

Abstract
Early-onset cataract and Alzheimer’s disease occur with high frequency in Down syndrome (trisomy 21), the most common chromosome duplication in human live births. Previously, we used in vivo and lens organ culture models to demonstrate Alzheimer pathophysiology in oxidative stress-related lens degeneration. Currently, well-characterized Alzheimer transgenic mouse models are used to extend these findings. Here, we report on mice carrying a complete copy of a wild-type human AβPP (hAβPP) gene from the Down syndrome critical region on chromosome 21. hAβPP mice produce fiber cell membrane defects similar to those described in human cataracts and increased age-related lens degeneration. hAβPP expression and mRNA alternative splicing in human and mouse lens and cornea favor longer, potentially more amyloidogenic forms. Endogenous mouse AβPP expression is increased in transgenic lenses, consistent with the cycle of oxidative stress proposed in the mechanism of Alzheimer pathophysiology. Alternative splicing previously designated as neuron-specific occurs in human lens and cornea, and is maintained by hAβPP expressed in mouse tissues. These present data implicate AβPP in fiber cell formation and in early-onset cataracts in Down syndrome. Finally, our findings provide further support for our hypothesis that Alzheimer pathophysiology contributes to the cataract formation that is increasing in the aging population.
Evidence indicates that Alzheimer pathophysiology contributes to the loss of mental function and visual clarity that can occur during aging. In trisomy 21 (Down Syndrome, DS), a condition with features of premature aging, virtually all individuals develop Alzheimer’s disease (AD) in their fifth decade 1 and a high percentage develop cataracts beginning in childhood. 2,3 Two factors have been cited that implicate Alzheimer precursor protein (AβPP) in DS Alzheimer pathophysiology. First, the AβPP gene is located within the DS critical region of human chromosome 21 and mouse chromosome 16. 4,5 Second, AβPP is not normally maximally expressed and therefore susceptible to gene dosage effects. 4 In addition, Alzheimer-related presenilin and co-expressed Notch receptor proteins play a role at most, if not all, stages of eye development in Drosophila and vertebrates. 6,7
In DS brain tissue, AβPP expression is 4- to 5-fold greater than normal, rather than the 50% expected from one extra gene copy. 8 This is consistent with an oxidative stress mechanism for Alzheimer pathophysiology by contributing to AβPP gene induction and further augmenting expression. 9,10 Alzheimer pathophysiology is linked with physiological oxidative stress principally due to the increased formation of β-amyloid (Aβ) peptides. Amyloid disease proteins like Aβ are increasingly understood as producing deleterious oxidative effects via interactions with metal ions. 11-13 For example, human Aβ is more deleterious than mouse Aβ due to additional metal-binding histidine residues. Transition metals, in particular copper, influence aggregation and confer superoxide dimutase (SOD) enzyme activity on amyloid disease proteins leading to an inappropriate production of hydrogen peroxide. 11-14 Moreover, Aβ turnover is on the order of 3 hours in brain tissues, 15 highlighting the need for regulation of AβPP expression and Aβ formation. These factors contribute to a significant elevation of AβPP and Aβ in DS brain tissue. In addition to expression levels, AβPP mRNA alternative splicing is affected in DS and AD, as well as in normal aging, such that longer transcripts and protein isoforms linked with increased deleterious Aβ production are favored. 16-19
Early-onset AD also occurs in non-trisomy individuals, however at a much lower rate. The majority of gene mutations linked with familial early-onset AD occur in presenilin genes that mediate γ-secretase (protease) AβPP cleavage and increase Aβ. 20,21 Less frequent mutations occur in AβPP itself near Aβ proteolytic cleavage sites, and accessory genes including apolipoproteins have been identified that predispose one to AD. 22 In addition to AβPP proteolysis, presenilins have a broader role that includes Notch and receptor tyrosine kinase proteolytic processing. 23
AβPP has a diverse set of biological activities ascribed to its various functional protein domains These include a putative transcription activator domain, 24 interaction site for JNK signaling proteins, 25 and a non-amyloidogenic copper-binding domain. 26 Human and mouse AβPP genes contain 18 exons, of which exons 7, 8, and 15 are alternatively spliced. The presence of exons 7 and 15 in mature AβPP mRNA correlates with increased Aβ production and pathophysiology in vivo and in vitro. 18,19 Exon 7 encodes a KPI domain implicated in cell differentiation, 27 and exon 15 disrupts a chondroitin sulfate modification site for forming appican 28 that is linked with protein sorting, cell adhesion, and axon sprouting in neurons. 28,29
In mammalian lenses, AβPP, Aβ, presenilins, and Notch receptors are expressed and processed in equatorial epithelial and cortical fiber cells. 10,30-33 Previously, we demonstrated increased levels of AβPP and Aβ in cortical fiber cells in cultured intact rat and monkey lenses exposed to oxidative stress. Concurrent with higher AβPP and Aβ levels we demonstrated the activation of classic oxidative stress-related cell signaling pathways and AP-1 (Jun/Fos) transcription factor binding to cognate DNA sites. 10 In another study, systemic oxidative stress was produced using a thiamine (vitamin B1) deprivation regimen in mice that models Wernike-Korsakoff encephalopathy and produces Alzheimer-associated brain pathology. There we demonstrated lens fiber cell degeneration with locally increased distributions of AβPP, Aβ, and presenilin proteins. 32
In the present study we examine lenses from transgenic mice that model the AβPP gene dosage imbalance that occurs in DS. Human AβPP (hAβPP) transgenic mice were engineered by introducing a copy of the yeast artificial chromosome B142F9 containing ∼400 kilobases of DNA from the wild-type human AβPP locus on human chromosome 21 into mouse chromosomes. 34-36 This study describes hAβPP mice 34 (gift of B. Lamb, Cleveland, OH) that carry one copy of the hAβPP gene locus with all exon coding sequences, all intron intervening sequences, and much native 5′ promoter and 3′ flanking DNA, allowing for alternative hAβPP transcript splicing in mouse tissues. We describe changes in morphology and increased age-related fiber cell degeneration in hAβPP transgenic mouse lenses that suggest a role for hAβPP gene dosage effects in the high levels of early-onset cataract observed in DS individuals.
Materials and Methods
Transgenic Animals and Tissue Preparation
hAβPPtg/tg transgenic mice 34 and age-matched wild-type C57-black mice were obtained from Charles River Laboratories (Wilmington, MA). Human eye research donor material was obtained after retina removal (gift of M. Zarbin, Newark, NJ). Histological sections were prepared by members of the Ocular Services Branch of the National Eye Institute (NEI) (Bethesda, MD) from enucleated eyes fixed in 4% paraformaldehyde (Fisher, Pittsburgh, PA) in phosphate-buffered saline (pH 7.4) and embedded in paraffin. 10 RNA and protein from eye tissues were solubilized in Trizol (Invitrogen, Carlsbad, CA) or “strong lysis buffer” (Cytosignal, Mamhead, UK) respectively. Lens fiber cell structure in 12 wt and 12 hAPPtg/tg lenses was observed in paraffin thin sections stained with hematoxylin and eosin using standard blue/green immunofluorescence filters. Membrane organization is visible by fluorescence microscopy due to intrinsic eosin fluorescence, a fluorescein-related dye.
Analysis of AβPP Expression and Alternative Splicing
cDNA was produced from total RNA using AMV reverse transcriptase (Invitrogen) and polydT oligonucleotide primers. The resultant cDNAs were amplified with PCR kits (Invitrogen) using the following 5′ to 3′ primers. Mouse glyceraldehyde dehydrogenase (GAPDH): TCCACCACCCTGTTGCTGTAGC, CCACAGTCCATGCCATCACTGC; mouse AβPP exons 6–9: AGTAGAAGTCGCCGAAGAGGAG, CTCGTCCCCGGGTGTCTCCAGG; mouse AβPP exons 13–17: TGCTCTACAATGTCCCTGCGG, ACCATGAGTCCGATGATGGCG; human AβPP exons 6–9: AAGTAGTAGAAGTAGCAGAG, ATTCTCATCCCCAGGTGTCTC; and human AβPP exons 13–17: TACAACGTGCCTGCAGTGGCC, AAGGTGATGACGATCACTGTC. Amplified products from at least four lenses were resolved on agarose gels, purified, and their sequences determined to confirm identifications (Molecular Resource Facility, UMDNJ-New Jersey Medical School).
Immunopreciptiation and Western Blotting
Total lens protein solubilized in “strong lysis buffer” was immunopreciptitated with pan-specific mouse monoclonal AβPP antibody, clone 22C11, using an Immunocatcher kit (Cytosignal). Subsequently, immunoprecipitates were resolved on acrylamide gels and blotted to filter paper. Identical blots were probed with pan-specific 22C11 or KPI-specific (Sigma, St. Louis, MO) antibodies to qualitatively assess AβPP isoforms. Aβ was detected using an affinity purified human Aβ(1–16) specific monoclonal antibody (Biosource, Camarillo, CA) on Western blots of synthetic human Aβ peptides (Sigma) and total lens proteins from wt and hAβPP mice resolved on 20% SDS-PAGE gels with 5% urea included in all buffers.
Results
To first establish hAβPP transgene expression in mouse lenses and to compare the native human AβPP mRNA splicing pattern with the hAβPP transgene, we purified total RNA from ocular tissues for analysis by RT-PCR. Figure 1 ▶ demonstrates the pattern of mRNA splicing for native AβPP expression in wt mouse (Figure 1A) ▶ and human (Figure 1B) ▶ tissues. Using PCR primers corresponding to exons 6 and 9, we determined that the predominant forms of alternatively spliced mRNAs in wt mouse and human lens and cornea contain exon 7 +/− exon 8 which encode AβPP 751 and 770 amino acid proteins. In contrast, the major AβPP mRNA expressed in brain (present data and reference 37) and retina is the shorter 695 amino acid encoding transcript (exon 6 to 9 splicing).
Figure 1.

Human AβPP transgene expression and alternative mRNA splicing in mouse ocular tissues. A: RT-PCR detection of mouse AβPP using exon 6 and 9 primers in mouse tissues. B: RT-PCR analysis of human AβPP exons 6 to 9 in human lens and cornea. C: hAβPP transgene expression in hAβPPtg/tg mouse tissues. D: Duplicate immunoprecipitations of total AβPP protein from hAβPPtg/tg mouse lenses subsequently probed with anti-KPI domain (exon 7) or pan-AβPP-specific antibodies to detect AβPP isoforms. E: Human Aβ peptides detected in hAβPPtg/tg and wt mouse lenses. F and G: Exon 15 alternative splicing in human, transgenic mouse, and wt mouse lenses using exon 13 and 17 primers. GAPDH-amplified products are included for semi-quantitative comparison.
The amplified products shown in Figure 1C ▶ demonstrates hAβPP transgene expression in mouse tissues. The alternative splicing pattern for hAβPP expressed in mouse tissues appears quite similar to the pattern observed in human eyes. The longer human AβPP transcripts again predominate in transgenic lens and cornea, and higher levels of hAβPP 695 transcripts were present in transgenic mouse retina. As expected, no hAβPP mRNA was present in wt mouse lenses. When we examined AβPP protein expression in hAβPPtg/tg mouse lenses we found the protein expression pattern corresponded well with the mRNA splicing patterns we identified. Figure 1D ▶ demonstrates that predominantly longer forms of AβPP protein are present in hAβPPtg/tg mouse lenses using KPI/exon7- and pan-specific antibodies. In addition, human Aβ was present in transgenic but not wt mouse lenses (Figure 1E) ▶ .
A second region of AβPP encoded by exons 14, 15, and 16 also undergoes alternative splicing and is linked with increased Alzheimer pathophysiology. 17 The junction of exons 14 and 16 encodes a chondroitin sulfate addition site for producing appican. 28,29 We detected species differences between the human and mouse in the splicing of AβPP exon 15 in lens. Moreover, the human and mouse splicing patterns were separately maintained in transgenic animals that co-express the mouse and human genes (Figure 1F) ▶ . Normal human lens and cornea predominantly express AβPP with exon 15 and this was also the case in transgenic eye tissues. This exon 15 splicing pattern was previously identified as unique to neuronal cells in humans. 17 In contrast, mouse AβPP transcripts +/− exon 15 are present in wt and hAβPPtg/tg lenses at comparable levels. These results indicate that nucleotide sequences residing at the human and mouse exon 14, 15, and 16 intron/exon boundaries are recognized and used differently by lens RNA splicing machinery in human or mouse lenses.
Alzheimer pathophysiology can be initiated by a variety of factors including oxidative stress. In lens organ cultures oxidative stress stimulates AβPP expression and increases Aβ peptides. 10 In turn, Aβ can contribute additional oxidative stress to form a “vicious cycle.” 38 We used semi-quantitiative RT-PCR to begin examining one aspect of this model in the lens (Figure 1, F and G) ▶ . Mouse AβPP transcripts assayed in RT-PCR reactions using wt and hAβPPtg/tg transgenic mouse lenses are greater in transgenic lenses relative to GAPDH expression.
Lens Fiber Cell Defects and Age-Related Degeneration in hAβPP Mice
Consistent phenotypic differences were observed in hAβPPtg/tg mice. In contrast to the regular fiber cell organization and membrane morphology observed in wt lenses, individual fiber cells in hAβPPtg/tg mice are irregularly shaped and the fiber cell arrays are misaligned. Fluorescence photomicrographs of cross-sections of lens fiber cells are presented in Figure 2 ▶ . The lens regions shown in each of these photomicrographs are located within the lens transition zone, where fiber cell organelle loss occurs, and outside the lens center referred to as the fetal nucleus. Fibers in the fetal nucleus are formed during embryogenesis. Fiber cell morphology in a wt 16- month-old mouse lens is shown in Figure 2, A and E ▶ . Figure 2B ▶ demonstrates the irregular shape and alignment of lens fibers cell arrays in hAβPPtg/tg mouse lenses of the same age. In addition to changes in fiber cell shape and alignment, numerous bodies with circular profiles in thin section were present in all transgenic lens histological sections. The perimeters of these objects have the same staining properties as surrounding fiber cell membranes suggesting that they are also membrane-bound. Panels C and D in Figure 2 ▶ demonstrate that these objects retain their circular profiles in sections cut at increasingly oblique angles, consistent with these objects being spherical in three dimensions. However, these membrane bodies may also represent cross-sections of finger-like projections described by others in senescent human and monkey lenses, 39,40 and further experiments will determine whether these objects may have a multilamellar makeup similar to those demonstrated in the fetal nucleus of human nuclear cataracts. 41 Numerous circles were present in hAβPPtg/tg lenses examined in transgenic mice 6 months or older and not in wt controls. Twelve hAβPPtg/tg and twelve 16-month-old control lenses were examined. Membrane circles in hAβPPtg/tg mouse lens fibers are ∼1 to 5 μm in diameter, a dimension that would allow them to interact with visible light and potentially contribute to lens opacification.
Figure 2.
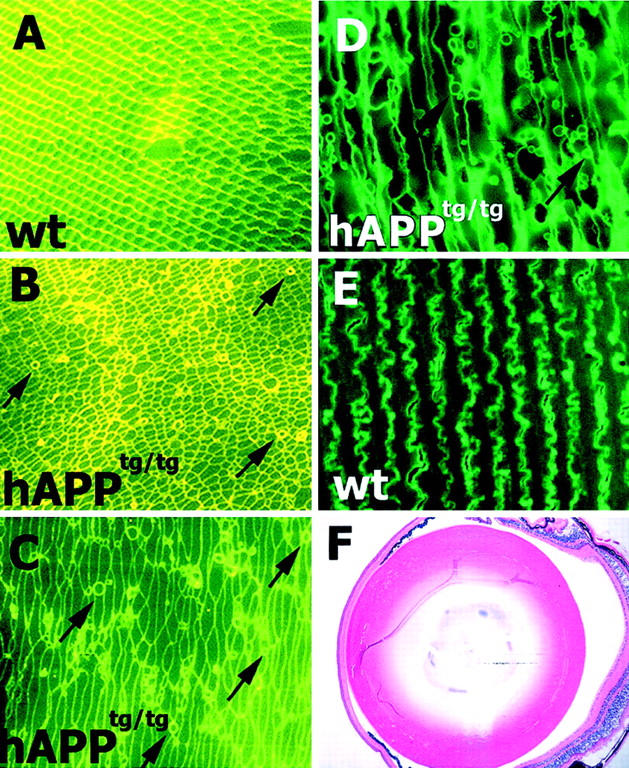
Lens fiber cell membrane abnormalities in hAβPP mice. Fluorescence photomicrographs of lens fibers in 16 month-old wt and hAβPPtg/tg mice stained with hematoxylin and eosin (a fluorescein-related dye). A and E: wt and B–D and F: hAβPPtg/tg lenses. A and B: Perpendicular fiber cell cross sections showing fiber cell arrays. C, D, and E: Lens fibers in oblique and longitudinal sections. Circular cross-sections indicated with arrows are ∼1 to 5 μm in diameter. Regions shown are 250 to 325 μm from the perimeter (Lenses are ∼2.5 mm in diameter).
In addition to abnormalities in fiber cell membrane morphology, hAβPPtg/tg mice exhibit increased age-related fiber cell degeneration and cortical plaque formation. Lenses from 6-month-old hAβPPtg/tg (Figure 3A) ▶ and wt (Figure 3B) ▶ and from 16-month-old hAβPP mice (Figure 3, C and D) ▶ were examined in paraffin sections stained with hematoxylin and eosin. The transgenic mouse lens in Figure 3A ▶ contains swollen cortical fiber cells and lens plaques (areas of apparent greater density). In addition, fiber cell nuclei are disorganized in the outer cortical region in comparison with wt lenses. However, the lens degeneration phenotype was variable in severity and onset. An example of this variability is demonstrated in photomicrographs of the left eye (Figure 3C) ▶ and right eye (Figure 3D) ▶ from the same transgenic animal. Although both lenses are affected, one eye has considerably more swollen and disorganized lens fiber cells containing plaques and flocculent material. Of interest was our observation that little or no perturbations in cornea and retina structure were apparent in hAβPPtg/tg transgenic animals suggesting the lens has the greatest sensitivity to Alzheimer pathophysiology in this human AβPP gene dosage model.
Figure 3.
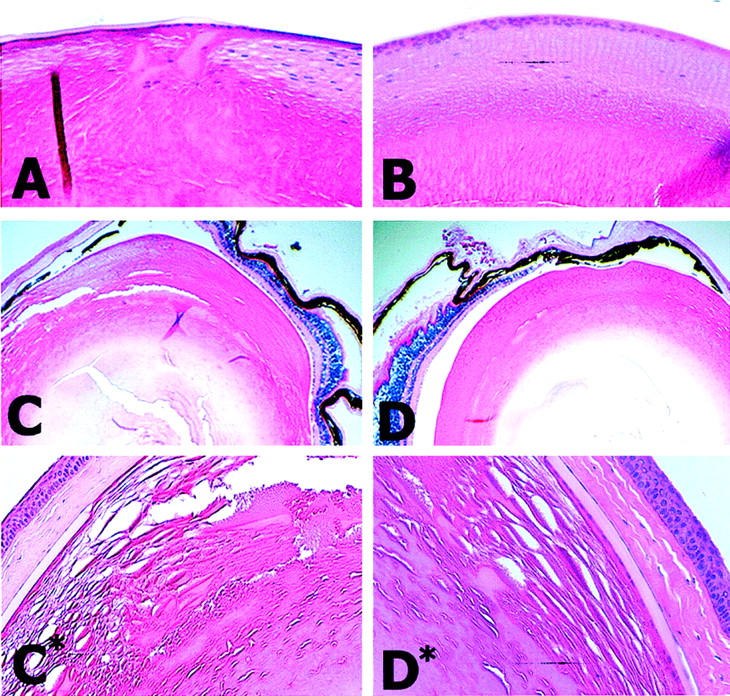
Lens degeneration and cortical plaque formation in hAβPP mice. Hematoxylin and eosin stained hAβPP and wt mouse eyes lenses viewed under bright-field illumination. A: 6-month-old hAβPPtg/tg transgenic mouse. B: 6-month-old wt mouse lens. C and C*: Left eye from a 16-month-old hAβPPtg/tg transgenic mouse. D and D*: Right eye from a 16-month-old hAβPPtg/tg transgenic mouse. Mouse lenses are ∼2.5 mm in diameter.
Discussion
The present study demonstrates that mice carrying a complete copy of a genomic human AβPP gene, in addition to the native AβPP gene, express both genes in mouse ocular tissues. Human and mouse lenses predominantly express longer and potentially more deleterious alternatively spliced forms of AβPP. However, the human and mouse AβPP genes undergo different alternative splicing regulation and these splicing patterns are separately maintained in transgenic mouse tissues.
When lens fiber cell morphology in mice that express a copy of the human AβPP gene was examined we observed that hAβPPtg/tg mouse lens fiber cells exhibit differences in membrane morphology. Fiber cells are irregularly shaped, not aligned in regular arrays, and consistently contain numerous circular membrane-bound bodies in fiber cells outside the lens fetal nucleus. In histological sections, these objects have circular profiles in perpendicular sections as well as in more longitudinal sections. These circles or spheres may be related to similar structures identified in the lens nucleus of senescent human and other primate lenses. 39-41 In addition, similar membrane-bound bodies were described in the central fetal nucleus of lenses from a glutathione peroxidase gene-knockout mice that also model oxidative stress in the lens. 42 In our preliminary examination of hAβPPtg/tg fetal lens sections we did not observe any similar structures in day 15 post-conception fetal hAβPPtg/tg lenses, suggesting these membrane-bound bodies form after initial fiber cell differentiation begins. This observation is consistent with our localization of membrane-bound bodies in adult lenses outside the central fetal nucleus.
The present data describing morphological abnormalities in hAβPP mice suggests these changes result from inappropriate lens development or trans-differentiation attributed to increased AβPP gene dosage that also produces the more deleterious human Aβ peptides 11-14 in mouse tissues. The present findings suggest that increased human AβPP and Aβ expression in lenses can contribute to morphological changes, as well as protein aggregation effects, 43 that lead to cataract formation.
When considering the mechanism of increased AβPP dosage in membrane morphology, a second example comes from Drosophila studies. Flies have been genetically engineered to overexpress an extra copy of the Drosophila AβPP gene. 44 Such animals exhibit a dramatic increase in membrane synaptic boutons in neuronal cells. These boutons have the expected increased distribution of synapsin and other neuronal markers on their membrane surface. Synaptic boutons bear a cursory resemblance to lens membrane circles described here insofar as both involve increased formation of membrane out-pouchings related to AβPP dosage.
The present study identified consistent changes in lens fiber cell morphology in hAβPP mice, however, we observed variability in the lens degeneration phenotype, even in eyes from the same animal. The present data agree with the variability in lens degeneration described in the classic human AβPP Tg2675 mouse model of AD and lens degeneration phenotypes presented by others. 45,46 Tg2675 mice express an AβPP cDNA with the Swedish familial AD mutation driven by the prion promoter 47 that also directs lens gene expression. 48 In constrast, the present study describes mice carrying a complete copy of the wild-type human AβPP gene expressed by native human AβPP promoter sequences. Taken together, these findings suggest that additional factors are involved in the age-related lens degeneration phenotype in models of lens Alzheimer pathophysiology. Finally, details concerning the relationships between fiber cell degeneration, aberrant lens membrane formations, and Aβ oxidative stress remain to be determined.
In addition to the fundamental role Alzheimer-related proteins have in ocular lens development and maintenance, the presence of predominantly longer, potentially more deleterious AβPP transcripts in human lens may indicate an inherent vulnerability to Alzheimer pathophysiology. In summary, the present data provide further evidence for a role for Alzheimer pathophysiology in oxidative stress-related, and age-related cataract formation, and that AβPP gene dosage effects may contribute to early-onset cataracts in DS.
Acknowledgments
We thank J. Samuel Zigler, Jr. (National Institutes of Health), Debbie Carper (National Institutes of Health) Patricia Farnsworth (New Jersey Medical School), Betty Jean Wagner (New Jersey Medical School) and Robert Donnelly (Molecular Resource Facility, New Jersey Medical School) for support and advice throughout this project. We are grateful to Nicole Newman for her assistance in lens preparations.
Footnotes
Address reprint requests to Peter H. Frederikse, Ph.D., UMDNJ-New Jersey Medical School, 185 S. Orange Avenue, MSB H-645 Newark, NJ 07103. E-mail: frederph@umdnj.edu.
Supported in part by National Institutes of Health Grant EY-12377 (to P.H.F.).
References
- 1.Teller J, Russo C, DeBusk L, Angelini G, Zaccheo D, Dagna-Bricarelli F, Scartezzini P, Bertolini S, Mann D, Tabaton M, Gambetti P: Presence of soluble amyloid β-peptide precedes amyloid plaque formation in Down’s syndrome. Nat Med 1996, 2:93-95 [DOI] [PubMed] [Google Scholar]
- 2.Lott I: Down’s syndrome, aging, and Alzheimer’s disease: a clinical review. Ann NY Acad Sci 1982, 396:15-27 [DOI] [PubMed] [Google Scholar]
- 3.da Cunha R, Moreira J: Ocular findings in Down’s syndrome. Am J Ophthalmol 1996, 122:236-244 [DOI] [PubMed] [Google Scholar]
- 4.Reeves R, Irving N, Moran T, Wohn A, Kitt C, Sisodia S, Schmidt C, Bronson R, Davisson M: A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 1995, 11:177-184 [DOI] [PubMed] [Google Scholar]
- 5.Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, Butler AC: Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann Neurol 1998, 43:380-383 [DOI] [PubMed] [Google Scholar]
- 6.Artavanis-Tsakonas S, Rand, M, Lake J: Notch signaling: cell fate control and signal integration in development. Science 1999, 284:770-776 [DOI] [PubMed] [Google Scholar]
- 7.Kurata S, Go M, Artavanis-Tsakonas S, Gehring W: Notch signaling and the determination of appendage identity. Proc Natl Acad Sci USA 2000, 97:2117-2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beyreuther K, Pollwein P, Multhaup G, Monning U, Konig G, Dyrks T, Schubert W, Masters C: Regulation and expression of the Alzheimer’s β/A4 amyloid protein precursor in health, disease, and Down’s syndrome. Ann NY Acad Sci 1993, 695:91-102 [DOI] [PubMed] [Google Scholar]
- 9.Behl C: Amyloid β-protein toxicity and oxidative stress in Alzheimer’s disease. Cell Tissue Res 1997, 290:471-480 [DOI] [PubMed] [Google Scholar]
- 10.Frederikse P, Garland D, Zigler J, Jr, Piatigorsky J: Oxidative stress increases production of β-amyloid precursor protein and β-amyloid (Aβ) in mammalian lenses, and Aβ has toxic effects on lens epithelial cells. J Biol Chem 1996, 271:10169-10174 [DOI] [PubMed] [Google Scholar]
- 11.Lynch T, Cherny R, Bush A: Oxidative processes in Alzheimer’s disease: the role of Aβ-metal interactions. Exp Gerontol 2000, 35:445-451 [DOI] [PubMed] [Google Scholar]
- 12.Brown D: Copper and prion disease. Brain Res Bull 2000, 55:165-173 [DOI] [PubMed] [Google Scholar]
- 13.Cherny R, Atwood C, Xilinas M, Gray D, Jones W, McLean C, Barnham K, Volitakis I, Fraser F, Kim Y, Huang X, Goldstein L, Moir R, Lim J, Beyreuther K, Zheng H, Tanzi R, Masters C, Bush A: Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30:665-676 [DOI] [PubMed] [Google Scholar]
- 14.Bush A: Metals and neuroscience. Curr Opin Chem Biol 2000, 4:184-191 [DOI] [PubMed] [Google Scholar]
- 15.Dovey H, John V, Anderson J, Chen L, de Saint Andrieu P, Fang L, Freedman S, Folmer B, Goldbach E, Holsztynska E, Hu K, Johnson-Wood K, Kennedy S, Kholodenko D, Knops J, Latimer L, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Nietz J, Quinn K, Sacchi K, Seubert P, Shopp G, Thorsett E, Tung J, Wu J, Yang S, Yin C, Schenk D, May P, Altstiel L, Bender M, Boggs L, Britton T, Clemens J, Czilli D, Dieckman-McGinty D, Droste J, Fuson K, Gitter B, Hyslop P, Johnstone E, Li W, Little S, Mabry T, Miller F, Audia J: Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J Neurochem 2001, 76:173-181 [DOI] [PubMed] [Google Scholar]
- 16.Palmert M, Golde T, Cohen M, Kovacs D, Tanzi R, Gusella J, Usiak M, Younkin L, Younkin S: Amyloid protein precursor messenger RNAs: differential expression in Alzheimer’s disease. Science 1988, 241:1080-1084 [DOI] [PubMed] [Google Scholar]
- 17.Sandbrink R, Monning U, Masters C, Beyreuther K: Expression of the APP gene family in brain cells, brain development, and aging. Gerontology 1997, 43:119-131 [DOI] [PubMed] [Google Scholar]
- 18.Higgins L, Catalano R, Quon D, Cordell B: Transgenic mice expressing human β-APP751, but not mice expressing β-APP695, display early Alzheimer’s disease-like histopathology. Ann NY Acad Sci 1993, 695:224-227 [DOI] [PubMed] [Google Scholar]
- 19.Ho L, Fukuchi K, Younkin S: The alternatively spliced Kunitz protease inhibitor domain alters amyloid β protein precursor processing and amyloid β protein production in cultured cells. J Biol Chem 1996, 271:30929-30934 [DOI] [PubMed] [Google Scholar]
- 20.Hutton M, Perez-Tur J, Hardy J: The presenilins and Alzheimer’s disease. Essays Biochem 1998, 33:33117-33131 [DOI] [PubMed] [Google Scholar]
- 21.Selkoe D: Presenilin, Notch, and the genesis and treatment of Alzheimer’s disease. Proc Natl Acad Sci USA 2001, 98:11039-11041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertram L, Tanzi RE: Of replications and refutations: the status of Alzheimer’s disease genetic research. Curr Neurol Neurosci Rep 2001, 1:442-450 [DOI] [PubMed] [Google Scholar]
- 23.Lee H, Jung K, Huang Y, Bennett L, Lee J, Mei L, Kim T: Presenilin-dependent γ-secretase-like intra-membrane cleavage of ErbB4. J Biol Chem 2002, 270:2419-2422 [DOI] [PubMed] [Google Scholar]
- 24.Cao X, Sudhof T: A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293:115-120 [DOI] [PubMed] [Google Scholar]
- 25.Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis J, Nishimoto I: c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer’s amyloid precursor protein with JNK. J Neurosci 2001, 21:6597-6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White A, Reyes R, Mercer J, Camakaris J, Zheng H, Bush A, Multhaup G, Beyreuther K, Masters C, Cappai R: Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res 1999, 842:439-444 [DOI] [PubMed] [Google Scholar]
- 27.Akaaboune M, Allinquant B, Farza H, Roy K, Magoul R, Fiszman M, Festoff B, Hantai D: Developmental regulation of amyloid precursor protein at the neuromuscular junction in mouse skeletal muscle. Mol Cell Neurosci 2000, 15:355-367 [DOI] [PubMed] [Google Scholar]
- 28.Pangalos M, Shioi J, Efthimiopoulos S, Wu A, Robakis N: Characterization of appican, the chondroitin sulfate proteoglycan form of the Alzheimer amyloid precursor protein. Neurodegeneration 1996, 5:445-451 [DOI] [PubMed] [Google Scholar]
- 29.Wu A, Pangalos M, Efthimiopoulos S, Shioi J, Robakis N: Appican expression induces morphological changes in C6 glioma cells and promotes adhesion of neural cells to the extracellular matrix. J Neurosci 1999, 17:4987-4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao Z, Cepko C: The expression and function of Notch pathway genes in the developing rat eye. J Neurosci 1997, 17:1425-1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frederikse P, Zigler J, Jr: Presenilin expression in the ocular lens. Curr Eye Res 1998, 17:947-952 [DOI] [PubMed] [Google Scholar]
- 32.Frederikse P, Farnsworth P, Zigler J, Jr: Thiamine deficiency in vivo produces fiber cell degeneration in mouse lenses. Biochem Biophys Res Commun 1999, 258:703-707 [DOI] [PubMed] [Google Scholar]
- 33.Mitsiadis A, Gayet O, Zhang N, Carroll P: Expression of Deltex1 during mouse embryogenesis: comparison with Notch1, 2, and 3 expression. Mech Dev 2001, 109:399-403 [DOI] [PubMed] [Google Scholar]
- 34.Lamb B, Sisodia S, Lawler A, Slunt H, Kitt C, Kearns W, Pearson P, Price D: Gearhart J: Introduction and expression of the 400 kilobase amyloid precursor protein gene in transgenic mice. Nat Genet 1993, 5:22-30 [DOI] [PubMed] [Google Scholar]
- 35.Buxbaum J, Christensen J, Ruefli A, Greengard P, Loring J: Expression of APP in brains of transgenic mice containing the entire human APP gene. Biochem Biophys Res Commun 1993, 197:639-645 [DOI] [PubMed] [Google Scholar]
- 36.Pearson B, Choi T: Expression of the human β-amyloid precursor protein gene from a yeast artificial chromosome in transgenic mice. Proc Natl Acad Sci USA 1993, 90:10578-10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rockenstein E, McConlogue L, Tan H, Power M, Masliah E, Mucke L: Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J Biol Chem 1995, 270:28257-28267 [DOI] [PubMed] [Google Scholar]
- 38.Gervais F, Xu D, Robertson G, Vaillancourt J, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman M, Clarke E, Zheng H, Van Der Ploeg L, Ruffolo S, Thornberry N, Xanthoudakis S, Zamboni R, Roy S, Nicholson D: Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-β precursor protein and amyloidogenic A-β peptide formation. Cell 1999, 97:395-406 [DOI] [PubMed] [Google Scholar]
- 39.Boyle D, Takemoto L: Finger-like projections of plasma membrane in the most senescent fiber cells of human lenses. Curr Eye Res 1998, 17:1118-1123 [DOI] [PubMed] [Google Scholar]
- 40.Kuszak J, Ennesser C, Umlas J, Macsai-Kaplan M, Weinstein R: The ultrastructure of fiber cells in primate lenses: a model for studying membrane senescence. J Ultrastruct Mol Struct Res 1988, 100:60-74 [DOI] [PubMed] [Google Scholar]
- 41.Gilliland K, Freel C, Lane C, Fowler W, Costello M: Multilamellar bodies as potential scattering particles in human age-related nuclear cataracts. Mol Vis 2000, 7:120-130 [PubMed] [Google Scholar]
- 42.Reddy V, Giblin F, Lin L, Dang L, Unakar N, Musch D, Boyle D, Takemoto L, Ho Y, Knoernschild T, Juenemann A, Lutjen-Drecoll E: Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice. Invest Ophthalmol Vis Sci 2001, 42:3247-3255 [PubMed] [Google Scholar]
- 43.Bush A, Goldstein L: Specific metal-catalysed protein oxidation reactions in chronic degenerative disorders of ageing: focus on Alzheimer’s disease and age-related cataracts. Novartis Found Symp 2001, 235:26-38 [DOI] [PubMed] [Google Scholar]
- 44.Torroja L, Packard M, Gorczyca M, White K, Budnik V: The Drosophila β-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci 1999, 19:7793-7803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldstein L, Muffat J, Cherny R, Faget K, Coccia F, Fraser F, Masters C, Tanzi R, Chylack L, Jr, Bush A: Aβ peptides in human and amyloid bearing transgenic mouse lens implications for Alzheimer’s disease and cataracts. Invest Ophthalmol Vis Sci 2001, 42:S299 [Google Scholar]
- 46.Goldstein L, Muffat J, Cherny R, Ericsson J, Coccia, F., Faget K, Delalle I, Hedley-Whyte T, Chylack L, Bush A: The Alzheimer’s diseas β-amyloid peptide promotes lens protein aggregation and cataract formation. November2001. Soc for Neurosci, San Diego, CA (Prog 128.4)
- 47.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G: Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274:99-102 [DOI] [PubMed] [Google Scholar]
- 48.Frederikse P, Zigler J, Jr, Farnsworth P, Carper D: Prion protein expression in mammalian lenses. Curr Eye Res 2000, 20:137-143 [PubMed] [Google Scholar]