | Novel protein |
| Int 13 | 6322 | p53 | AK021163 | None | None | KIAA0996 | Coiled-coil, CG-NAP protein |
| Int 14 | 6534 | p53 | AK015267 | None | None | None | None |
Analysis of RNA from Tumors
To show that the genes located at the MMTV proviral insertion sites are up-regulated as a result of proviral insertion, we performed PCR analysis of cDNAs derived from RNA from tumors 6098, 6536, 6509, and 6509. Tumor 6098 had a proviral insertion in the promoter region of Plzf, a zinc finger protein for which mRNA transcripts are undetectable by RT-PCR in normal mammary tissue (Figure 5A ▶ , lane 2), but are detectable at apparently low levels in embryo RNA (Figure 5B ▶ , lane 3). Analysis of Plzf transcript in tumor 6098 shows detectable levels of expression of the gene, suggesting activation of the locus by the MMTV proviral insertion (Figure 5A ▶ , lane 1).
Figure 5.

RT-PCR analysis of total RNA. A: PCR with primers for Plzf (lanes 1 to 3), Olig1 (lanes 4 to 6), and Tfap2β (lanes 7 to 9). Key to samples: B, brain RNA; E, embryo RNA; K, kidney RNA; N, normal mammary gland RNA; and T, tumor RNA from tumor 6098 (lane 1), tumor 6536 (lane 4), and 6509 (lane 7). Bottom panel in each section depicts RT-PCR for β-actin, which serves as a control for equal loading of RNA. B: PCR with primers for Fgfr2. Diagram to left is a schematic of the Fgfr2 gene, showing the location of the primers used in the reactions shown (not to scale). Primer combinations included those for exon 7 and exon 10, exon 10 and exon 11, and exon11 and exon 15. The small arrows indicate the primers. MMTV insertion site and direction are shown in Fgfr2.
Tumor 6536 shows an insertion at 3′ UTR of the transcription factor Olig1. RT-PCR analysis of RNA from this tumor revealed the presence of Olig1 transcripts in tumor 6536 and brain, but not in normal mammary gland (Figure 5A ▶ , lanes 4 to 6). The levels of β-actin detected by RT-PCR suggest that equal amounts of RNA were analyzed in each lane (Figure 5A ▶ , lanes 4 to 6, bottom).
Tumor 6509 has an insertion in intron 1 within the coding region of transcription factor Tfap2β. By our RT-PCR analysis, Tfap2β expression is not detectable in normal mammary but is abundantly expressed in kidney tissue (Figure 5A ▶ , lanes 8 and 9). Such transcripts are detectable by RT-PCR in tumor 6509 RNA, consistent with proviral activation of the locus. β-actin analysis indicates that essentially similar amounts of RNA were used in each reaction. Tumor 6509 also has an insertion in intron 10 within Fgfr2, which encodes a receptor tyrosine kinase. Because of this location, we used primers in exons 7, 10, 11, and 15 for RT-PCR to detect transcripts in the tumor RNA that included exons either 5′ or 3′ of the insertion site, or both (Figure 5B) ▶ . Using brain cDNA as a positive control, we assessed the presence of transcripts that encompass exons 7 and 10, 10 and 11, 11 and 15 (Figure 5B ▶ , lane 12, top three panels, respectively). Using primers for exons 7 and 10, which are 5′ of the proviral insertion, we detect a small amount of transcript in tumor 6509 (Figure 5B ▶ , lane 10, top) but none in normal mammary gland (Figure 5B ▶ , lane 11, top). Using primers for exons 10 and 11, which reside on either side of the proviral insertion, we fail to detect a transcript in the tumor sample, suggesting that transcription terminates at the provirus. With primers for 11 and 15, which are 3′ of the insertion, we observe abundant transcript in both tumor and brain samples, and a faint band in normal mammary gland (Figure 5B ▶ , lanes 10 to 12, third panel from top). This is consistent with the activation of a transcript that encompasses the 3′ part of the gene, perhaps initiating at a cryptic promoter downstream of the proviral insertion, but upstream of exon 11. All of the experiments are repeated three times. The levels of β-actin was similar in all of the samples, suggesting that identical amounts of RNA were analyzed for each sample (Figure 5B ▶ , lanes 10 to 12, bottom).
We performed Northern blot to analyze the level of transcript of Cd44 in tumor 6505, which has a proviral insertion in exon 8. We documented two transcripts, of 3.4 and 1 kb in length, that were unique to tumor 6505, and were not detectable in three other tumors (6118, 6116, and 6509), or in normal mammary gland or brain (Figure 6) ▶ . These data suggest proviral activation of transcripts from this gene.
Figure 6.
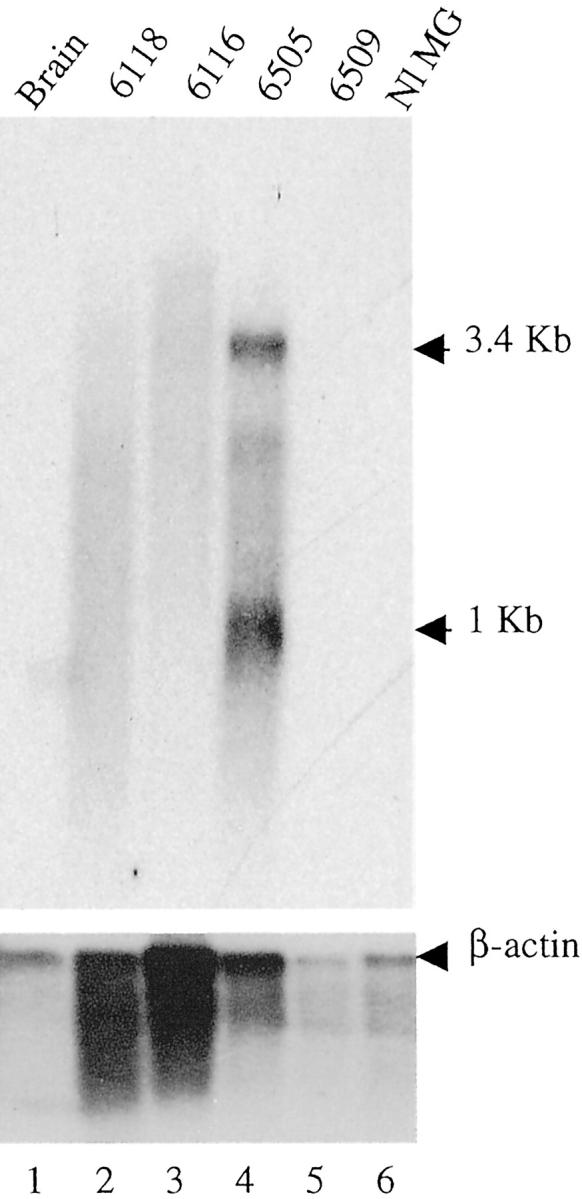
Northern blot analysis for Cd44 (top) and β-actin (bottom). The sizes (in kb) of the transcripts are indicated to the right. The samples analyzed are indicated on the top of each lane. The numbers indicate the number of the tumor. Normal mammary gland is analyzed in lane 6 (Nl MG).
Discussion
Four significant conclusions emerge from this work: 1) that FVB background significantly affects MMTV-induced mammary tumors in four ways: shortens their latency, increases their frequency, causes a shift to a more poorly differentiated tumor histology, and heightens the likelihood of pulmonary metastasis; 2) that in N2 backcross mice but not in F1 C3H/HeN × FVB hybrid mice, p53172H accelerates MMTV-induced mammary tumors; 3) that MMTV proviral insertions at Fgf8 were observed in 4 of 25 MMTV+, p53172H+ mammary tumors arising in N2 mice, suggesting cooperativity between p53172H and Fgf8 activation; and 4) that MMTV proviral tagging has identified a wide assortment of genes as sites of proviral insertion, providing clues regarding the types of genes that can cooperate with p53172H in mammary tumorigenesis.
Our experiments show that the degree of FVB contribution to strain background can have a profound influence on MMTV-induced mammary tumorigenesis (Table 4) ▶ : in F1 hybrid mice, the incidence was 100% and the latency 217 days (range, 113 to 314 days); in N2 mice the incidence was 86% and the latency 269 days (213 to 322 days); and from previous reports on inbred C3H/HeN mice, the incidence was 62.5 to 98% and the latency 300 to 320 days. 26,27 From the work of Leder and co-workers, it is clear that MMTV-infected pure FVB background mice develop mammary tumors (P. Leder, personal communication). However, the latency and incidence have not been reported. It appears clear from these experiments that the FVB strain enhances the MMTV-induced mammary tumor phenotype. This is consistent with what we have reported previously with studies of MMTV-induced acceleration of tumorigenesis in ErbB2 transgenic FVB × C3H/HeN F1 mice. 5 The mechanism by which FVB can hasten tumor formation is not clear; because of the low numbers of mice in each cohort, the mapping of quantitative trait loci is not possible. Because none of the N2 mice exhibited metastases, it is likely that multiple FVB alleles are needed concomitantly for this phenotype: if one FVB allele was needed, one half (seven mice) of the N2 animals should show metastases; with two alleles, one would expect three to four mice to have metastases. The fact that none showed metastases suggests that three or more FVB alleles may be required. To map such loci is beyond the scope of the present study.
MMTV-induced mammary tumorigenesis is determined by virus strain, hormonal factors, and genetic background. 28 Productive infection of the mammary epithelium and tumor induction depends on repeated cycles of pregnancy, parturition, and lactation: the incidence is lowest in virgin mice and highest in multiparous mice. 29 Thus, differences in fecundity between C3H and FVB mice may contribute to the shortened latency of mammary tumor onset in the F1 mice relative to N2 mice. However, confirmation of this will require further study. Other possible explanations include a genetically determined difference in efficiency of MMTV replication in F1 versus N2 mice, which would lead to higher rates of infection in the mammary epithelium.
The ability of p53172H to cooperate in MMTV tumorigenesis is dependent on the genetic background of the mice: no effect was observed in the FVB × C3H/N F1 hybrid mice, but an effect was seen in both N2 and N4/5 of backcross mice. This achieved statistical significance in the N2 cohort, but not in the N4/5 cohort, most likely because of the low numbers of mice. The reason for the inability of p53172H to accelerate MMTV-induced mammary tumorigenesis in F1 mice, while being able to do so in N2 animals, is not clear. In our previous study we observed a tumor-accelerating effect with p53172H in combination with ErbB2 in FVB mice. Thus we consider it unlikely that the tumor-promoting effect of p53172H is abrogated in F1 mice derived from FVB. We consider it more likely that the FVB provides genes that heighten the mutagenic effect of MMTV, thus obviating the need for the p53172H transgene.
Typical tumors for MMTV infection as reported by Dunn 30 and by Sass and Dunn 14 in C3H, but not FVB mice, are seen in mice with high (>87%) C3H background (Figure 2A) ▶ . The cytology as well as the tumor incidence, the tumor latency and the frequency of lung metastases changes toward more malignant growth if either the FVB part of the background is elevated (Figure 2C) ▶ , or the p53172H transgene is present (Figure 2D) ▶ . However, the effect of the background is more profound than the effect of the p53172H transgene. Previous studies with p53172H+ErbB2 bitransgenic mice demonstrated that the p53172H transgene induced aneuploidy in all of the tumors analyzed, which was reflected histologically in higher grade neoplasms, relative to ErbB2-induced tumors lacking p53172H. 4 In this study, histological analysis of mammary tumors arising in transgenic versus nontransgenic mice revealed mild histopathological differences between the two groups except for the F1 generation (Table 5 ▶ ; Figure 2, A to D ▶ ). Tumors in F1 mice were equally poorly differentiated regardless of the transgene status.
One surprising finding is the occurrence of tumors in female mice from an MMTV-negative dam (groups N4/5 and N4/5.TG). These mice never acquire the highly oncogenic milk-transmitted MMTV. They do retain Mtv1, an endogenous MMTV of low oncogenic potential that can induce late-onset tumors. 29 This is likely equivalent to the nodule-inducing virus (NIV), 31,32 an endogenous, rarely oncogenic virus expressed at low level, which is present in C3H/HeOuJ (RD Cardiff, unpublished data). This virus is known to induce hyperplastic alveolar nodules and rarely, adenosquamous tumors. The tumorigenesis seems not to be promoted by a mutant p53 or by high FVB contribution to the genetic background (Table 5) ▶ .
From this study, the origin of the clinical and cytological differences between N2 and N4/5 tumors is not clear. They may be because of the reduced FVB background. This is supported by the mild cytological differences between F1 and N2 tumors and by our previous study. 5 However, because N4/5 mice have 43.75% or 46.9%, respectively, of the C3H/HeOuJ genes, N4/5 tumors may have genes modifying tumor susceptibility such as the HeOuJ substrain typical LPS mutation, 13 which affects the host response.
Given the p53172H-induced acceleration of MMTV tumorigenesis in the N2 cohort, we examined the sites of proviral insertion by Southern blotting and inverse PCR to identify genes that can cooperate with p53172H in mammary tumorigenesis. A remarkable finding in our analysis of nontransgenic and transgenic N2 tumors is that we found insertions at 18 known genes that have not been previously identified as sites of MMTV insertion, 8 in nontransgenic tumors and 10 in transgenic tumors. The proteins encoded by these are involved in diverse cellular activities. Seven of the 18 protein products are involved in cell signaling, five in regulation of transcription, three in glycoprotein or glycolipid synthesis, one in the extracellular matrix, and one in regulation of intracellular potassium. Histopathological analysis of these tumors did not reveal specific phenotypes for the identified insertion sites (Figure 2D) ▶ . In contrast, mammary tumors in genetically engineered mice frequently have signature phenotypes, which allow the diagnosis of the transgene 33 or of the signaling pathway activated by the transgene. 34 To identify such specific phenotypes, in most cases more than four tumors per insertion site would be required. In addition, MMTV-induced mammary tumors frequently have areas of several differentiations within the same tumor. This could be because of either several virus insertion sites within the same tumor or to the activation of pluripotent differentiation pathways as observed in tumors from genetically engineered mice with an activation of Wnt or Fgf signaling.
Among nontransgenic tumors, we found one insertion at Cdh23, a newly identified member of the large family of cadherin genes, 35,36 which conceivably could deregulate the Wnt signaling pathway. In nontransgenic tumor 6509 there is a proviral insertion in the first intron of Tfap2b, which encodes the transcription factor AP2β known to transcriptionally regulate Erbb2 transcription. 37,38 This is very likely an important contributing event to mammary tumorigenesis. Interestingly, Tfap2a has been found to be a site of ecotropic murine leukemia virus proviral insertion in myeloid tumors arising in CasBrE mice (R Delwel and colleagues, personal communication), further implicating this family of proteins in oncogenesis. Other proviral insertion genes in nontransgenic tumors include several involved in signal transduction. These include Fgfr2, Pdgfra, Irs4, Bart1 and Nod1. Bart1 encodes a 19-kd protein that is likely an effector protein for ARL2, a Ras-like GTPase of unknown function. 39 Nod1 encodes a bifunctional protein that can induce apoptosis via a CARD domain, but can also promote cell survival through the nuclear factor-κB pathway. 40
Among the 23 p53172H transgenic tumors for which we obtained useful iPCR data, we found four insertions at Fgf8, indicating cooperativity between Fgf signaling and a dominant-negative mutant p53 allele. Cells with mutant p53 have a selective advantage and can survive this mitotic stimulation. 41 In transgenic tumors we found a proviral insertion in Wnt1, consistent with previous finding of cooperativity between Wnt1 and p53 loss in mice. 42
We identified Cd44 and Galgt1 as proviral insertion sites in transgenic tumors. These genes appear to promote tumorigenesis by altering the invasive and angiogenic properties of tumors. 43,44 Pde1b, which was also a site of proviral insertion in a p53 transgenic tumor, also appears to have a role in tumor cell invasiveness and angiogenesis. 45-48 Our data provide the first genetic evidence linking Cd44, Galgt1, and Pde1b to malignancy.
p53172H transgenic tumor 6115 (Figure 2D) ▶ had an insertion at Uncx4.1 which encodes a paired homeobox transcription factor that in normal murine development plays an important role in proliferation and condensation of the lateral presomitic mesenchyme. 49,50 In the developing somite, Uncx4.1 appears to be regulated by the Notch signaling pathways, because diminished or absent expression of Uncx4.1 in the somite is seen in mutants in a ligand (Dll1), receptor (Notch1), or downstream signaling factor (RBPJκ) in the Notch pathway. 51 Because the Notch pathway has been implicated in mammary cancer in mouse, and because it appears that Uncx4.1 is regulated by Notch signaling components, it is likely that Uncx4.1 is a downstream effector in this pathway in mammary tumorigenesis. Information on the nuclear targets of Notch critical for oncogenesis have eluded investigators for some time. The data of Ronchini and colleagues 52 and Dumont and colleagues 53 and our data may provide important new clues. It will be of interest to see if Uncx4.1 is activated in mammary tumors with MMTV insertions at Notch1 or Notch4.
p53172H transgenic tumor 6536 had insertion at both Olig1 and Olig2, two linked genes encoding helix-loop-helix proteins expressed in oligodendrocytes. Olig1 and Olig 2 are basic helix-loop-helix transcription factors that are associated with development of oligodendrocytes in the vertebrate central nervous system. 54,55 Olig2 was also identified as Bhlhb1, the gene at t(14;21)(q11.2;q22) in human T-cell acute lymphoblastic lymphoma, 56 in which the gene is transcriptionally activated by fusion to the T cell receptor-α locus. The finding that both Olig1 and Olig2 are sites of insertion in one mammary tumor suggests that they cooperate in oncogenic transformation; because both encode bHLH proteins, which are known to function as heterodimers, one possibility is that Olig1 and Olig2 dimerize to induce changes in gene expression, leading to tumor formation. The role that these proteins play in cellular growth control is not known.
In summary, proviral insertions in p53172H transgenic tumors occur at genes involved in mitogenic signal transduction (Fgf8, Pdgfra, Wnt1), in tumor cell invasiveness and angiogenesis (CD44, Pde1b, Galgt1) and downstream of Notch (Uncx4.1). We suggest that these insertions confer a selective advantage on tumor cells, and thus provide functions not provided by mutant p53. It is likely that deregulation of similar pathways occurs in the course of breast tumorigenesis in humans.
Acknowledgments
We thank Greg Shackleford for providing the hybridization probes and Pinki Chakraborty for her excellent technical assistance.
Footnotes
Address reprint requests to Archibald S. Perkins, Department of Pathology, Yale University School of Medicine, P.O. Box 208023, New Haven, CT 06520-8023. E-mail: archibald.perkins@yale.edu.
Supported by the United States Department of the Army (individual grant 96-1-6242, DAAD to A. R.), the National Institutes of Health (U42-RR14905), and the National Cancer Institute (R01 CA89140-01).
References
- 1.Armitage P, Doll R: The age distribution of cancer and a multistage theory of carcinogenesis. Br J Cancer 1954, 8:1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwan H, Pecenka V, Tsukamoto A, Parslow T, Guzman R, Lin T, Muller W, Lee F, Leder P, Varmus H: Transgenes expression the Wnt-1 and int-2 proto-oncogenes cooperate during mammary carcinogenesis in doubly transgenic mice. Mol Cell Biol 1992, 12:147-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elenbaas B, Spirio L, Koerner F, Fleming M, Zimonjic D, Donaher J, Popescu N, Hahn W, Weinberg R: Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 2001, 15:50-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li B, Rosen J, McMenamin-Balano J, Muller W, Perkins A: Neu/ErbB2 cooperates with p53–172H during murine mammary tumorigenesis in transgenic mice. Mol Cell Biol 1997, 17:3155-3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zelazny E, Anagnostopolous A-M, Coleman A, Perkins A: Cooperating oncogenic events in murine mammary tumorigenesis: assessment of ErbB2, mutant p53, and MMTV. Exp Mol Pathol 2001, 70:183-193 [DOI] [PubMed] [Google Scholar]
- 6.Nusse R: Insertional mutagenesis in mouse mammary tumorigenesis. Curr Top Microbiol Immunol 1991, 171:44-65 [DOI] [PubMed] [Google Scholar]
- 7.Callahan R, Smith G: MMTV-induced mammary tumorigenesis: gene discovery, progression to malignancy and cellular pathways. Oncogene 2000, 19:992-1001 [DOI] [PubMed] [Google Scholar]
- 8.Dievart A, Beaulieu N, Jolicoeur P: Involvement of Notch1 in the development of mouse mammary tumors. Oncogene 1999, 18:5973-5981 [DOI] [PubMed] [Google Scholar]
- 9.Gray D, McGrath C, Jones R, Morris V: A common mouse mammary tumor virus integration site in chemically induced precancerous mammary hyperplasias. Virology 1986, 148:360-368 [DOI] [PubMed] [Google Scholar]
- 10.Durgam V, Tekman R: The nature and expression of Int5, a novel MMTV integration locus gene in carcinogen-induced mammary tumors. Cancer Lett 1994, 87:178-186 [DOI] [PubMed] [Google Scholar]
- 11.Marchetti A, Buttitta F, Miyazaki S, Gallahan D, Smith G, Callahan R: Int6, a highly conserved, widely expressed gene, is mutated by mouse mammary tumor virus in mammary neoplasia. J Virol 1995, 69:1932-1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia M, Wellinger R, Vessaz A, Digglemann H: A new site of integration for mouse mammary tumor virus proviral DNA common to BALB/cf(C3H) mammary and kidney adenocarcinomas. EMBO J 1986, 5:127-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Outzen H, Corrow D, Shultz L: Attenuation of exogenous murine mammary tumor virus virulence in the C3H/HeJ mouse substrain bearing the Lps mutation. J Natl Cancer Inst 1985, 75:917-923 [DOI] [PubMed] [Google Scholar]
- 14.Sass B, Dunn T: Classification of mouse mammary tumors in Dunn’s miscellaneous group including recently reported types. J Natl Cancer Inst 1979, 62:1287-1293 [PubMed] [Google Scholar]
- 15.Mucenski ML, Gilbert DJ, Taylor BA, Jenkins NA, Copeland NG: Common sites of viral integration in lymphomas arising in AKXD recombinant inbred mouse strains. Oncogene Res 1987, 2:33-48 [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning, A Laboratory Manual ed 2 1989. Cold Spring Harbor Laboratory Press Cold Spring Harbor
- 17.Feinberg AP, Vogelstein B: A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 1983, 132:6-13 [DOI] [PubMed] [Google Scholar]
- 18.Church GM, Gilbert W: Genomic sequencing. Proc Natl Acad Sci USA 1984, 81:1991-1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fung Y-K, Shackleford G, Braown A, Sanders G, Varmus H: Nucleotide sequence and expression in vitro of cDNA derived from mRNA of int-1, a provirally activated mouse mammary oncogene. Mol Cell Biol 1985, 5:3337-3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Shackleford G: Murine Wnt10a and Wnt10b: cloning and expression in developing limbs, face, and skin of embryos and in adults. Oncogene 1996, 13:1537-1544 [PubMed] [Google Scholar]
- 21.Shackleford G, MacArthur C, Kwan H, Varmus H: Mouse mammary tumor virus infection accelerates mammary carcinogenesis in Wnt-1 transgenic mice by insertional activation of int-2/Fgf-3 and hst/Fgf-4. Proc Natl Acad Sci USA 1993, 90:740-744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacArthur C, Shankar D, Shackleford G: Fgf-8, activated by proviral insertion, cooperates with the Wnt-1 transgene in murine mammary tumorigenesis. J Virol 1995, 69:2501-2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naor D, Sionov R, Ish-Shalom D: CD44: structure, function, and association with the malignant process. Adv Cancer Res 1997, 71:241-319 [DOI] [PubMed] [Google Scholar]
- 24.Li J, Shen H, Himmel KL, Dupuy AJ, Largaespada DA, Nakamura T, Shaughnessy JD, Jr, Jenkins NA, Copeland NG: Leukaemia disease genes: large-scale cloning and pathway predictions. Nat Genet 1999, 23:348-353 [DOI] [PubMed] [Google Scholar]
- 25.Aoyama A, Nagayoshi S, Saga S, Malavasi-Yamashiro J, Yokoi T, Takenaka T, Miyaishi O, Lu J, Imai M, Tomita T, Hoshino M: Genetic resistance to mammary tumorigenesis in a mouse strain with high murine mammary tumor virus expression. Cancer Lett 1987, 36:119-123 [DOI] [PubMed] [Google Scholar]
- 26.Vaage J, Smith G, Asch B, Teramoto Y: Mammary tumorigenesis and tumor morphology in four C3H sublines with or without exogenous mammary tumor virus. Cancer Res 1986, 45:945-977 [PubMed] [Google Scholar]
- 27.Kurachi H, Okamoto S, Oka T: Evidence for the involvement of the submandibular gland epidermal growth factor in mouse mammary tumorigenesis. Proc Natl Acad Sci USA 1985, 82:5940-5943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilgers J, Bentvelzen P: Interaction between viral and genetic factors in murine mammary cancer. Adv Cancer Res 1979, 26:143-195 [DOI] [PubMed] [Google Scholar]
- 29.Teich N, Wyke J, Mak T, Bernstein A, Hardy W: Pathogenesis of retrovirus-induced disease. Weiss R Teich N Varmus H Coffin J eds. RNA Tumor Viruses. 1982, :785-998 Cold Spring Harbor Laboratory Press Cold Spring Harbor [Google Scholar]
- 30.Dunn T: Mammary tumors. Homburger F eds. The Physiopathology of Cancer. 1959, :38-84 Hoeber-Harper New York [Google Scholar]
- 31.Puma J, Fanning T, Young L, Cardiff R: Identification of a unique mouse mammary tumor virus in the BALB/cNIV mouse strain. J Virol 1982, 43:158-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young L, DeOme K, Blair P, Pitelka D, Cardiff R: Development and characterization of the BALB/cNIV mouse strain. Cancer Res 1984, 44:4333-4336 [PubMed] [Google Scholar]
- 33.Cardiff R, Sinn E, Muller W, Leder P: Transgenic oncogene mice. Tumor phenotype predicts genotype. Am J Pathol 1991, 139:495-501 [PMC free article] [PubMed] [Google Scholar]
- 34.Rosner A, Miyoshi K, Landesman-Bollag E, Xu X, Seldin D, Moser A, MacLeod C, Shyamala G, Gillgrass A, Cardiff R: Pathway pathology: histological differences between ErbB/Ras and Wnt pathway transgenic mammary tumors. Am J Pathol 2002, 161:1087-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiPalma F, Holme R, Bryda E, Belyantseva I, Pellegrino R, Kachar B, Steel K, Noben-Trauth K: Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in Waltzer, the mouse model for Usher syndrome type 1D. Nat Genet 2001, 27:103-107 [DOI] [PubMed] [Google Scholar]
- 36.Bolz H, von Brederlow B, Ramirez A, Bryda E, Kutsche K, Nothwang H, Seeliger M, Cabrera M, Vila M, Molina O, Gal A, Kubisch C: Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet 2001, 27:108-112 [DOI] [PubMed] [Google Scholar]
- 37.Turner B, Zhang J, Gumbs A, Maher M, Kaplan L, Carter D, Glazer P, Hurst H, Haffty B, Williams T: Expression of AP-2 transcription factors in human breast cancer correlates with the regulation of multiple growth factor signalling pathways. Cancer Res 1998, 58:5466-5472 [PubMed] [Google Scholar]
- 38.Bosher J, Williams T, Hurst H: The developmentally regulated transcription factor AP-2 is involved in c-erbB-2 overexpression in human mammary carcinoma. Proc Natl Acad Sci USA 1995, 92:744-747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharer J, Kahn R: The ARF-line 2 (ARL2)-binding protein, BART. J Biol Chem 1999, 274:27553-27561 [DOI] [PubMed] [Google Scholar]
- 40.Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Nunez G: Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kB. J Biol Chem 1999, 274:14560-14567 [DOI] [PubMed] [Google Scholar]
- 41.Lowe S, Jacks T, Housman D, Ruley H: Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci USA 1994, 91:2026-2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donehower L, Godley L, Aldaz C, Pyle R, Shi Y, Pinkel D, Gray J, Bradley A, Medina D, Varmus H: Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev 1995, 9:882-895 [DOI] [PubMed] [Google Scholar]
- 43.Naot D, Sionov R, Ish-Shalom D: CD44: structure, function, and association with the malignant process. Adv Cancer Res 1997, 71:241-319 [DOI] [PubMed] [Google Scholar]
- 44.Manfredi M, Lim S, Claffey K, Seyfried T: Gangliosides influence angiogenesis in an experimental mouse brain tumor. Cancer Res 1999, 59:5392-5397 [PubMed] [Google Scholar]
- 45.Ogasawara M, Murata J, Kamitani Y, Hayashi K, Saiki I: Inhibition by vasoactive intestinal polypeptide (VIP) of angiogenesis induced by murine colon 26-L5 carcinoma cells metastasized in liver. Clin Exp Metastasis 1999, 17:283-291 [DOI] [PubMed] [Google Scholar]
- 46.McCawley L, Li S, Benavidez M, Halbleib J, Wattenberg E, Hudson L: Elevation of intracellular cAMP inhibits growth factor-mediated matrix metalloproteinase-9 induction and keratinocyte migration. Mol Pharmacol 2000, 58:145-151 [DOI] [PubMed] [Google Scholar]
- 47.Maruno K, Absood A, Said S: Vasoactive intestinal peptide inhibits human small-cell lung cancer proliferation in vitro and in vivo. Proc Natl Acad Sci USA 1998, 95:14373-14378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim D, Lerner A: Type 4 cyclic adenosine monophosphate phosphodiesterase as a therapeutic target in chronic lymphocytic leukemia. Blood 1998, 92:2484-2494 [PubMed] [Google Scholar]
- 49.Leitges M, Neidhardt L, Haenig B, Herrmann B, Kispert A: The paired homeobox gene Uncx4.1 specifies pedicles, transverse processes and the proximal ribs of the vertebrate column. Development 2000, 127:2259-2267 [DOI] [PubMed] [Google Scholar]
- 50.Mansouri A, Voss A, Thomas T, Yokota Y, Gruss P: Uncx4.1 is required for the formation of the pedicles and proximal ribs and acts upstream of Pax9. Development 2000, 127:2251-2258 [DOI] [PubMed] [Google Scholar]
- 51.Barrantes I, Elia A, Wunsch K, DeAngelis M, Mak T, Rossant J, Conlon R, Gossler A, de la Pompa J: Interaction between Notch signalling and Lunatic fringe during somite boundary formation in the mouse. Curr Biol 1999, 9:470-480 [DOI] [PubMed] [Google Scholar]
- 52.Ronchini C, Capobianco A: Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Mol Cell Biol 2001, 21:5925-5934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dumont E, Fuchs K, Bommer G, Christoph B, Kremmer E, Kempkes B: Neoplastic transformation by Notch is independent of transcriptional activation by RBP-J signalling. Oncogene 2000, 19:556-561 [DOI] [PubMed] [Google Scholar]
- 54.Zhou Q, Wang S, Anderson D: Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 2000, 25:331-343 [DOI] [PubMed] [Google Scholar]
- 55.Lu Q, Yuk D, Alberta J, Zhu Z, Pawlitzky I, Chan J, McMahon A, Stiles C, Rowitch D: Sonic hedgehog-regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 2000, 25:317-329 [DOI] [PubMed] [Google Scholar]
- 56.Wang J, Jani-Sait S, Escalon E, Carroll A, de Jong P, Kirsch I, Aplan P: The t(14;21)(q11.2;q22) chromosomal translocation associated with T-cell acute lymphoblastic leukemia activates the BHLHB1 gene. Proc Natl Acad Sci USA 2000, 97:3497-3502 [DOI] [PMC free article] [PubMed] [Google Scholar]