

Abstract
Juvenile myositis is a heterogeneous group of systemic autoimmune diseases, in which clinical and serologic subgroups result in subsets of patients with distinct clinical manifestations, disease courses, immunogenetic associations, responses to therapy, and prognoses. A newly identified autoantibody of unknown specificity, anti-p155, is myositis-associated and seen in up to 20 – 30% of juvenile and adult DM patients. HLA DRB1*0301 and its linked allele DQA1*0501 have been identified as the major immunogenetic risk factor for juvenile and adult DM in both European- and African- American patients, and DQA1*0301 is an additional risk factor in European American patients. Several DQA1 alleles also are protective for juvenile DM. Environmental risk factors are poorly understood, but growing evidence suggests a role for infectious agents and ultraviolet radiation. The current therapy of juvenile DM consists of corticosteroids and other immunosuppressive agents, with the adjunctive treatment of cutaneous manifestations and rehabilitation. Therapeutic trials of biologic agents, including anti-TNFα and anti-CD20, may aid in developing promising new therapies for these disorders.
Keywords: Juvenile dermatomyositis, autoantibodies, immunogenetics, environmental factors, treatment
Take Home Messages
The juvenile myositis syndromes are a heterogeneous group of systemic autoimmune diseases in which clinical and serologic subgroups result in subsets with distinct clinical features, disease courses, responses to therapy and prognoses. Anti-p155, a new dermatomyositis (DM)-associated autoantibody, is present in up to 30% of juvenile DM patients.
The pathogenesis involves an immune attack on the muscle microvasculature with activation and inhibition of angiogenesis, as well as endothelial activation. Activation of interferon and antigen presentation pathways is also an important part of disease expression.
The primary immunogenetic risk factors are HLA DRB1 and DQA1 alleles, with a number of shared factors with adult DM.
Environmental factors are poorly understood, but growing evidence suggests a role for infectious agents, ultraviolet radiation and possibly maternal microchimerism.
Therapy is primarily immunosuppressive, including a prolonged course of corticosteroid therapy, but promising trials of biologic agents may change future therapeutic paradigms.
The idiopathic inflammatory myopathies (IIM) are a rare group of systemic connective tissue diseases with the hallmark of chronic muscle inflammation and weakness of unknown cause. Approximately one-fifth of the cases of IIM have onset during childhood, with an annual incidence for juvenile IIM of 2.5–5 cases per million population (1). The peak age of onset for juvenile dermatomyositis (DM) is approximately 7 years with a broad distribution throughout childhood; one-quarter have onset prior to age 5 years (2). Girls are preferentially affected, even pre-pubertally (2).
Subgroups of Myositis
The division of the myositis syndromes into clinicopathologic or serologic groups apparently results in more homogeneous subsets, in terms of distinctive epidemiologic, genetic, and prognostic features. The most common clinicopathologic subset, representing up to 85% of children with IIM is juvenile DM, in which Gottron’s papules or heliotrope rashes are present; (3). The two other major subsets of juvenile IIM are polymyositis (PM), in which these characteristic rashes are absent, and overlap myositis, including patients with either PM or DM who also meet criteria for another connective tissue disease. Inclusion body myositis and cancer-associated myositis, often defined as myositis occurring within two years of a diagnosis of cancer, are seen almost exclusively in adults with myositis. Some of the other subsets, such as granulomatous, eosinophilic, focal, proliferative, and orbital myositis, are quite rare and while they are pathologically distinct entities, it remains unclear whether they are clinically and prognostically distinct (3).
Juvenile DM has several clinical features that may be more characteristic of the juvenile than adult form of disease. These include the presence of dystrophic calcification, with calcium deposits in the skin, subcutaneous tissue, or muscle, in up to 30% of patients, which is associated with poorer functional outcomes (4); cutaneous or gastrointestinal ulceration, seen in up to 10% of patients, which is the result of tissue ischemia from the underlying vasculopathy and portends a severe course of illness; and acquired lipodystrophy, seen in up to 10% of patients, which may include generalized or partial fat loss and is often associated with calcinosis.
The pathogenesis of DM involves perivascular infiltration of B and CD4+ T lymphocytes and dendritic cells in the affected muscle. There is a loss of capillaries, due to complement mediated damage, and evidence for increased new vessel formation, particularly in juvenile DM muscle. From microarray experiments, in some cases confirmed by immunohistochemistry or real-time PCR, a number of promoters and inhibitors of angiogenesis are over-expressed in the affected muscle tissue (5;6). There is also increased expression of genes promoting endothelial differentiation and activation, as well as classical and alternative complement pathway regulators that facilitate angiogenesis in the muscle tissue of adult DM patients (5). Leukocyte adhesion molecules are also over-expressed, with ICAM-1 particularly on the vessels of juvenile DM patients (7). Immune dysregulation is also a key part of the pathogenesis, with upregulation of interferon α/β inducible genes and genes upregulated in a Type I interferon response, as well as genes involved in antigen presentation, suggesting either viral initiation of disease or activation of plasmacytoid dendritic cells (8).
Clinical Usefulness of the Myositis Autoantibodies
Defined autoantibodies have been identified in up to 70% of adult and 40% of juvenile IIM patients (3;9). Some of these are autoantibodies are found frequently in IIM patients but also are seen in other conditions (the myositis-associated autoantibodies, or MAAs), and some are autoantibodies unique to the IIM (the myositis-specific autoantibodies, or MSAs). The MSAs appear to be antigen-driven, arising months prior to the onset of myositis, correlating in titer with disease activity, and sometimes disappearing after prolonged remission (10). These autoantibodies bind to conserved conformational epitopes on cytoplasmic and nuclear ribonucleoproteins involved in basic cellular mechanisms, such as translation and transcription, and they inhibit the functions of the targeted antigens in vitro. There is no direct evidence that the MSAs play a pathogenic role, but recent data suggest that their disease specificity may be related to their increased expression in affected muscle of patients with myositis, including the regenerating myoblasts (11).
Testing for MAAs and MSAs using immunoprecipitation methodology is available as a reliable technique in a few commercial and research laboratories, and can be useful in diagnosing atypical patients and in predicting certain clinical features, immunogenetics, responses to therapy, and prognosis (Table 1). Although these associations were first identified in adult IIM patients (9), a smaller number of juvenile IIM subjects with MSAs have now been studied and appear to show similar features (3). New autoantibodies are being identified in adult and juvenile DM patients, and immunogenetic associations for a number of autoantibodies have been discovered (12) (Table 1).
Table 1.
Associations of the Myositis-specific and Myositis-associated Autoantibodies.
| Autoantibody | Associations and Comments |
|---|---|
| Myositis-Specific Autoantibody | |
| Anti-synthetase | Acute onset of polymyositis (PM), dermatomyositis (DM) or overlap myositis in the spring with high frequency of symmetric non-erosive arthritis, interstitial lung disease, fever, mechanic's hands, Raynaud's phenomenon; moderate response to therapy, myositis flare with tapering of therapy; seen in 20–25% of adult and 5–10% of juvenile myositis cases; immunogenetic association with HLA A*01, B*08, Cw*0701, DRB1*0301, DQA1*0501(9;15). |
| Anti-signal recognition particle (SRP) | Very acute onset of severe PM with frequent myalgias, severe weakness and very high creatine kinase levels; some adults more frequently have cardiac involvement and palpitations, and are African American females; poor response to therapy; seen in <5% of adult and juvenile myositis patients (9;13). |
| Anti-Mi-2 | Classic DM; mild to moderate weakness with V and shawl sign rashes, and cuticular overgrowth associated in adults; good response to therapy; seen in 5–10% of adult and 5% of juvenile myositis patients; immunogenetic association with HLA DRB1*0701, DQA1*0201(9;15). |
| Myositis-Associated Autoantibodies | |
| Anti-p155 | A newly identified autoantibody present in 20–30% of adult and juvenile DM patients, including those with DM associated with another connective tissue disease and cancer-associated DM Clinically distinct from patients with anti-synthetase antibodies, but high frequency of V-sign rash and cuticular overgrowth may not make this distinct from other patients with DM; immunogenetic association with HLA DQA1*0301 (12). |
| Anti-PM-Scl | Scleroderma/myositis overlap syndromes, mild myositis, high incidence of arthritis, Raynaud's, interstitial lung disease, calcinosis, sicca; associated with HLA A*0101, B*08, DRB1*0301, DQA1*0501 (15). |
| Anti-Ku | Scleroderma/myositis overlap syndromes, frequent Raynaud's, arthralgias, and reflux |
| Anti-Ro | Seen in 12% of adult idiopathic inflammatory myopathies (IIM) overall, but 17% of subjects with myositis overlap syndromes; associated with anti-Jo-1 antibodies; immunogenetic association is B*0801, Cw*0701, DRB1*0301, DQA1*0501(9;15). |
| Anti-La | Seen 8% of adult IIM, but 19% of subjects with myositis overlap syndromes; immunogenetic association is B*0801, Cw*0701, DRB1*0301 (9;15). |
| Anti-U1RNP | Myositis overlap syndromes |
| Anti-U2RNP | Scleroderma/myositis overlap syndromes |
| Anti-U3RNP | Myositis overlap syndromes |
| None of the above | A heterogeneous group of patients requiring further study and classification (myositis-specific and myositis-associated antibody negative) |
Adult and juvenile IIM patients with anti-synthetase autoantibodies tend to have a characteristic clinical presentation of fevers, small joint arthritis, interstitial lung disease, Raynaud’s phenomenon, mechanic’s hands, and severe PM or DM. They have a moderately good response to therapy, especially to corticosteroids and methotrexate, but their illness will frequently flare with tapering medications (Table 1) (9). Adult IIM patients with anti-synthetase autoantibodies have a 25% 5-year mortality rate, with death primarily due to interstitial lung disease (9). Adults and children with anti-signal recognition particle (SRP) autoantibodies tend to have a very acute onset of severe PM, with both proximal and distal weakness, very high serum levels of creatine kinase, frequent myalgias and muscle atrophy, poor responses to therapy, and extremely poor prognoses (9;13). Patients with anti-SRP autoantibodies are often bed-bound and sometimes require nasogastric tube feeding during severe flares of disease activity. They usually recover very slowly, and sometimes do not improve beyond a bed-bound or wheel chair-bound state despite combination immunosuppressive therapy, Some adults with anti-SRP have tended to be African American and also have frequent cardiac involvement, which has not been apparent in the children (9). Anti-Mi-2 autoantibodies are directed against a nuclear helicase, are DM-specific, and define a group of patients with a much better prognosis from the other MSAs. In addition to the classic Gottron's papules and heliotrope rash, adults with anti-Mi-2 autoantibodies frequently have V-sign, shawl-sign, and cuticular overgrowth (9). Children with anti-Mi2 autoantibodies all have DM, but they appear to have V- and shawl-sign rashes and cuticular overgrowth less frequently than their adult counterparts, and may not have features that are clinically distinguishable from other juvenile DM patients (3). Most patients with anti-Mi-2 autoantibodies respond well to oral and topical corticosteroids, hydroxychloroquine, and photoprotective measures, but some require methotrexate or other immunosuppressive agents to induce a complete response.
Recently, a new autoantibody, anti-p155, has been identified in 20 – 30% of adult and juvenile DM patients, thus characterizing some of these patients previously reported as MSA and MAA negative. Anti-p155 has relative specificity for primary DM, although DM patients with overlap myositis and cancer-associated myositis patients also may have this autoantibody, and rarely patients with other autoimmune diseases, such as lupus (12). The clinical features of adults with this autoantibody are distinct from those of anti-synthetases, but V-and shawl-sign rashes are commonly present, and it may not be clinically distinct from other patients with DM. Anti-p155 autoantibodies are also associated with HLA DQA1*0301, distinct from the HLA associations of other autoantibodies.
Immunogenetic Risk and Protective Factors
Genes encoding human MHC Class I (HLA-A, -B, -Cw) and Class II (HLA-DR, -DQ, -DP) molecules are the most polymorphic in the human genome and among the strongest and most consistently identified genetic factors associated with the development of autoimmune diseases. Recent work has demonstrated the association of the northwestern European 8.1 ancestral haplotype (containing HLA-A*0101; B*0801; Cw*0701; DRB1*0301; DQA1*0501) with the development of all forms of adult IIM in European American (EA) patients, with the MHC Class II DRB1*0301 allele as the strongest risk factor in the haplotype (14). In addition, several novel HLA factors have associated distinctly with different clinicopathologic and serologic groups of IIM patients (14;15) (Table 1).
Similarly, in children with juvenile DM, the same major immunogenetic risk factors have been identified, including HLA DRB1*0301 and its linked allele DQA1*0501 for both European and African American patients, with DQA1*0301 as an additional risk factor for European Americans (16). Related peptide binding motifs have also found to be secondary risk factors for juvenile and adult DM. Both adult and juvenile DM have a number of protective factors in European Americans, including DQA1*0201, *0101, and *0102 (16). A number of shared HLA risk and protective factors for both juvenile and adult DM suggest common elements in their pathogeneses. In Japanese patients, DRB1*15021, rather than DRB1*0301, may be a risk factor for juvenile DM (17).
Potential Environmental Factors
A number of environmental factors have been associated with the IIM, including infectious agents and non-infectious agents, such as ultraviolet light, physical exertion, psychological stress, medications, and vaccines (Table 2). Most are not proven environmental risk factors, but reports suggest a temporal association to the onset of the IIM. For some, case controlled epidemiologic studies, cases of de-challenge/re-challenge, and biomarker assays help in strengthening the possible association with illness onset. Of the infectious agents, Group A streptococcus and influenza have strongest evidence of association with onset of juvenile IIM from case-controlled epidemiologic studies. We found no increase in parvovirus B19 seroconversion, or evidence of an increase in the viral genome in the serum or muscle tissue of patients recently diagnosed with juvenile DM compared to a group of matched healthy children (18). For coxsackie B virus, the evidence is mixed, with a case-controlled study not supporting an association with onset of juvenile DM and mixed reports of isolation of the virus from the affected muscle (19;20).
Table 2.
Environmental Factors Associated with the Onset of Juvenile Myositis.
| Environmental Factor | Association and Comments |
|---|---|
| Infectious Agents | |
| Group A Streptococcus | Associated with onset of juvenile PM in case controlled study (34). Case reports of temporal association to onset of juvenile DM, with elevated titers present at time of myositis onset. Peripheral blood T lymphocytes from juvenile DM patients react with streptococcal M5 protein and a myositis peptide with homology to M5 (35). |
| Influenza A | Increased viral titers in juvenile PM patients compared to controls suggest an association (36). Isolation of the virus in juvenile DM muscle has not been confirmed (20). |
| Enteroviruses | High frequency of antibody to coxsackievirus B in juvenile DM patients ≤7 yeas of age and healthy controls (19). Variability in detecting coxsackievirus in muscle biopsies by PCR, suggest virus may not be associated with onset of juvenile DM (20). Echovirus infection associated with juvenile DM and X-linked hypogammaglobulinemia, with T lymphocytic perivascular infiltration of muscle, often responsive to intravenous gammaglobulin therapy (37). |
| Parvovirus | In a case controlled study, no association of parvovirus B19 with onset of juvenile DM, including similar rates of seroconversion in patients and controls, and similar viral load in the plasma and muscle (18). |
| Other infections | Case reports of other infectious agents temporally related to the onset of juvenile DM and PM include hepatitis B, HIV, toxoplasmosis, borrelia burgdorferi, but these need additional supporting evidence to confirm if they are associated (reviewed in (3;38)). |
| Non-Infectious Agents | |
| Ultraviolet light | Photosensitive skin rashes in juvenile DM patients, and anecdotes of disease onset or flare following intense sun exposure. Adult DM patients also have increased sensitivity to UV B as demonstrated by an abnormally low minimal erythema UV B dose (21). Correlation of proportion of DM and anti-Mi-2 autoantibodies with surface UV light exposure (22). Increased apoptosis and surface expression of autoantigens in keratinocytes treated with UV radiation (23). |
| Emotional stress | Greater frequency of emotional stress associated with the onset of adult DM/PM in a case controlled study (39). |
| Physical exertion | Heavy physical exertion associated with the onset of adult DM/PM in a case controlled study (39). |
| Microchimerism | Increased numbers and frequency of maternal cells in the peripheral blood T lymphocytes and affected muscle tissue of juvenile DM patients (24;25). Chimeric cells may become immunoreactive against host juvenile DM lymphocytes, activated to produce interferon-gamma (25). Relationship to initiation of disease is unclear. |
| Other non-infectious agents | Anecdotal case reports of a temporal association of certain non-infectious agents with onset of juvenile DM, including vaccines, medications (penicillamine and caritcaine), and growth hormone, suggest these as potential environmental factors that require additional confirmation (reviewed in (3;38)). With growth hormone therapy, symptom resolution with de-challenge and their reappearance with re-challenge further supports the potential association. |
Abbreviations: DM, dermatomyositis; PM, polymyositis
Growing evidence suggests a role for ultraviolet radiation (UV) in the onset of DM. DM patients have a number of photosensitive rashes and anecdotally, illness may exacerbate following sun exposure. Adult DM patients also have increased sensitivity to UV B as demonstrated by an abnormally low minimal erythema UV B dose (21). An increase in the proportion of DM relative to PM and an increase in the proportion of patients with the DM-associated Mi-2 autoantibody in areas of the world with higher surface UV radiation suggest a relationship between UV light exposure and the development of DM (22). Keratinocytes damaged by UV radiation also undergo apoptosis, with increased autoantigen expression on the surface of such cells (23).
We have been interested in the influence of perinatal factors on the onset of illness later in childhood. Specifically, the trafficking of maternal cells to the fetus during pregnancy, known as maternal microchimerism, is increased in frequency and degree in the peripheral blood T lymphocytes and affected muscle from patients with juvenile IIM compared to their unaffected siblings and healthy control children (24;25). Chimeric cells may become immunoreactive against host juvenile DM lymphocytes, activated to produce interferon-gamma (25). Maternal chimerism has also been observed in children with autoimmune thyroid disease and neonatal lupus. The role of maternal microchimerism in pathogenesis of juvenile DM is being further examined.
Therapeutic Approaches to the Juvenile IIM
The therapy of juvenile IIM remains based primarily on the use of immunosuppressive agents, with daily oral corticosteroid therapy in doses of 1–2 mg/kg/day of prednisone administered until the disease achieves complete clinical remission, as the mainstay of therapy (Figure 1). Early introduction of intravenous pulse methylprednisolone and/or methotrexate have resulted in less corticosteroid toxicity, with apparent ability to reduce oral prednisone more rapidly, decrease the frequency of calcinosis and develop a more rapid response to therapy (26;27). Treatment of the cutaneous manifestations with topical agents and hydroxychloroquine, use of photoprotective measures, administration of calcium and vitamin D for prevention of osteoporosis, and implementation of a graded physical therapy program are important adjunctive first-line therapies (3). For patients with severe myositis, poor prognostic signs or a modest response to initial therapy, intravenous immunoglobulin, cyclosporine and azathioprine are often used, based on uncontrolled trials in pediatric patients and several controlled trials in adult IIM (3;28). One open label study suggested moderate improvement in myositis disease activity, including muscle strength and function, with administration of intravenous bolus cyclophosphamide in juvenile DM patients with refractory disease (29). Case series and open label studies in adult patients support use of mycophenolate mofetil and tacrolimus for patients with refractory disease (30;31).
Figure 1. Therapeutic algorithm for the treatment of juvenile dermatomyositis.
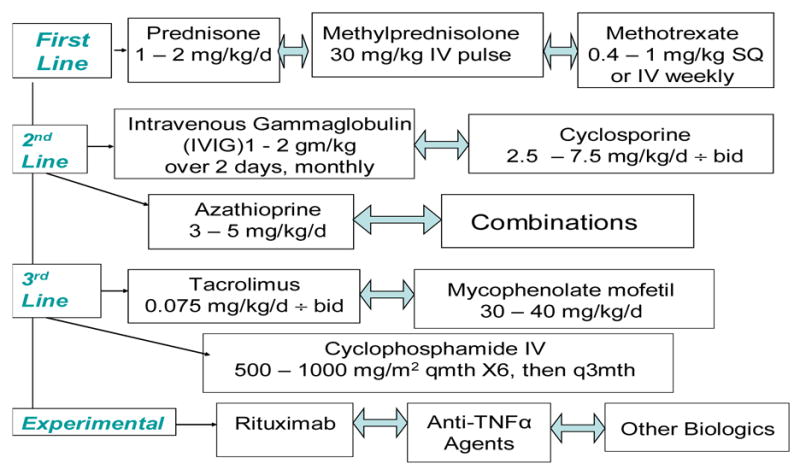
Adjunctive therapies not pictured include photoprotective measures, physical therapy, calcium and vitamin D to prevent osteoporosis, and topical therapies, as well as hydroxychloroquine, to treat the skin rashes.
Clinical trials for biologic therapies are currently in progress. An open-label study of etanercept in ten juvenile DM patients suggested modest benefit primarily to the cutaneous features (32). Case reports of infliximab in adult patients appear promising, and two randomized controlled trials of anti-TNFα therapies currently in progress should aid in determining efficacy of these agents. Rituximab, directed against anti-CD20 on B lymphocytes, appears promising in a pilot study of 7 adult DM patients, with improvement in muscle strength, as well as enzymes, skin rashes and other parameters of disease (33). Randomized controlled trials of these new therapies, currently in progress, should aid in determining the efficacy of these agents.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mendez EP, Lipton R, Ramsey-Goldman R, Roettcher P, Bowyer S, Dyer A, et al. US incidence of juvenile dermatomyositis, 1995–1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum. 2003;49(3):300–305. doi: 10.1002/art.11122. [DOI] [PubMed] [Google Scholar]
- 2.Pachman LM, Lipton R, Ramsey-Goldman R, Shamiyeh E, Abbott K, Mendez EP, et al. History of infection before the onset of juvenile dermatomyositis: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis Rheum. 2005;53(2):166–172. doi: 10.1002/art.21068. [DOI] [PubMed] [Google Scholar]
- 3.Rider LG, Miller FW. Classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 1997;23:619–655. doi: 10.1016/s0889-857x(05)70350-1. [DOI] [PubMed] [Google Scholar]
- 4.Huber AM, Lang B, LeBlanc CM, Birdi N, Bolaria RK, Malleson P, et al. Medium- and long-term functional outcomes in a multicenter cohort of children with juvenile dermatomyositis. Arthritis Rheum. 2000;43(3):541–549. doi: 10.1002/1529-0131(200003)43:3<541::AID-ANR9>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 5.Nagaraju K, Rider LG, Fan C, Chen YW, Mitsak M, Rawat R, et al. Endothelial cell activation and neovascularization are prominent in dermatomyositis. J Autoimmune Dis. 2006;3:2. doi: 10.1186/1740-2557-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fall N, Bove KE, Stringer K, Lovell DJ, Brunner HI, Weiss J, et al. Association between lack of angiogenic response in muscle tissue and high expression of angiostatic ELR-negative CXC chemokines in patients with juvenile dermatomyositis: possible link to vasculopathy. Arthritis Rheum. 2005;52(10):3175–3180. doi: 10.1002/art.21303. [DOI] [PubMed] [Google Scholar]
- 7.Sallum AM, Kiss MH, Silva CA, Wakamatsu A, Vianna MA, Sachetti S, et al. Difference in adhesion molecule expression (ICAM-1 and VCAM-1) in juvenile and adult dermatomyositis, polymyositis and inclusion body myositis. Autoimmun Rev. 2006;5(2):93–100. doi: 10.1016/j.autrev.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168(8):4154–4163. doi: 10.4049/jimmunol.168.8.4154. [DOI] [PubMed] [Google Scholar]
- 9.Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70:360–374. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Miller FW, Waite KA, Biswas T, Plotz PH. The role of an autoantigen, histidyl-tRNA synthetase, in the induction and maintenance of autoimmunity. Proc Natl Acad Sci U S A. 1990;87:9933–9937. doi: 10.1073/pnas.87.24.9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;201(4):591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Targoff I, Shamim E, Sherry D, Wallace C, Haftel H, Miller F, et al. A novel dermatomyositis-associated protein directed against a 155 kd protein. Arthritis Rheum. 2000;43(suppl):S175. [Google Scholar]
- 13.Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50(1):209–215. doi: 10.1002/art.11484. [DOI] [PubMed] [Google Scholar]
- 14.O'Hanlon TP, Carrick DM, Arnett FC, Reveille JD, Carrington M, Gao X, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1 and -DQA1 allelic profiles and motifs define clinicopathologic groups in Caucasians. Medicine (Baltimore) 2005;84:338–349. doi: 10.1097/01.md.0000189818.63141.8c. [DOI] [PubMed] [Google Scholar]
- 15.O'Hanlon TP, Carrick DM, Targoff IN, Arnett FC, Reveille JD, Carrington M, et al. HLA-A, -B, -DRB1 and -DQA1 allelic profiles for the idiopathic inflammatory myopathies: Distinct immunogenetic risk and protective factors distinguish European American patients with different myositis autoantibodies. Medicine. 2006;85:111–127. doi: 10.1097/01.md.0000217525.82287.eb. [DOI] [PubMed] [Google Scholar]
- 16.Mamyrova G, Carrick D, O'Hanlon T, Malley J, Reed A, Sherry DWC, et al. Relative importance of immunogenetic risk and protective factors for the development of juvenile dermatomyositis (JDM) in European American (EA) patients. Arthritis and Rheumatism. 2005;52 (9):S308–S309. [Google Scholar]
- 17.Tomono N, Mori M, Nakajima S, Miyamae T, Ito S, Mitsuda T, et al. HLA-DRB1*15021 is the predominant allele in Japanese patients with juvenile dermatomyositis. J Rheumatol. 2004;31(9):1847–1850. [PubMed] [Google Scholar]
- 18.Mamyrova G, Rider LG, Haagenson L, Wong S, Brown KE. Parvovirus B19 and onset of juvenile dermatomyositis. JAMA. 2005;294(17):2170–2171. doi: 10.1001/jama.294.17.2170. [DOI] [PubMed] [Google Scholar]
- 19.Pachman LM, Hayford JR, Hochberg MC, Pallansch MA, Chung A, Daugherty CD, et al. New-onset juvenile dermatomyositis: Comparisons with a healthy cohort and children with juvenile rheumatoid arthritis. Arthritis Rheum. 1997;40:1526–1533. doi: 10.1002/art.1780400822. [DOI] [PubMed] [Google Scholar]
- 20.Pachman LM, Litt DL, Rowley AH, Hayford JR, Caliendo J, Heller S, et al. Lack of detection of enteroviral RNA or bacterial DNA in magnetic resonance imaging-directed muscle biopsies from twenty children with active untreated juvenile dermatomyositis. Arthritis Rheum. 1995;38:1513–1518. doi: 10.1002/art.1780381019. [DOI] [PubMed] [Google Scholar]
- 21.Dourmishev L, Meffert H, Piazena H. Dermatomyositis: comparative studies of cutaneous photosensitivity in lupus erythematosus and normal subjects. Photodermatol Photoimmunol Photomed. 2004;20(5):230–234. doi: 10.1111/j.1600-0781.2004.00115.x. [DOI] [PubMed] [Google Scholar]
- 22.Okada S, Weatherhead E, Targoff IN, Wesley R, Miller FW. Global surface ultraviolet radiation intensity may modulate the clinical and immunologic expression of autoimmune muscle disease. Arthritis Rheum. 2003;48:2285–2293. doi: 10.1002/art.11090. [DOI] [PubMed] [Google Scholar]
- 23.Casciola-Rosen L, Rosen A. Ultraviolet light-induced keratinocyte apoptosis: a potential mechanism for the induction of skin lesions and autoantibody production in LE. Lupus. 1997;6(2):175–180. doi: 10.1177/096120339700600213. [DOI] [PubMed] [Google Scholar]
- 24.Artlett CM, Ramos R, Jiminez SA, Patterson K, Miller FW, Rider LG. Chimeric cells of maternal origin in juvenile idiopathic inflammatory myopathies. Childhood Myositis Heterogeneity Collaborative Group Lancet. 2000;356(9248):2155–2156. doi: 10.1016/s0140-6736(00)03499-1. [DOI] [PubMed] [Google Scholar]
- 25.Reed AM, McNallan K, Wettstein P, Vehe R, Ober C. Does HLA-dependent chimerism underlie the pathogenesis of juvenile dermatomyositis? J Immunol. 2004;172(8):5041–5046. doi: 10.4049/jimmunol.172.8.5041. [DOI] [PubMed] [Google Scholar]
- 26.Fisler RE, Liang MG, Fuhlbrigge RC, Yalcindag A, Sundel RP. Aggressive management of juvenile dermatomyositis results in improved outcome and decreased incidence of calcinosis. J Am Acad Dermatol. 2002;47:505–511. doi: 10.1067/mjd.2002.122196. [DOI] [PubMed] [Google Scholar]
- 27.Ramanan AV, Campbell-Webster N, Ota S, Parker S, Tran D, Tyrrell PN, et al. The effectiveness of treating juvenile dermatomyositis with methotrexate and aggressively tapered corticosteroids. Arthritis Rheum. 2005;52(11):3570–3578. doi: 10.1002/art.21378. [DOI] [PubMed] [Google Scholar]
- 28.Reed AM, Lopez M. Juvenile dermatomyositis: recognition and treatment. Paediatr Drugs. 2002;4(5):315–321. doi: 10.2165/00128072-200204050-00004. [DOI] [PubMed] [Google Scholar]
- 29.Riley P, Maillard SM, Wedderburn LR, Woo P, Murray KJ, Pilkington CA. Intravenous cyclophosphamide pulse therapy in juvenile dermatomyositis. A review of efficacy and safety. Rheumatology (Oxford) 2004;43(4):491–496. doi: 10.1093/rheumatology/keh082. [DOI] [PubMed] [Google Scholar]
- 30.Edge JC, Outland JD, Dempsey JR, Callen JP. Mycophenolate mofetil as an effective corticosteroid-sparing therapy for recalcitrant dermatomyositis. Arch Dermatol. 2006;142(1):65–69. doi: 10.1001/archderm.142.1.65. [DOI] [PubMed] [Google Scholar]
- 31.Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52(8):2439–2446. doi: 10.1002/art.21240. [DOI] [PubMed] [Google Scholar]
- 32.Miller ML, Smith RL, Abbott KA, Sinacore JM, Pachman LM. Use of etanercept in chronic juvenile dermatomyositis. Arthritis Rheum. 2002;46(suppl):S306. [Google Scholar]
- 33.Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52(2):601–607. doi: 10.1002/art.20849. [DOI] [PubMed] [Google Scholar]
- 34.Koch MJ, Brody JA, Gillespie MM. Childhood polymyositis: a case-control study. Am J Epidemiol. 1976;104:627–631. doi: 10.1093/oxfordjournals.aje.a112341. [DOI] [PubMed] [Google Scholar]
- 35.Massa M, Costouros N, Mazzoli F, De BF, La CA, Le T, et al. Self epitopes shared between human skeletal myosin and Streptococcus pyogenes M5 protein are targets of immune responses in active juvenile dermatomyositis. Arthritis Rheum. 2002;46(11):3015–3025. doi: 10.1002/art.10566. [DOI] [PubMed] [Google Scholar]
- 36.Koch M, Brody JA, Nemo GJ, Sever JL. Antibody levels to parainfluenza, rubella, measles, and influenza A virus in children with polymyositis. Arthritis Rheum. 1975;18:353–355. doi: 10.1002/art.1780180410. [DOI] [PubMed] [Google Scholar]
- 37.Rudge P, Webster AD, Revesz T, Warner T, Espanol T, Cunningham-Rundles C, et al. Encephalomyelitis in primary hypogammaglobulinaemia. Brain. 1996;119:1–15. doi: 10.1093/brain/119.1.1. [DOI] [PubMed] [Google Scholar]
- 38.Reed AM, Ytterberg SR. Genetic and environmental risk factors for idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:891–916. doi: 10.1016/s0889-857x(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 39.Lyon MG, Bloch DA, Hollak B, Fries JF. Predisposing factors in polymyositis-dermatomyositis: results of a nationwide survey. J Rheumatol. 1989;16:1218–1224. [PubMed] [Google Scholar]