

Abstract
Porcine membranoproliferative glomerulonephritis type II in piglets of the Norwegian Yorkshire breed is considered the first animal model of human dense deposit disease. Porcine dense deposit disease is caused by the absence of the complement regulator factor H in plasma. Here we report the molecular basis for this absence. Single nucleotide exchanges at position C1590G and T3610G in the coding region of the factor H gene result in amino acid exchanges at nonframework residues L493V and I1166R that are located within SCR 9 and SCR 20, respectively. Apparently the L493V mutation represents a polymorphism whereas the I1166R causes the physiological consequences a block in protein secretion. Expression analysis shows comparable mRNA levels for factor H in liver tissue derived from both affected and healthy animals. In affected piglets, factor H protein is detected in increased amounts in liver cells. Factor H accumulates inside the hepatocytes and is not released as shown by Western blot analysis and immunohistochemistry. These data demonstrate that single amino acid exchanges of two nonframework amino acids either alone or in combination block protein secretion of factor H. This observation is also of interest for other human diseases in which factor H is involved, such as human factor H-associated form of hemolytic uremic syndrome.
Membranoproliferative glomerulonephritis type II (MPGN II) is a relatively rare kidney disease that is characterized by glomerular capillary wall thickening, mesangial cell proliferation, and an increase in mesangial matrix size. Heavy dense deposits of complement are found in the mesangium and along the capillary wall. The lack or inactivation of complement factor H in plasma causes damage in the glomerular membrane of the kidney and can cause MPGN type II, as well as type III, as demonstrated in animals as well as in humans. 1-3
Factor H-deficient pigs of the Norwegian Yorkshire breed serve as a model for MPGN II. 4 The disease is inherited in an autosomal recessive pattern with complete penetrance as shown by mating experiments. 5 Affected animals die of renal failure early in life. The kidneys of such animals show extensive glomerular hypercellularity and profound thickening of the glomerular capillary wall. The glomerular basement membranes are thickened because of large amounts of dense deposits. In addition excessive complement activation is observed in affected piglets, as demonstrated by low plasma C3, elevated levels of plasma terminal complement complex, and massive deposits of complement within the glomerular basement membranes and mesangial matrix. 6 This disease is because of the deficiency of the complement regulator factor H in plasma of the affected piglets as demonstrated by enzyme-linked immunosorbent assay and Western blotting. 7 That factor H deficiency causes the renal defects is further confirmed by transfusion experiments, as substitution of factor H purified from pig plasma, reduced complement activation and resulted in a prolonged survival of the animals. 4 Despite these detailed studies the molecular basis of this factor H deficiency is still unclear.
Deficiencies of factor H have been reported in several cases causing a wide panel of defects ranging from recurrent microbial infections, glomerular effects, and hemolytic uremic syndrome. 8-19 The molecular basis for factor H deficiency has been investigated only in one patient with inherited factor H deficiency and collagen type III glomerulopathy. 15,20 In this case single nucleotide exchanges occurring in one allele causing exchange of C518R and the other allele affecting C941Y cause exchanges of the highly conserved Cys residues within SCR 9 and SCR 16. The absence of essential cysteine residues affects disulfide bond formation and as a consequence the mutated protein is retained in the endoplasmic reticulum and secretion is blocked. 21 Similarly a case of MPGN type II was reported for a patient who developed autoantibodies against the factor H protein at an age older than 30 years. 22 Thus factor H deficiency and inactivation of factor H function by autoreactive antibodies cause related symptoms and glomerulopathy.
The multifunctional and multicomponent protein factor H acts as a central regulator of the complement systems and thus represents an essential part of innate immunity. 23 Factor H is an abundant 150-kd single-chain plasma glycoprotein that is composed of 20 separately folding protein domains, termed “short consensus repeats” (SCR). 24 Factor H is the major fluid phase complement regulator and in human plasma an additional soluble protein exists, the factor H-like protein-1 (FHL-1), also termed reconectin (regulator of complement and fibronectin-like adhesion protein) that is derived from the factor H gene by means of alternative splicing. 25
Here we report the molecular basis of factor H deficiency in pigs of the Norwegian Yorkshire breed. Single nucleotide exchanges of the factor H molecule that cause amino acid exchanges within SCR 9 and SCR 20 are detected. Expression analysis show comparable steady state mRNA levels in liver cells derived from affected and healthy animals. On the protein level, factor H is detected in liver cells, however the protein accumulates intracellularly and is not released. Apparently the mutation in SCR 9 represents a polymorphism, whereas the I1166R mutation causes the physiological effect of a block in protein secretion.
Materials and Methods
Materials
Molecular Biology
Unless otherwise stated reagents were obtained from Sigma (Taufkirchen, Germany) and all labware was from Greiner (Solingen, Germany). Restriction enzymes and Taq DNA polymerase were purchased from Amersham Pharmacia Biotech (Freiburg, Germany) and tissue extraction and lysis buffer PE-LB containing Protease Arrest Inhibitor were from CellConcepts (Umkirch, Germany). SuperScriptII reverse transcriptase and oligo(dT) primer were obtained from Life Technologies (Karlsruhe, Germany). The TOPO-T/A cloning kit was purchased from Invitrogen (Groningen, The Netherlands). The QIAprep miniprep spincolumns, the QIAquick gel extraction kit, and the QIAquick PCR purification kit were obtained from Qiagen (Hilden, Germany). The High Pure RNA tissue kit was purchased from Roche (Mannheim, Germany). The Protran nitrocellulose membrane was obtained from Schleicher & Schuell (Dassel, Germany) and the BigDye terminator kit for sequencing reaction was obtained from Applied Biosystems (Langen, Germany).
Tissues and Sera
Preparation of plasma from factor H-deficient and normal pigs were described earlier. 2 Fresh liver tissues from diseased and healthy animals were snap-frozen in liquid nitrogen immediately after slaughter of the animals and stored at −70°C until used.
Antibodies and Proteins
Production of antiserum anti-pig FH (rabbit) has been described earlier. 4 Anti-human SCR1-4 (rabbit) antiserum was produced by Eurogentec (Seraing, Belgium) using a recombinantly expressed fragment for immunization. Horseradish peroxidase-conjugated secondary antibody anti-rabbit IgG (goat) was obtained from DAKO (Glostrup, Denmark). Fluorescein isothiocyanate-coupled secondary anti-rabbit IgG (goat) antibody was obtained from ICN (Eschwege, Germany). Bicinchoninic acid (BCA) protein assay reagents were obtained from Pierce (Rockford, IL).
Technical Equipment
Sequencing reactions were separated using an ABI PRISM 377 and polymerase chain reactions (PCRs) were performed using a GeneAmp 9700 thermocycler both obtained from Applied Biosystems (Langen, Germany). BCA protein assay was performed in enzyme-linked immunosorbent assay plates and evaluated at 570 nm with a MRX microplate reader purchased from Dynatech (Denkendorf, Germany). The laser microscope system, Leica TCS (Leica, Heidelberg, Germany), was used for confocal microscopy.
Methods
RNA Isolation and Reverse Transcription PCR
Total RNA was isolated using the High Pure tissue RNA kit. Frozen pig liver tissues from homozygous healthy and homozygous factor H-deficient pigs were ground to powder under constant addition of liquid nitrogen using mortar and pestle. Aliquots, representing ∼10 mg of tissue were added to 400 μl of lysis buffer and further homogenized using syringe and needle. An average yield of 5 μg of total RNA was achieved by this method. This procedure integrates a DNAse I digestion step in the RNA isolation process. Reverse transcription was performed using Superscript II reverse transcriptase (RT), oligo(dT) primer, and 5 μg of total RNA. PCR analysis was performed using 1 μl of cDNA as a template or 1 μl of total RNA in RT-minus control reaction. Primers specific for porcine factor H were derived from the published porcine small intestine cDNA library 26 expressed sequence tag (EST) sequence (GenBank accession no. F22851) using EST-5For (CCACCCCCTCAACTCCTCAATGG) and EST-3Rev (GGGTCCAACCAGTGAAAATGATGCA). Additional primers for pig factor H were derived according to the sequence of porcine factor H (GenBank accession no. AJ278470; Hegasy et al, in preparation) pig FH-0For (GCAGAACCTGACACAGTGTTG) and pig FH-7Rev (TGCCAAGTTTAATGAGATGC). Primers specific for β-actin were pβ-actFor (ACGGGCAGGTCATCACCATC) and pβ-actRev (ACACGGAGTACTTGCGCTCG) and hβ-actFor (TGACGGGGTCACCCACACTGTGCCCATCTA) and hβ-actRev (GGCTCCATCCTGGCCTCGCTGTCC). The amplification protocol used was as follows: 95°C for 5 minutes of denaturation, 30 amplification cycles at 95°C for 30 seconds, 58°C for 30 seconds, 72°C for 1 minute, followed by 72°C for 10 minutes. PCR products were resolved on 1% agarose gels.
Amplification, Cloning, and Sequencing of Pig cDNA
Two overlapping fragments were amplified spanning the entire coding domain of pig factor H cDNA, covering the 5′-untranslated region to four bases upstream of the poly(A) tail. A 2241-bp N-terminal fragment was amplified using primer pig FH-0For and primer EST-3Rev, and a 2030-bp C-terminal fragment was amplified using primers EST-5For and pig FH-7Rev. PCR conditions for amplification were as follows: 95°C for 5 minutes of denaturation, 35 amplification cycles of 95°C for 1 minute 30 seconds, 56°C for 1 minute 30 seconds, 72°C for 1 minute 15 seconds, followed by 72°C for 8 minutes. The product was purified with QIAquick PCR purification columns and directly used for sequencing. Both fragments were ligated into TOPO-T/A vector, transformed into One-Shot cells and individual clones were sequenced.
Total Liver Protein Isolation and Analysis
Approximately 100 μg of tissue powder from liver slices obtained from healthy and diseased animals were homogenized in 1.5 ml of tissue extraction buffer PE-LB using an Ultraturrax. Samples were centrifuged at 120,000 × g at 4°C for 1 hour. The supernatant was collected and protein concentrations were determined using the BCA protein assay. The assay was performed in a 96-multiwell plate in triplicate and serial dilutions of bovine serum albumin were used as standard. Assays were incubated at 37°C for 45 minutes and then read at 570 nm.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis and Western Blot Analysis
Approximately equal amounts of total protein extract obtained from liver tissues and sera were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis under nonreducing conditions. Proteins were either visualized by silver staining or electroblotted onto nitrocellulose membrane as described. 27 Membranes were blocked with 5% dried milk in phosphate-buffered saline (PBS) for 30 minutes and incubated overnight either with anti-pig FH (rabbit) or anti-human SCR1-4 (rabbit) antisera both diluted 1:400 in PBS with 2% dried milk. Secondary antibody was a horseradish peroxidase-conjugated anti-rabbit IgG (goat) antibody diluted 1:1000 in PBS and 2% dried milk and incubated for 3 hours. The membranes were developed in chromogenic solution (45 ml PBS, 5 ml methanol, 15 mg 4-chloro-1-naphtol, and 50 μl H2O2).
Immunofluorescence Staining and Confocal Laser Microscopy
Cryosections of liver tissue were obtained from healthy and diseased pigs with a cryostat and transferred to l-lysine-coated glass slides. All further processing was performed in a moist chamber. Cryosections were covered with fixative solution containing 1% formaldehyde and 0.1% glutaraldhyde in PBS for 5 minutes and then washed three times with PBS. Sections were blocked with PBS containing 3% bovine serum albumin for 15 minutes and washed again. Cryosections were incubated overnight at 4°C with anti-pig FH (rabbit) antiserum, diluted 1:80 in PBS (3% bovine serum albumin). As a negative control sections were incubated with PBS (3% bovine serum albumin) in the absence of antiserum. After three washing steps with PBS containing 0.05% Tween, a secondary fluorescein isothiocyanate-coupled anti-rabbit-IgG (goat) antibody diluted 1:50 in PBS (3% bovine serum albumin) was incubated for 1 hour. Sections were washed three times with PBS, air-dried, mounted in anti-fade medium, and covered with coverslips. Slides were kept in the dark at 4°C. Slides were examined by microscopy using a Leica TCS confocal laser-scanning microscope system. Images were acquired with ×10, ×20, and oil immersed ×63 lenses, standard fluorescein isothiocyanate filter settings and without any integration along the z axis. A four-frame-averaging process was performed at a resolution of 512 × 512.
Results
Mutations of the Pig Factor H cDNA
Factor H cDNA was amplified in two fragments using cDNA derived from liver tissue of healthy and diseased animals of the Norwegian Yorkshire breed. Both the N-terminal and C-terminal fragments were sequenced in both directions and the complete sequence was assembled. The sequence was compared to the porcine factor H cDNA sequence (GenBank accession no. AJ278470; Hegasy et al, in preparation) derived from a normal pig, breeding type Deutsches Mastschwein. Comparison of the healthy animal of the Norwegian Yorkshire breed and the Deutsches Mastschwein breed showed a single base exchange G2939A that is silent and does not lead to a change of the amino acid sequence. The same set of primers was used to amplify factor H cDNA from a diseased animal and revealed two mutations: a C to G exchange at position 1590; C1590G and a T to G exchange at position 3610 T3610G (Figure 1) ▶ . Both mutations cause amino acid exchanges at position L493V in SCR 9 and at position I1166R in SCR 20. No double peaks were detected at these positions in the PCR product-sequencing chromatogram. The mutations were confirmed in cloned PCR products from additional amplification reactions. These mutations cause single amino acid exchanges, but apparently do not seem to affect the overall protein structure. Consequently we were interested to further analyze the reasons for the absence of factor H in plasma of diseased animals.
Figure 1.
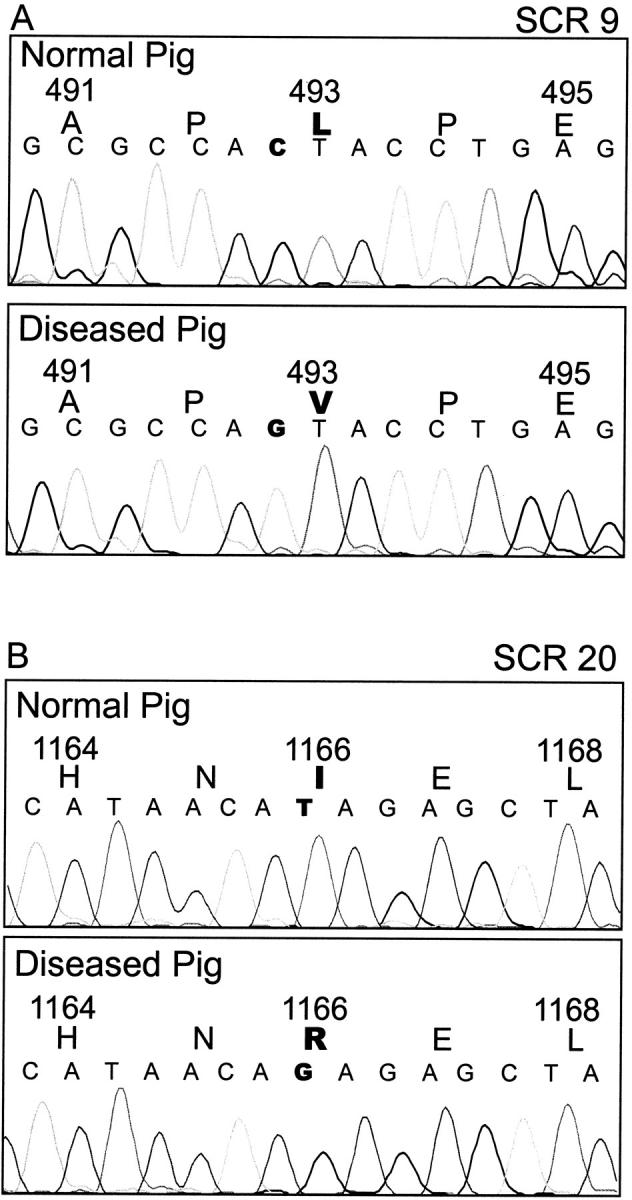
Nucleotide sequence of factor H cDNA products derived from healthy and diseased pigs. cDNA was derived from liver mRNA from healthy and diseased pigs by reverse transcription. A: A C1590G base exchange. This mutation causes an amino acid exchange L493V in SCR 9. B: A T3610G base exchange. This mutation causes an amino acid exchange I1166R in SCR 20. RNA was isolated three times from two different animals, and three cDNA preparations were used for three distinct RT-PCR experiments and in each case the sequences of 6 to 10 clones were determined.
Expression of Factor H mRNA
To confirm expression of the mutated factor H molecules on the RNA level we performed RT-PCR analysis using RNA isolated from liver tissue of both healthy and affected littermates. Homozygosity for plasma factor H deficiency was shown by plasma levels and mating experiments as described earlier. 2,3 Primers specific for factor H generated the predicted 291-bp fragment, which was detected with similar intensities in healthy and affected animals (Figure 2 ▶ , compare lanes 1 and 5). A similar amount of cDNA was used for amplification as demonstrated by the two β-actin bands of 288 bp and 660 bp, which were generated by two different sets of primers. Thus in healthy and diseased animals comparable factor H mRNA expression levels are demonstrated.
Figure 2.

RNA expression in healthy and factor H-deficient animals. RNA isolated from pig liver tissue derived from healthy (lanes 1 to 4) and diseased pigs (lanes 5 to 8) was reverse-transcribed and the corresponding cDNAs were used as templates for PCR amplification using pig factor H-specific primers EST-5For and EST-3Rev and two sets of β-actin-specific primers. The specific factor H product is shown for healthy (lane 1) and for diseased animals (lane 5). The two β-actin-specific products for healthy (lanes 2 and 3) and diseased animals (lanes 6 and 7) show that equal amounts of cDNA were used. RT-minus control reactions generated no products (lanes 4 and 8). The mobility of DNA size markers in bp is indicated.
Factor H Protein Levels in Plasma and Liver Extract
To further confirm expression on the protein level we compared factor H protein levels in plasma and liver tissue of both healthy and diseased animals by Western blotting. This approach shows the presence of the 150-kd factor H protein in plasma derived from healthy animals (Figure 3A ▶ , lane 1) and the absence of this protein in plasma of factor H-deficient pigs (Figure 3A ▶ , lane 2). However, a smaller protein of ∼28 kd is detected. Liver is the major source for plasma factor H and consequently we compared factor H levels in liver tissues. In tissue derived from healthy animals factor H was detected as a single 150-kd band (Figure 3A ▶ , lane 3) and similarly a protein of identical mobility was present in tissue derived from diseased animals (Figure 3A ▶ , lane 4). Apparently the intensity of the 150-kd factor H band is higher in tissue derived from diseased animals as compared to control animals. In addition a protein of higher molecular mass is detected with this antiserum. To further confirm these results identical blots were developed using an antiserum specific for the N-terminal region of human factor H. This approach shows again the absence of factor H in plasma of diseased animals and confirms the presence of this protein in liver tissue derived from affected animals (Figure 3B ▶ , lanes 2 and 4). The data are representative, as similar or identical amounts of protein are loaded as confirmed by silver staining (Figure 3C) ▶ and BCA protein assay (data not shown). These experiments show that mutated factor H is synthesized in the liver of diseased animals, and suggest a block in protein secretion that causes intracellular accumulation of the protein.
Figure 3.
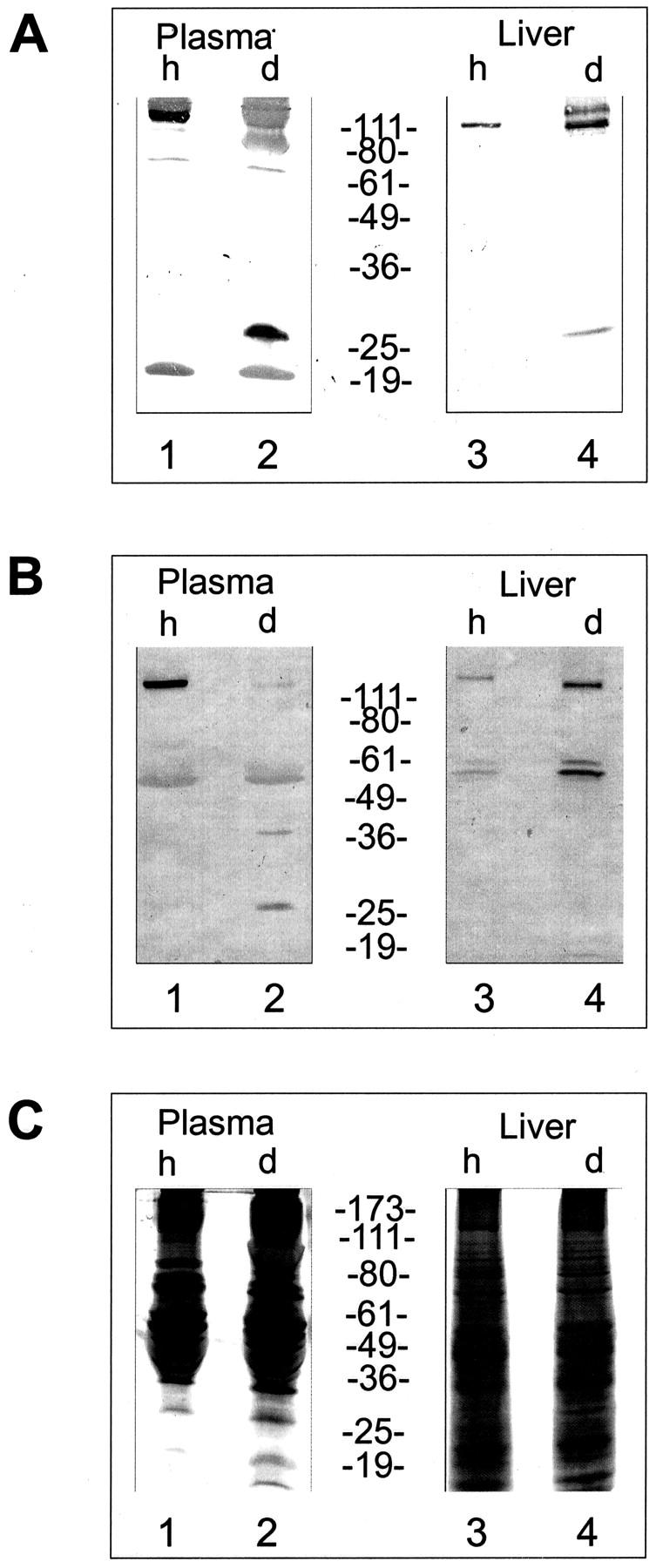
Protein expression levels in plasma and liver derived from healthy and diseased pigs. Plasma or liver protein extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nylon membranes. The corresponding blots were analyzed with antiserum raised against pig (A) or human (B) factor H. Plasma derived from healthy pigs is shown in lane 1, that of deficient pigs in lane 2. Protein extract derived from liver tissue of healthy animals is shown in lane 3 and of affected factor H-deficient animals is shown in lane 4. Comparable amounts of protein were loaded as indicated by silver staining (C). The mobility of the marker proteins in kd is indicated.
Visualization of Factor H by Immunofluorescence
Expression analysis showing intracellular synthesis of factor H in diseased animals and the absence of the protein in plasma suggests a defect in protein secretion. Immunohistochemistry was used to assay the presence and distribution of factor H in liver tissue derived from healthy and diseased animals. Fluorescence signals were visualized by confocal laser microscopy. Tissue derived from healthy animals showed the presence of factor H particularly in the septa and the portal triad (Figure 4A) ▶ but only a low intensity of staining was detected within the hepatic lobule. Staining intensity of the parenchyma was generally low and appeared to decrease with distance from the central vein (Figure 4B) ▶ . Rather little or no protein was detectable intracellularly (Figure 4C) ▶ . This pattern of distribution is clearly in agreement with that of a secreted protein. Staining reactions are considered specific because no signal was detected in the absence of the first antibody (data not shown).
Figure 4.
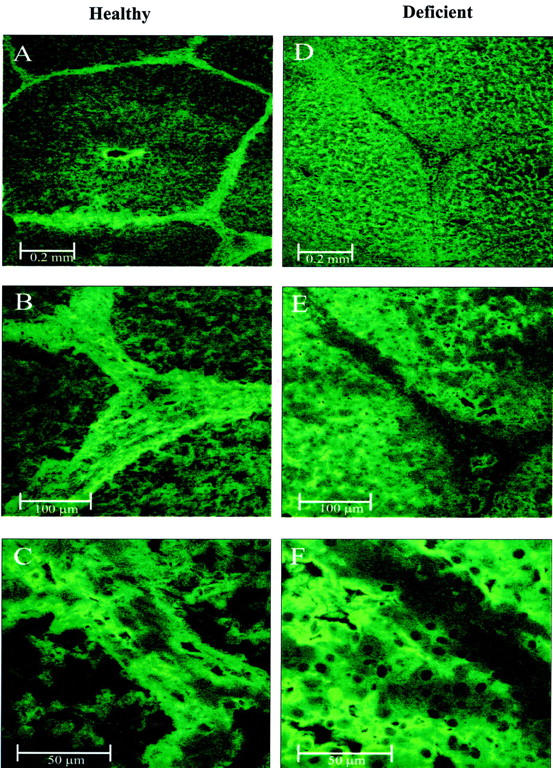
Confocal images of liver tissue derived from healthy and factor H-deficient pigs. Liver tissues were stained with antiserum specific for pig factor H and a secondary fluorescein isothiocyanate-coupled antibody. Fluorescence staining was visualized by confocal laser microscopy. Images on left (A–C) show sections obtained from healthy animals and images on the right (D–F) show sections obtained from diseased animals. A: Septa, a portal triad, and a central vein. B and C: Higher magnifications of the portal triad shown in the lower right corner of A. D: Septa and a portal triad of a diseased animal. E and F: Higher magnifications of the triad in the center of D.
Tissue derived from diseased animals showed strong parenchyma staining, but no staining of the connective tissue surrounding the hepatic lobule (Figure 4D) ▶ . In higher magnification, intracellular staining and absence of extracellular protein can be seen (Figure 4E) ▶ . Hepatocytes were stained strongly intracellularly, but the nucleus was omitted from staining (Figure 4F) ▶ . A clearly different distribution of factor H is detected in tissue derived from diseased and healthy animals. This distribution pattern is in agreement with the biochemical data and confirms synthesis and intracellular presence of factor H and a block in protein secretion of the mutated protein.
Discussion
Factor H-deficient pigs of the Norwegian Yorkshire breed that lack complement factor H in plasma are considered an ideal animal model to study the pathophysiology of MMPGN II. These factor H-deficient piglets have been studied in great detail, but the molecular cause of the disease has not yet been described. Here we report two nucleotide sequence alterations in the factor H gene of the affected pigs: a C1590G and a T3610G substitution. Both mutations cause single amino acid exchanges within SCR 9 position L493V and within SCR 20 position I1166R. Based on sequence alignment it appears that the mutation at position 493 represents a polymorphic change. All other species, such as human, 24 mouse, 28 rat, 29 cow, 30 and the sand bass 31 have a Val (V) at this particular position. In contrast, isoleucine (I) residue at position 1166 is conserved in pig, human, mouse, rat, and sand bass. Thus the amino acid exchange at position 1166 is the functional relevant alteration. This change occurs within SCR 20 of the protein, a domain that is central for factor H function. 32,33 The replacement of a neutral isoleucine residue with a positively charged amino acid could affect factor H processing or activity. In diseased animals factor H expression occurs on the mRNA level, as demonstrated by RT-PCR analysis. In addition in liver cells of diseased animals factor H protein is present and enriched intracellularly. Thus the I1166R mutation, which alters a nonframework amino acid appears to cause a block in protein release and results in intracellular accumulation of the mutated protein. This study provides the explanation for the molecular defect causing FH deficiency in Norwegian Yorkshire pigs and is the first report demonstrating that exchanges of nonframework amino acid residues cause a block in protein secretion. This result is also of interest for the mechanisms of other diseases in which factor H is involved, eg, hemolytic uremic syndrome (HUS). A factor H-associated form of HUS has been described in which patients show mutations in the factor H gene. 34-38
The relevance of framework Cys residues has also been confirmed for the SCR-containing factor XIIIb protein where mutations of essential Cys residues similar to that reported for human factor H, cause a block of protein secretion. 39 Framework Cys residues form disulfide bonds, which are essential for processing and function. Apparently in the mutations reported here for the pigs nonframework residues also affect the release of factor H.
Hereditary factor H deficiency has been reported in several human cases causing a wide panel of defects ranging from recurrent microbial infections, glomerular effects, and HUS. Pathologies belonging to the group of primary glomerular lesions has been reported in six human cases. 10,11,15 The renal pathologies of three cases have been classified to membranoproliferative glomerulonephritis, 11 two cases to atypical intramembranous dense deposit disease, 10 and one case to collagen type III glomerulopathy. 15 For the patient who suffered from collagen type III glomerulopathy, a 2-year-old Native American, the genetic defect and the molecular pathophysiology have been described. Each factor H allele shows a mutation in a conserved framework cysteine residue, occurring at position [C518R] within SCR 9 and [C941Y] within SCR 16. 20,21 Corresponding mutant proteins were synthesized and both mutant forms were retained inside the cells in the endoplasmic reticulum. 20,21 The half life of the mutants was increased and both proteins degraded relative slowly. This result has been interpreted that disruption of essential framework Cys residues affect disulfide bond formation, block protein secretion, and cause intracellular protein accumulation. A role of factor H for glomerular basement membrane alteration is concluded from a patient who developed autoantibodies to factor H. 40 In this case the disease occurred later in life and correlated with the appearance of autoantibodies. These antibodies bind to SCR 3, which includes the complement regulatory region of factor H and consequently affects factor H function. Apparently this inactivation allows unrestricted complement activation, which fully explains the low C3 and the high terminal complement components in plasma of factor H-deficient individuals.
In addition a factor H-associated form of HUS has emerged, which is because of mutations in the factor H gene. 34-36 The vast majority of these patients show heterozygous mutations. So far intracellular protein accumulation has not been assayed in any patient that has a mutated factor H gene. The effect of the nonframework amino acid mutations characterized in this work for the factor H-deficient pigs gives a new explanation for the defective pathway in factor H-associated HUS and opens new ways for therapeutic approaches to the associated diseases. 41
Footnotes
Address reprint requests to Peter F. Zipfel, Department of Infection Biology, Hans Knoell Institute for Natural Products Research, Beutenbergstr. 11a, D-07745 Jena, Germany. E-mail: zipfel@pmail.hki-jena.de.
References
- 1.Ault BH: Factor H and the pathogenesis of renal diseases. Pediatr Nephrol 2000, 14:1045-1053 [DOI] [PubMed] [Google Scholar]
- 2.West CD: Nephritic factors predispose to chronic glomerulonephritis. Am J Kidney Dis 1994, 24:956-963 [DOI] [PubMed] [Google Scholar]
- 3.West CD, McAdams AJ: Glomerular paramesangial deposits: association with hypocomplementemia in membranoproliferative glomerulonephritis types I and III. Am J Kidney Dis 1998, 31:427-434 [DOI] [PubMed] [Google Scholar]
- 4.Hogasen K, Jansen JH, Mollnes TE, Hovdenes J, Harboe M: Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiency. J Clin Invest 1995, 95:1054-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogasen K, Jansen JH, Harboe M: Eradication of porcine factor H deficiency in Norway. Vet Rec 1997, 140:392-395 [DOI] [PubMed] [Google Scholar]
- 6.Jansen JH: Porcine membranoproliferative glomerulonephritis with intramembranous dense deposits (porcine dense deposit disease). APMIS 1993, 101:281-289 [DOI] [PubMed] [Google Scholar]
- 7.Jansen JH, Hogasen K, Mollnes TE: Extensive complement activation in hereditary porcine membranoproliferative glomerulonephritis type II (porcine dense deposit disease). Am J Pathol 1993, 143:1356-1365 [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson RA, Winterborn MH: Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin Exp Immunol 1981, 46:110-119 [PMC free article] [PubMed] [Google Scholar]
- 9.Wyatt RJ, Julian BA, Weinstein A, Rothfield NF, McLean RH: Partial H (beta 1H) deficiency and glomerulonephritis in two families. J Clin Immunol 1982, 2:110-117 [DOI] [PubMed] [Google Scholar]
- 10.Levy M, Halbwachs-Mecarelli L, Gubler MC, Kohout G, Bensenouci A, Niaudet P, Hauptmann G, Lesavre P: H deficiency in two brothers with atypical dense intramembranous deposit disease. Kidney Int 1986, 30:949-956 [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Larrea C, Dieguez M, Enguix A, Domiguez O, Marin B, Gomez F: A familial deficiency of complement factor H. Biochem Soc Trans 1987, 15:648-649 [Google Scholar]
- 12.Brai M, Misiano G, Maringhini S, Cutaja I, Hauptmann G: Combined homozygous factor H and heterozygous C2 deficiency in an Italian family. J Clin Immunol 1988, 8:50-56 [DOI] [PubMed] [Google Scholar]
- 13.Nielsen HE, Christensen KC, Koch C, Thomsen BS, Heegaard NH, Tranum-Jensen N: Hereditary, complete deficiency of complement factor H associated with recurrent meningococcal disease. Scand J Immunol 1989, 30:711-718 [DOI] [PubMed] [Google Scholar]
- 14.Pichette V, Querin S, Schurch W, Brun G, Lehner-Netsch G, Delage JM: Familial hemolytic-uremic syndrome and homozygous factor H deficiency. Am J Kidney Dis 1994, 24:936-941 [DOI] [PubMed] [Google Scholar]
- 15.Vogt BA, Wyatt RJ, Burke BA, Simonton SC, Kashtan CE: Inherited factor H deficiency and collagen type III glomerulopathy. Pediatr Nephrol 1995, 9:11-15 [DOI] [PubMed] [Google Scholar]
- 16.Fijen CA, Kuijper EJ, Te Bulte M, van de Heuvel MM, Holdrinet AC, Sim RB, Daha MR, Dankert J: Heterozygous and homozygous factor H deficiency states in a Dutch family. Clin Exp Immunol 1996, 105:511-516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohali M, Shalev H, Schlesinger M, Katz Y, Kachko L, Carmi R, Sofer S, Landau D: Hypocomplementemic autosomal recessive hemolytic uremic syndrome with decreased factor H. Pediatr Nephrol 1998, 12:619-624 [DOI] [PubMed] [Google Scholar]
- 18.Rougier N, Kazatchkine MD, Rougier JP, Fremeaux-Bacchi V, Blouin J, Deschenes G, Soto B, Baudouin V, Pautard B, Proesmans W, Weiss E, Weiss L: Human complement factor H deficiency associated with hemolytic uremic syndrome. J Am Soc Nephrol 1998, 9:2318-2326 [DOI] [PubMed] [Google Scholar]
- 19.Taylor CM: Hemolytic-uremic syndrome and complement factor H deficiency: clinical aspects. Semin Thromb Hemost 2001, 27:185-190 [DOI] [PubMed] [Google Scholar]
- 20.Ault BH, Schmidt BZ, Fowler NL, Kashtan CE, Ahmed AE, Vogt BA, Colten HR: Human factor H deficiency. Mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem 1997, 272:25168-25175 [DOI] [PubMed] [Google Scholar]
- 21.Schmidt BZ, Fowler NL, Hidvegi T, Perlmutter DH, Colten HR: Disruption of disulfide bonds is responsible for impaired secretion in human complement factor H deficiency. J Biol Chem 1999, 274:11782-11788 [DOI] [PubMed] [Google Scholar]
- 22.Meri S, Koistinen V, Miettinen A, Tornroth T, Seppala IJ: Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis. J Exp Med 1992, 175:939-950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zipfel PF, Hellwage J, Friese MA, Hegasy G, Jokiranta ST, Meri S: Factor H and disease: a complement regulator affects vital body functions. Mol Immunol 1999, 36:241-248 [DOI] [PubMed] [Google Scholar]
- 24.Ripoche J, Day AJ, Harris TJ, Sim RB: The complete amino acid sequence of human complement factor H. Biochem J 1988, 249:593-602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zipfel PF, Jokiranta TS, Hellwage J, Koistinen V, Meri S: The factor H protein family. Immunopharmacology 1999, 42:53-60 [DOI] [PubMed] [Google Scholar]
- 26.Wintero AK, Fredholm M, Davies W: Evaluation and characterization of a porcine small intestine cDNA library: analysis of 839 clones. Mamm Genome 1996, 7:509-517 [DOI] [PubMed] [Google Scholar]
- 27.Skerka C, Hellwage J, Weber W, Tilkorn A, Buck F, Marti T, Kampen E, Beisiegel U, Zipfel PF: The human factor H-related protein 4 (FHR-4). A novel short consensus repeat-containing protein is associated with human triglyceride-rich lipoproteins. J Biol Chem 1997, 272:5627-5634 [DOI] [PubMed] [Google Scholar]
- 28.Kristensen T, Tack BF: Murine protein H is comprised of 20 repeating units, 61 amino acids in length. Proc Natl Acad Sci USA 1986, 83:3963-3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demberg T, Pollok-Kopp B, Gerke D, Gotze O, Schlaf G: Rat complement factor H: molecular cloning, sequencing and quantification with a newly established ELISA. Scand J Immunol 2002, 56:149-160 [DOI] [PubMed] [Google Scholar]
- 30.Soames CJ, Day AJ, Sim RB: Prediction from sequence comparisons of residues of factor H involved in the interaction with complement component C3b. Biochem J 1996, 315:523-531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kemper C, Zipfel PF, Gigli I: The complement cofactor protein (SBP1) from the barred sand bass (Paralabrax nebulifer) mediates overlapping regulatory activities of both human C4b binding protein and factor H. J Biol Chem 1998, 273:19398-19404 [DOI] [PubMed] [Google Scholar]
- 32.Prodinger WM, Hellwage J, Spruth M, Dierich MP, Zipfel PF: The C-terminus of factor H: monoclonal antibodies inhibit heparin binding and identify epitopes common to factor H and factor H-related proteins. Biochem J 1998, 331:41-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S: Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J Biol Chem 2000, 275:27657-27662 [DOI] [PubMed] [Google Scholar]
- 34.Caprioli J, Bettinaglio P, Zipfel PF, Amadei B, Daina E, Gamba S, Skerka C, Marziliano N, Remuzzi G, Noris M: The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol 2001, 12:297-307 [DOI] [PubMed] [Google Scholar]
- 35.Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, Tielemans CL, Goodship JA, Goodship TH: Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet 2001, 68:485-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Caballero D, Gonzalez-Rubio C, Gallardo ME, Vera M, Lopez-Trascasa M, Rodriguez DC, Sanchez-Corral P: Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet 2001, 68:478-484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zipfel PF: Hemolytic uremic syndrome: how do factor H mutants mediate endothelial damage? Trends Immunol 2001, 22:345-348 [DOI] [PubMed] [Google Scholar]
- 38.Taylor CM: Complement factor H and the haemolytic uraemic syndrome. Lancet 2001, 358:1200-1202 [DOI] [PubMed] [Google Scholar]
- 39.Hashiguchi T, Ichinose A: Molecular and cellular basis of deficiency of the b subunit for factor XIII secondary to a Cys430-Phe mutation in the seventh Sushi domain. J Clin Invest 1995, 95:1002-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jokiranta TS, Solomon A, Pangburn MK, Zipfel PF, Meri S: Nephritogenic lambda light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol 1999, 163:4590-4596 [PubMed] [Google Scholar]
- 41.Remuzzi G, Ruggenenti P, Codazzi D, Noris M, Caprioli J, Locatelli G, Gridelli B: Combined kidney and liver transplantation for familial haemolytic uraemic syndrome. Lancet 2002, 359:1671-1672 [DOI] [PubMed] [Google Scholar]