

Abstract
The leading cause of premature death in smokers is cardiovascular disease. Diabetics also suffer from increased cardiovascular disease. This results, in part, from the hypercoagulable state associated with these conditions. However, the molecular cause(s) of the elevated risk of cardiovascular disease and the prothrombotic state of smokers and diabetics remain unknown. It is well known that oxidative stress is increased in both conditions. In smokers, it is established that oxidation of methionine residues takes place in α1-antitrypsin in lungs and that this leads to emphysema. Thrombomodulin is a key regulator of blood clotting and is found on the endothelium. Oxidation of methionine 388 in thrombomodulin is known to slow the rate at which the thrombomodulin-thrombin complex activates protein C, a protein which, in turn, degrades the factors which activate thrombin and lead to clot formation. In analogy to the cause of emphysema, it is hypothesized that oxidation of this methionine is elevated in smokers relative to non-smokers and, perhaps, in conditions such as diabetes that impose oxidative stress on the body. Evidence for the hypothesis that such an oxidation and concomitant reduction in activated protein C levels would lead to elevated cardiovascular risk is presented.
Introduction
Cardiovascular disease is the most common cause of premature death in smokers [1]. Smoking related cardiovascular diseases are the cause of 140,000 premature deaths annually in the United States [2]. The most common cardiovascular diseases in smokers are the thrombotic arterial occlusive diseases, in particular myocardial infarction and stroke. While narrowing of arteries from atherosclerosis is an important component of these diseases, equally important is the fact that the blood of smokers is much more prone to clot than that of non-smokers. While these facts are well established, the molecular origin of this prothrombotic state has remained unclear, despite intensive research. We present here a hypothesis for the molecular root of this hypercoagulability. Further, we believe that this hypothesis is equally plausible for explaining the molecular origin of a similar prothrombotic state [3–5] and increased cardiovascular risk in diabetics and may also explain, in part, why elevated levels of homocysteine are a risk factor for heart disease.
Thrombomodulin is a key regulatory protein in hemostasis
Central to our hypothesis is thrombomodulin. Detailed reviews of hemostasis and blood coagulation in general [6–11] and thrombomodulin in particular [12–15] have been recently published and will not be repeated at length here. Briefly however, thrombomodulin is critically important in regulation of clotting. Thrombomodulin was isolated initially by Esmon’s group in 1981 [16]. Thrombomodulin serves, as the name implies, to regulate the activity of thrombin. In complex with thrombin it activates protein C, which degrades key factors in the clotting cascade [17]. Deficiency in protein C or activated protein C is well established as increasing the risk of thrombosis [17, 18]. Low levels of thrombomodulin are a well established risk factor for heart disease [19]. Without a doubt, thrombomodulin plays a key role in slowing or stopping clotting.
While superficially it may seem contradictory, more recent discoveries have shown that thrombomodulin [20–22], again in complex with thrombin, activates thrombin activatable fibrinolysis inhibitor (TAFI) [23], which stabilizes clots [24], Reflection on this point emphasizes the critical role thrombomodulin plays in clotting regulation, since it controls both the rate at which clots form and the rate at which clots breakdown. As we shall see, a critical question is the molecular mechanism by which thrombomodulin strikes a balance between promoting clot formation or degradation.
Thrombomodulin is found anchored on the luminal surface of the endothelium. In rats, it is found predominantly in the lungs, at much higher levels than even other highly vascularized organs such as liver or kidney [25, 26]. Thrombomodulin does undergo endocytosis with subsequent degradation, but significant amounts of thrombomodulin are cleaved from the surface to circulate in the blood before being cleared through the urine [27]. High levels of thrombomodulin in plasma have been reported in a variety of conditions including diabetes, lupus, pre-eclampsia, and disseminated intravascular coagulation [12] and are believed to serve as a good marker of endothelial damage [28, 29]. Several small studies have found that levels of plasma thrombomodulin are not correlated with the incidence of cardiovascular disease [30–32] but work on a larger study population has found that high levels, taken to be indicative of higher levels of thrombomodulin expression, appear protective [19].
Oxidation of methionine 388 is critical in the regulation of thrombomodulin activity
In 1992 a paper was published showing that the oxidation of a single methionine, residue 388, destroyed most of the activity of the thrombomodulin-thrombin complex in proteolytically activating protein C [33]. Oxidation of other methionines in the protein did not appear to alter activity [34]. Substitution of the methionine with a leucine result in a mutant capable of activating protein C, without sensitivity to oxidation.
Numerous studies [35–38] have shown that binding and activation of thrombin only requires the 81 amino acid fragment of thrombomodulin corresponding to EGF domains 4 and 5, although domain 6 increases the Km of thrombomodulin for thrombin by a factor of 10 without altering the kcat of the thrombomodulin-thrombin complex for protein C [39]. A thrombomodulin fragment consisting of domains 5 and 6 does bind to thrombin, but the complex fails to activate protein C [40]. Met 388 is one of three residues linking domains 4 and 5 [34, 35].
This study was undertaken because an increased tendency toward coagulation is a common complication of inflammation, and thrombosis is an important contributor to death in inflammatory processes such as sepsis. The examination of methionine oxidation was a natural extension of various observations of regulation of activity by methionine oxidation wherein methionine is oxidized to the sulfoxide form by the reactive oxygen species generated by leukocytes and neutrophils during inflammation [41–45]. To cite just one example, it has been shown that the C5 component of the complement system, normally activated by proteolysis, can be activated by the oxidation of a specific methionine residue [46–48]. Active C5 triggers the complement cascade. These workers argue that leukocytes, which generate various oxygen radicals when active, may thus also activate the complement system to aid in the immune response.
In 2000, it was further shown that oxidation of Met388 in thrombomodulin has no effect on the clot stabilizing activation of TAFI by the thrombomodulin-thrombin complex [49]. Thus, oxidation of this methionine removes the downregulation of clotting by thrombomodulin with no effect on the coagulation enhancing regulatory capacity. While both of these effects have only been demonstrated in vitro, it seems likely that the combination of effects acts in vivo to increase the propensity to form clots whenever significant amounts of methionine oxidation have occurred. It is plausible that this methionine forms a crucial molecular switch which controls the balance between clot formation and breakdown. In the course of this work, this group reconfirmed the original observation that Met388 oxidation that oxidation of Met388 dramatically decreases the activation of protein C by the thrombomodulin-thrombin complex.
Even more recently the structural basis of the inactivation of thrombomodulin has been determined. Wood et al. [34, 50] solved the structure of thrombomodulin fragments by both NMR and x-ray crystallography. Structures of the unoxidized and oxidized forms showed clear structural differences in the fifth domain of thrombomodulin. Phenylalanine 376 packs against the hydrophobic methionine but occupies a substantially different position when the hydrophilic sulfoxide form is present, making it a key part of the conformational switch. These structural changes bury several residues which interact with thrombin in the structure of the thrombomodulin-thrombin complex [35].
Although the primary focus of this work by the Komives group at UCSD was structural, they also performed binding and activity assays [34]. Oxidation of Met388 increased the Km for thrombin from 140 ± 5 nM to 460 ± 70 nM. Oxidation left Km for the binding of the thrombomodulin-thrombin complex with protein C unchanged within experimental error. However, kcat for the activation of protein C by the thrombomodulin-thrombin complex dropped from 5.0 ± 0.1 s−1 to 1.4 ± 0.1 s−1. Specific activity fell by almost an order of magnitude, the oxidized form showing just 15% of the activity of the unoxidized form.
We note again that this effect is not likely a fluke unique to humans since this methionine is conserved in all thrombomodulin genes that have been sequenced, as shown in Figure 1. Phenylalanine 376, the other half of the conformational switch, is also conserved.
Figure 1.

Alignment of thrombomodulin genes sequenced to date in the region of interest. Squirrel monkey, rhesus monkey, and chimpanzee thrombomodulin genes are virtually identical to human, including at Met388, and are not shown. Methionine 388 is indicated in bold and is conserved. Note the conservation of phenylalanine 376 as described in the text as well as the cysteines, involved in disulfide bonds, and of asparagine 391, which has been shown to be glycosylated in humans.
This work has contributed to the acceptance of thrombomodulin as a key link between inflammation and coagulation [22, 51–53]. We argue here that a strong case can be made that oxidation of methionine 388 in thrombomodulin is important in a variety of other human diseases.
Is a decrease in thrombomodulin activity biologically relevant?
The question of biological relevance has been addressed by mutation of thrombomodulin in mice [54]. In the same numbering system as elsewhere in this paper, the glutamate at position 387, normally a glutamine side chain in humans, was substituted with a proline. This is, of course, immediately adjacent to the methionine of concern in the hypothesis put forward here. As might be expected, the ability of this mutant protein to activate protein C suffered, an estimated factor of about a thousand fold reduced efficiency in protein C activation with physiological concentrations of the various proteins.
Despite this virtual elimination of the activation of protein C, the mutant animals are viable, but are impaired in ways reminiscent of cardiovascular disease. The initial paper focused more on the creation of the strain and the reproductive effects, but the most notable observation from the perspective of cardiovascular disease is that mutant mice suffer from increased fibrin deposition in the heart and lungs [54], by as much as ten fold over wild-type in 3 to 6 month old mice [55]. Enhanced fibrin deposition has been linked to myocardial infarction [56]. That these mice are hypercoaguable was shown in subsequent work by an accelerated rate of platelet thrombus growth after FeCl3 injury to the carotid artery [57]. The time at which flow was reduced to 50% of the initial flow (t50) was reduced by approximately 22% [57]. Mice with the same mutation in a slightly different genetic background, when subjected to the same FeCl3 insult, showed complete thrombotic occlusion in 80% of the mutant animals, versus only 27% of the wild-type mice [58]. Similarly, surgical occlusion of the carotid artery resulted in extensive stasis-induced thrombosis, extending the entire length of the artery in many of the mutant mice. In wild-type mice occlusion was restricted to within less than 1 mm of the ligation [57]. These mice with reduced capability of thrombomodulin to activate protein C also exhibited increased sensitivity to lipopolysaccharide-induced septicemia. Injection of the wild-type LD50 dose of LPS resulted in 100% mortality in mutant mice [57]. Further, the mutant mice succumbed much earlier than the wild-type mice.
Methionine oxidation in proteins
This is an appropriate point at which to quickly review the literature on methionine oxidation more generally. Proteins are well known to be sensitive to oxidative damage, often with important biological effects. Protein oxidation has been suggested as a causative or contributory factor in many diseases [59]. Oxidized proteins have been found to increase in aged organisms, leading to the proposal that protein oxidation contributes to the aging process [60, 61].
Methionine, cysteine, tryptophan, tyrosine, and histidine residues are susceptible to oxidation. Cysteine and methionine are the most easily oxidized. The oxidation of the cysteine thiol to the disulfide form is a normal, beneficial, and familiar reaction. Oxidation of methionine may be less familiar, but still readily occurs. The oxidation to a sulfone can be accomplished by fairly strong oxidants, but it is the oxidation of methionine to the sulfoxide form (Figure 2) that concerns us here, as it has been shown to occur in a wide variety of proteins with both mild and strong oxidizing species, such as H2O2, hypochlorous acid, and superoxide [62–66].
Figure 2.
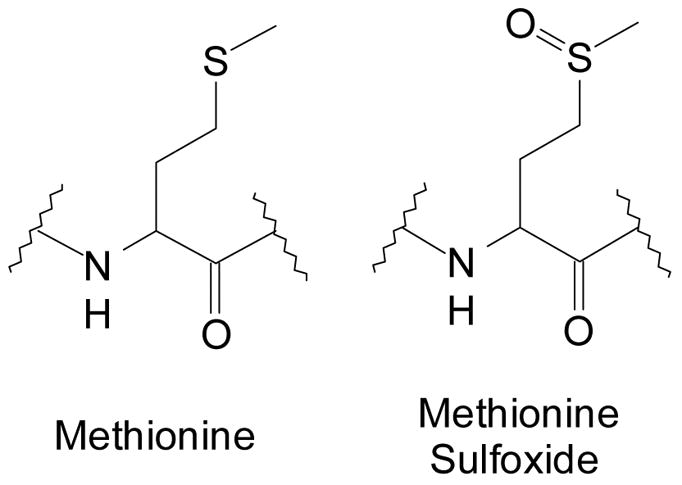
Structures of methionine and methionine sulfoxide residues.
Oxidants found naturally in biological systems [33, 44, 65–67], cigarette smoke [68–72], and ozone [73, 74] or other environmental oxidants [75, 76] have all been demonstrated to cause methionine sulfoxide formation in proteins and peptides. Methionine oxidation often reduces or eliminates biological activity [77, 78]. Methionine oxidation is therefore of serious concern when proteins are used as pharmaceuticals because oxidation, which may occur readily during processing or storage, often alters activity [79–82]. The alteration in activity is undoubtedly due to the considerable alteration in the character of the methionine. The side chain alters in size and geometry, but more importantly the sulfoxide is very polar and hydrophilic, with significant partial positive charge on the sulfur and negative charge on the oxygen. The oxygen is an excellent hydrogen bond acceptor. In contrast the reduced form is very non-polar and hydrophobic. This can readily lead to changes in the stability of different protein conformations or in the ability of a binding site to recognize another protein or substrate.
Reactive oxygen species have lately become more widely recognized as biologically important messengers [44, 83–87] and methionine is one likely target for oxidation by such species [44, 45, 88, 89]. Indeed, recent and somewhat surprising work has shown that the rate of reaction of methionine with hypochlorous acid is faster than the reaction with cysteine [90]. Methionine as a target of redox signaling is particularly interesting since sulfoxide formation is reversible. Indeed, all organisms have a variety of enzymes whose specific role to reduce methionine sulfoxide in proteins and peptides back to the thioether [91].
Thus, there are many well documented, biologically relevant examples of methionine oxidation in other proteins. Evidence that methionine oxidation is an important reversible regulator of biological activity is accumulating at a rapid pace. It is plausible that oxidation of methionine 388 in thrombomodulin plays an important role in the regulation of hemostasis. Whether or not oxidation of Met388 is an inappropriate activation of a normal signaling system or just coincidently deleterious is not strictly relevant to the hypothesis advanced here. It is clear however that Met388 oxidation causes a profound biological effect and that such effect is a reasonable result of methionine oxidation is well supported by analogy in other proteins.
Link to smoking
Tobacco smoke is a complex mixture, but includes many oxidizing species that impose significant oxidative stress on the body [92–100]. These oxidizing species include organic radicals and hydrogen peroxide, which can oxidize methionine. Further, smokers are known to have increased levels of immune system cells such as activated neutrophils in their lungs [101, 102], cells which in turn release still more oxidizing agents. In addition, it has been shown that smokers have elevated levels of iron in their lungs [103]. Iron catalyzes the Fenton reaction of ascorbate, simultaneously consuming this key antioxidant and producing oxidizing radicals [104].
Most importantly, there is a well established linkage between the disease of emphysema in smokers and methionine oxidation of another protein, α1-antitrypsin [43, 68, 105–108], Oxidation of either methionine 351 or 358 in the binding site of α1-antitrypsin destroys the protein’s ability to bind to and inhibit elastase [109], The degradation of elastin by elastase is an important step in enabling immune cells to infiltrate the site of an infection. The generation of reactive oxygen species by leukocytes and neutrophils during inflammation thus facilitates the immune response to infection. However, it is now indisputable that components of cigarette smoke can carry out this oxidation [71, 72, 110–112], leading to the inappropriate and chronic activation of elastase and, hence, causing emphysema.
Another common cause of emphysema is chronic exposure to mineral dust, such as in coal and hard rock miners. It has more recently been shown that mineral dusts can cause oxidation of methionine in α1-antitrypsin in vitro [113]. Even more convincingly, the ability of different dusts to cause breakdown of elastin in vivo in rats was correlated with the ability of the dust to oxidize methionine in the inhibitor in vitro. Thus, two apparently different causes of emphysema seem to have the same molecular origin: methionine oxidation.
Curiously, despite a much greater toll in human lives, the molecular cause of the hyperthrombotic state in smokers has remained unclear. There is growing evidence that free radical, oxidative damage to the endothelium is very important in the development of cardiovascular disease in smokers, although most attention seems focused on oxidative impairment of nitric oxide signaling [114, 115]. It is our belief that the precedent established for the cause of emphysema and the known effect of methionine oxidation upon thrombomodulin activity make it extremely likely that a similar oxidation is taking place in smokers, making it an important molecular root of their cardiovascular ills. In addition to their pronounced tendency to clot, there is growing evidence that a prothrombotic state contributes to atherogenesis [116–120], thus thrombomodulin oxidation may increase the risk of smokers for atherosclerosis as well.
We remind the reader that the predominant location for thrombomodulin is the lung [25, 26, 121], a location which obviously renders it even more vulnerable to oxidation by the witch’s brew of reactive oxidizing species in cigarette smoke and the high levels of activated immune cells caused by smoking. The linkage of low levels of thrombomodulin with increased risk of heart disease further strengthens our hypothesis [19]. We note again that while the levels of thrombomodulin in smokers and non-smokers have been examined, no group has ever examined thrombomodulin methionine oxidation in vivo in smokers or non-smokers.
The Fernández group published a paper strengthening the case for a linkage between smoking and thrombomodulin oxidation as an important molecular cause of their cardiovascular disease. Apparently reaching the same hypothesis that we present here, they tested the levels of activated protein C in non-smokers and smokers [122]. (Low levels of activated protein C have been found by others to be a strong, independent risk factor for venous thromboembolism [123, 124] and may be a risk factor for ischemic stroke [125].) Circulating levels of activated protein C were a very statistically significant 23.3% lower in smokers than nonsmokers. While other causes, such as reduced expression of protein C and increased degradation of protein C or of activated protein C can not be ruled out, one possible cause of low protein C levels is that smokers have reduced thrombomodulin activity, which could be due to methionine oxidation.
The circumstantial case for our hypothesis is strong. This hypothesis postulates a plausible molecular mechanism linking the oxidative stress imposed by smoking to the disruption of the key endothelial function of hemostasis, leading to thrombosis. Further, it is not a terribly speculative stretch to imagine that this oxidative modification of thrombomodulin may be a very useful biomarker for cigarette smoke exposure and for cardiovascular risk [56].
Oxidative stress is present in other conditions which are prone to thrombosis
Oxidative stress in general is linked to increased tendency to coagulate, but the molecular mechanism is clearly complex and significant factors remain unknown [115, 126–129]. We have already mentioned sepsis, which motivated the original study of methionine 388 oxidation. Three other conditions where oxidative stress and hypercoagulability are present bear mention in particular.
Diabetes is well known to cause oxidative stress, thought to be due to oxidation of glucose in the presence of transition metal ions with concomitant product of hydrogen peroxide and because increased metabolic flux through unusual pathways increases mitochondrial superoxide production [130–132]. Diabetics are equally well known to have an elevated risk of thrombosis [3–5, 133, 134] and cardiovascular disease [135]. The molecular causes are by no means fully understood. However, we found very interesting a recent paper reporting that activated protein C levels in type-2 diabetics are significantly depressed relative to normal controls [136].
Elevated homocysteine levels are a very strongly established marker for increased risk of heart disease and thrombosis [137, 138]. The molecular cause of thrombosis in individuals with hyperhomocysteinemia is unclear [139–143]. Oxidative stress is known to be present in individuals with elevated levels of homocysteine [144, 145]. There is some controversy over whether homocysteine is the cause of the oxidative stress (from disulfide formation in the presence of metals and oxygen and concomitant peroxide and thio radical formation [146]) or if it is just a marker of oxidative stress caused by an underlying folate deficiency [147]. However, all agree hyperhomocysteinemia is associated with oxidative stress, whether or not homocysteine directly causes it. It is less clear if our hypothesis fits this condition, since one study has reported that activated protein C levels are no different in individuals with and without elevated homocysteine [148] and another study failed to find increased activation of the coagulation system in healthy volunteers under methionine load to induce mild hyperhomocysteinemia [149]. Still, the appearance again of an increased risk of thrombosis in a condition associated with oxidative stress is interesting and suggestive.
Similarly, it is also known the patients with hyperthyroidism are subject to elevated levels of oxidative stress [150–152]. Such patients are also known to be at increased risk for thrombosis [153, 154]. Indeed, as many as 18% of patients with thryotoxicosis actually die from embolism [155]. We draw attention to a hypothesis that the cause of elevated levels of death from cardiovascular disease in end-stage renal failure, another condition closely linked with diabetes, is due to the elevated levels of oxidative stress known to exist in this condition [129, 156, 157]. Oxidative stress and an elevated tendency to coagulate [157, 158] are clearly present in this patient population. Lastly, oxidative stress and inflammation are clearly associated with one another in many diseases, including the classic inflammatory disease, arthritis [159]. It is intriguing that arthritics were recently shown to have an elevated risk of cardiovascular disease [160], although superficially there is no reason to link inflammation in the joints to cardiovascular problems. Arthritic patients appear to be prothrombotic as well [161, 162]. In short, oxidative stress or inflammation in general seems to increase cardiovascular risk and blood coagulability. Oxidation of methionine 388 in thrombomodulin may be a key molecular linkage between these disparate conditions and cardiovascular risk.
Thrombosis is the result of a multitude of factors and oxidative stress can have a multitude of effects
Lastly, we recognize that oxidative stress in general and smoking, diabetes, or hyperhomocysteinemia, in particular, have other significant health effects beyond hypercoagulability. We have discussed briefly above the development of emphysema through methionine oxidation and, to name just one other effect of oxidative stress, the role of lipid oxidation in LDL in atherothrombosis is becoming clearer. Again, although other important effects of oxidative stress and methionine oxidation have been identified, it seems certain that additional effects are waiting for identification.
Similarly, we wish to make clear that we recognize that many contributing factors, both environmental and genetic, are responsible for the medical conditions discussed in this paper. Many of these other causative agents are very well supported by huge amounts of evidence and we have touched lightly, if at all, on those factors. Even though important contributors to thrombotic disorders have been identified, it seems virtually certain our understanding of identified causes is incomplete and that other contributing factors remain to be identified. We have emphasized in the discussion above just one such possible factor disrupting hemostasis, and while the overall causes of cardiovascular disease are undoubtedly more complicated, we feel the case is strong that thrombomodulin Met388 oxidation is important in the health effects of smoking, diabetes, and other conditions that impose oxidative stress.
Acknowledgments
Our gratitude to Samuel Gellman for the conversations that sparked interest in methionine oxidation. We thank Martin Hauer-Jensen, Enrico Di Cera, and Elizabeth Komives for helpful discussion and criticism. This work was supported by the Arkansas Biosciences Institute and NIH grant 1R15HL078994-01A1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–7. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 2.Burns DM. Epidemiology of smoking-induced cardiovascular disease. Prog Cardiovasc Dis. 2003;46:11–29. doi: 10.1016/s0033-0620(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 3.Dunn EJ, Grant PJ. Type 2 diabetes: an atherothrombotic syndrome. Curr Mol Med. 2005;5:323–32. doi: 10.2174/1566524053766059. [DOI] [PubMed] [Google Scholar]
- 4.Aras R, Sowers JR, Arora R. The proinflammatory and hypercoagulable state of diabetes mellitus. Rev Cardiovasc Med. 2005;6:84–97. [PubMed] [Google Scholar]
- 5.Carr ME. Diabetes mellitus: a hypercoagulable state. J Diabetes Complications. 2001;15:44–54. doi: 10.1016/s1056-8727(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 6.Dahlback B. Blood coagulation and its regulation by anticoagulant pathways: genetic pathogenesis of bleeding and thrombotic diseases. J Intern Med. 2005;257:209–23. doi: 10.1111/j.1365-2796.2004.01444.x. [DOI] [PubMed] [Google Scholar]
- 7.Davidson CJ, Tuddenham EG, McVey JH. 450 million years of hemostasis. J Thromb Haemost. 2003;1:1487–94. doi: 10.1046/j.1538-7836.2003.00334.x. [DOI] [PubMed] [Google Scholar]
- 8.Norris LA. Blood coagulation. Best Pract Res Clin Obstet Gynaecol. 2003;17:369–83. doi: 10.1016/s1521-6934(03)00014-2. [DOI] [PubMed] [Google Scholar]
- 9.Lasne D, Jude B, Susen S. From normal to pathological hemostasis. Can J Anaesth. 2006;53:S2–11. doi: 10.1007/BF03022247. [DOI] [PubMed] [Google Scholar]
- 10.Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol. 2006;26:41–8. doi: 10.1161/01.ATV.0000193624.28251.83. [DOI] [PubMed] [Google Scholar]
- 11.Schenone M, Furie BC, Furie B. The blood coagulation cascade. Curr Opin Hematol. 2004;11:272–7. doi: 10.1097/01.moh.0000130308.37353.d4. [DOI] [PubMed] [Google Scholar]
- 12.Wu KK, Matijevic-Aleksic N. Thrombomodulin: a linker of coagulation and fibrinolysis and predictor of risk of arterial thrombosis. Ann Med. 2000;32 (Suppl 1):73–7. [PubMed] [Google Scholar]
- 13.Weiler H, Isermann BH. Thrombomodulin. J Thromb Haemost. 2003;1:1515–24. doi: 10.1046/j.1538-7836.2003.00306.x. [DOI] [PubMed] [Google Scholar]
- 14.van de Wouwer M, Collen D, Conway EM. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. 2004;24:1374–83. doi: 10.1161/01.ATV.0000134298.25489.92. [DOI] [PubMed] [Google Scholar]
- 15.Esmon CT. Crosstalk between inflammation and thrombosis. Maturitas. 2004;47:305–14. doi: 10.1016/j.maturitas.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 16.Owen WG, Esmon CT. Functional properties of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. 1981;256:5532–5. [PubMed] [Google Scholar]
- 17.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 18.Espana F, Medina P, Navarro S, Estelles A, Aznar J. Inherited abnormalities in the protein C activation pathway. Pathophysiol Haemost Thromb. 2002;32:241–4. doi: 10.1159/000073573. [DOI] [PubMed] [Google Scholar]
- 19.Salomaa V, Matei C, Aleksic N, et al. Soluble thrombomodulin as a predictor of incident coronary heart disease and symptomless carotid artery atherosclerosis in the Atherosclerosis Risk in Communities (ARIC) Study: a case-cohort study. Lancet. 1999;353:1729–34. doi: 10.1016/s0140-6736(98)09057-6. [DOI] [PubMed] [Google Scholar]
- 20.Mosnier LO, Meijers JC, Bouma BN. Regulation of fibrinolysis in plasma by TAFI and protein C is dependent on the concentration of thrombomodulin. Thromb Haemost. 2001;85:5–11. [PubMed] [Google Scholar]
- 21.Nesheim M. Thrombin and fibrinolysis. Chest. 2003;124:33S–9S. doi: 10.1378/chest.124.3_suppl.33s. [DOI] [PubMed] [Google Scholar]
- 22.Nesheim M, Wang W, Boffa M, Nagashima M, Morser J, Bajzar L. Thrombin, thrombomodulin and TAFI in the molecular link between coagulation and fibrinolysis. Thromb Haemost. 1997;78:386–91. [PubMed] [Google Scholar]
- 23.Marx PF. Thrombin-activatable fibrinolysis inhibitor. Curr Med Chem. 2004;11:2335–48. doi: 10.2174/0929867043364586. [DOI] [PubMed] [Google Scholar]
- 24.Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271:16603–8. doi: 10.1074/jbc.271.28.16603. [DOI] [PubMed] [Google Scholar]
- 25.Song YK, Liu D, Maruyama KZ, Takizawa T. Antibody mediated lung targeting of long-circulating emulsions. PDA J Pharm Sci Technol. 1996;50:372–7. [PubMed] [Google Scholar]
- 26.Ford VA, Stringer C, Kennel SJ. Thrombomodulin is preferentially expressed in Balb/c lung microvessels. J Biol Chem. 1992;267:5446–50. [PubMed] [Google Scholar]
- 27.Ishii H, Majerus PW. Thrombomodulin is present in human plasma and urine. J Clin Invest. 1985;76:2178–81. doi: 10.1172/JCI112225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boffa MC, Karmochkine M. Thrombomodulin: an overview and potential implications in vascular disorders. Lupus. 1998;7 (Suppl 2):S120–5. doi: 10.1177/096120339800700227. [DOI] [PubMed] [Google Scholar]
- 29.Ishii H, Uchiyama H, Kazama M. Soluble thrombomodulin antigen in conditioned medium is increased by damage of endothelial cells. Thromb Haemost. 1991;65:618–23. [PubMed] [Google Scholar]
- 30.Blann AD, McCollum CN. von Willebrand factor and soluble thrombomodulin as predictors of adverse events among subjects with peripheral or coronary atherosclerosis. Blood Coagul Fibrinolysis. 1999;10:375–80. doi: 10.1097/00001721-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Blann AD, McCollum CN, Lip GY. Relationship between plasma markers of endothelial cell integrity and the Framingham cardiovascular disease risk-factor scores in apparently healthy individuals. Blood Coagul Fibrinolysis. 2002;13:513–8. doi: 10.1097/00001721-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Nilsson TK, Hellsten G, Amiral J. Plasma thrombomodulin concentrations in relation to cardiovascular risk factors in a population sample. Blood Coagul Fibrinolysis. 1993;4:455–8. doi: 10.1097/00001721-199306000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Glaser CB, Morser J, Clarke JH, et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J Clin Invest. 1992;90:2565–73. doi: 10.1172/JCI116151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wood MJ, Becvar LA, Prieto JH, Melacini G, Komives EA. NMR structures reveal how oxidation inactivates thrombomodulin. Biochemistry. 2003;42:11932–42. doi: 10.1021/bi034646q. [DOI] [PubMed] [Google Scholar]
- 35.Fuentes-Prior P, Iwanaga Y, Huber R, et al. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 2000;404:518–25. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 36.Wood MJ, Sampoli Benitez BA, Komives EA. Solution structure of the smallest cofactor-active fragment of thrombomodulin. Nat Struct Biol. 2000;7:200–4. doi: 10.1038/73302. [DOI] [PubMed] [Google Scholar]
- 37.Nagashima M, Lundh E, Leonard JC, Morser J, Parkinson JF. Alanine-scanning mutagenesis of the epidermal growth factor-like domains of human thrombomodulin identifies critical residues for its cofactor activity. J Biol Chem. 1993;268:2888–92. [PubMed] [Google Scholar]
- 38.Sadler JE, Lentz SR, Sheehan JP, Tsiang M, Wu Q. Structure-function relationships of the thrombin-thrombomodulin interaction. Haemostasis. 1993;23 (Suppl 1):183–93. doi: 10.1159/000216927. [DOI] [PubMed] [Google Scholar]
- 39.White CE, Hunter MJ, Meininger DP, White LR, Komives EA. Large-scale expression, purification and characterization of small fragments of thrombomodulin: the roles of the sixth domain and of methionine 388. Protein Eng. 1995;8:1177–87. doi: 10.1093/protein/8.11.1177. [DOI] [PubMed] [Google Scholar]
- 40.Schenk-Braat EA, Morser J, Rijken DC. Identification of the epidermal growth factor-like domains of thrombomodulin essential for the acceleration of thrombin-mediated inactivation of single-chain urokinase-type plasminogen activator. Eur J Biochem. 2001;268:5562–9. doi: 10.1046/j.1432-1033.2001.02487.x. [DOI] [PubMed] [Google Scholar]
- 41.Brot N, Weissbach H. Biochemistry of methionine sulfoxide residues in proteins. Biofactors. 1991;3:91–6. [PubMed] [Google Scholar]
- 42.Drozdz R, Naskalski JW. Inactivation and denaturation of some proteins by enzyme system: myeloperoxidase, chloride and hydrogen peroxide. Folia Histochem Cytobiol. 1993;31:71–5. [PubMed] [Google Scholar]
- 43.Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci. 2000;899:191–208. doi: 10.1111/j.1749-6632.2000.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 44.Naskalski JW, Marcinkiewicz J, Drozdz R. Myeloperoxidase-mediated protein oxidation: its possible biological functions. Clin Chem Lab Med. 2002;40:463–8. doi: 10.1515/CCLM.2002.080. [DOI] [PubMed] [Google Scholar]
- 45.Levine RL, Moskovitz J, Stadtman ER. Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. IUBMB Life. 2000;50:301–7. doi: 10.1080/713803735. [DOI] [PubMed] [Google Scholar]
- 46.Vogt W, Damerau B, von Zabern I, Nolte R, Brunahl D. Non-enzymic activation of the fifth component of human complement, by oxygen radicals. Some properties of the activation product, C5b-like C5. Mol Immunol. 1989;26:1133–42. doi: 10.1016/0161-5890(89)90057-6. [DOI] [PubMed] [Google Scholar]
- 47.Vogt W, Hesse D. Oxidants generated by the myeloperoxidase-halide system activate the fifth component of human complement, C5. Immunobiology. 1994;192:1–9. doi: 10.1016/S0171-2985(11)80403-1. [DOI] [PubMed] [Google Scholar]
- 48.Discipio RG, Daffern PJ, Kawahara M, et al. Cleavage of human complement component C5 by cysteine proteinases from Porphyromonas (Bacteroides) gingivalis. Prior oxidation of C5 augments proteinase digestion of C5. Immunology. 1996;87:660–7. doi: 10.1046/j.1365-2567.1996.478594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Nagashima M, Schneider M, Morser J, Nesheim M. Elements of the primary structure of thrombomodulin required for efficient thrombin-activable fibrinolysis inhibitor activation. J Biol Chem. 2000;275:22942–7. doi: 10.1074/jbc.M001760200. [DOI] [PubMed] [Google Scholar]
- 50.Wood MJ, Helena Prieto J, Komives EA. Structural and functional consequences of methionine oxidation in thrombomodulin. Biochim Biophys Acta. 2005;1703:141–7. doi: 10.1016/j.bbapap.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 51.Opal SM. Interactions between coagulation and inflammation. Scand J Infect Dis. 2003;35:545–54. doi: 10.1080/00365540310015638. [DOI] [PubMed] [Google Scholar]
- 52.Esmon CT. Does inflammation contribute to thrombotic events? Haemostasis. 2000;30 (Suppl 2):34–40. doi: 10.1159/000054161. [DOI] [PubMed] [Google Scholar]
- 53.Esmon CT. Protein C pathway in sepsis. Ann Med. 2002;34:598–605. doi: 10.1080/078538902321117823. [DOI] [PubMed] [Google Scholar]
- 54.Weiler-Guettler H, Christie PD, Beeler DL, et al. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J Clin Invest. 1998;101:1983–91. doi: 10.1172/JCI2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christie PD, Edelberg JM, Picard MH, et al. A murine model of myocardial microvascular thrombosis. J Clin Invest. 1999;104:533–9. doi: 10.1172/JCI7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nesheim M. Myocardial infarction and the balance between fibrin deposition and removal. Ital Heart J. 2001;2:641–5. [PubMed] [Google Scholar]
- 57.Weiler H, Lindner V, Kerlin B, et al. Characterization of a mouse model for thrombomodulin deficiency. Arterioscler Thromb Vasc Biol. 2001;21:1531–7. doi: 10.1161/hq0901.094496. [DOI] [PubMed] [Google Scholar]
- 58.Dorffler-Melly J, de Kruif M, Schwarte LA, et al. Functional thrombomodulin deficiency causes enhanced thrombus growth in a murine model of carotid artery thrombosis. Basic Res Cardiol. 2003;98:347–52. doi: 10.1007/s00395-003-0416-9. [DOI] [PubMed] [Google Scholar]
- 59.Dean RT, Fu S, Stocker R, Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. Biochem J. 1997;324 ( Pt 1):1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–40. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 61.Friguet B. Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Lett. 2006;580:2910–6. doi: 10.1016/j.febslet.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 62.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–18. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 63.Richardson DE, Regino CA, Yao H, Johnson JV. Methionine oxidation by peroxymonocarbonate, a reactive oxygen species formed from CO2/bicarbonate and hydrogen peroxide. Free Radic Biol Med. 2003;35:1538–50. doi: 10.1016/j.freeradbiomed.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 64.Drozdz R, Naskalski JW, Sznajd J. Oxidation of amino acids and peptides in reaction with myeloperoxidase, chloride and hydrogen peroxide. Biochim Biophys Acta. 1988;957:47–52. doi: 10.1016/0167-4838(88)90155-0. [DOI] [PubMed] [Google Scholar]
- 65.Carr AC, Hawkins CL, Thomas SR, Stocker R, Frei B. Relative reactivities of N-chloramines and hypochlorous acid with human plasma constituents. Free Radic Biol Med. 2001;30:526–36. doi: 10.1016/s0891-5849(00)00495-0. [DOI] [PubMed] [Google Scholar]
- 66.Peskin AV, Winterbourn CC. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic Biol Med. 2001;30:572–9. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 67.Hong J, Schoneich C. The metal-catalyzed oxidation of methionine in peptides by Fenton systems involves two consecutive one-electron oxidation processes. Free Radic Biol Med. 2001;31:1432–41. doi: 10.1016/s0891-5849(01)00722-5. [DOI] [PubMed] [Google Scholar]
- 68.Gadek JE, Fells GA, Crystal RG. Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humans. Science. 1979;206:1315–6. doi: 10.1126/science.316188. [DOI] [PubMed] [Google Scholar]
- 69.Boudier C, Pelletier A, Pauli G, Bieth JG. The functional activity of alpha 1-proteinase inhibitor in bronchoalveolar lavage fluids from healthy human smokers and non-smokers. Clin Chim Acta. 1983;132:309–15. doi: 10.1016/0009-8981(83)90009-8. [DOI] [PubMed] [Google Scholar]
- 70.Maier KL, Leuschel L, Costabel U. Increased oxidized methionine residues in BAL fluid proteins in acute or chronic bronchitis. Eur Respir J. 1992;5:651–8. [PubMed] [Google Scholar]
- 71.Evans MD, Church DF, Pryor WA. Aqueous cigarette tar extracts damage human alpha-1-proteinase inhibitor. Chem Biol Interact. 1991;79:151–64. doi: 10.1016/0009-2797(91)90079-m. [DOI] [PubMed] [Google Scholar]
- 72.Evans MD, Pryor WA. Damage to human alpha-1-proteinase inhibitor by aqueous cigarette tar extracts and the formation of methionine sulfoxide. Chem Res Toxicol. 1992;5:654–60. doi: 10.1021/tx00029a010. [DOI] [PubMed] [Google Scholar]
- 73.Berlett BS, Levine RL, Stadtman ER. Comparison of the effects of ozone on the modification of amino acid residues in glutamine synthetase and bovine serum albumin. J Biol Chem. 1996;271:4177–82. doi: 10.1074/jbc.271.8.4177. [DOI] [PubMed] [Google Scholar]
- 74.Banerjee SK, Mudd JB. Reaction of ozone with glycophorin in solution and in lipid vesicles. Arch Biochem Biophys. 1992;295:84–9. doi: 10.1016/0003-9861(92)90491-e. [DOI] [PubMed] [Google Scholar]
- 75.Blaurock B, Hippeli S, Metz N, Elstner EF. Oxidative destruction of biomolecules by gasoline engine exhaust products and detoxifying effects of the three-way catalytic converter. Arch Toxicol. 1992;66:681–7. doi: 10.1007/BF01972618. [DOI] [PubMed] [Google Scholar]
- 76.Maier KL, Matejkova E, Hinze H, Leuschel L, Weber H, Beck-Speier I. Different selectivities of oxidants during oxidation of methionine residues in the alpha-1-proteinase inhibitor. FEBS Lett. 1989;250:221–6. doi: 10.1016/0014-5793(89)80725-2. [DOI] [PubMed] [Google Scholar]
- 77.Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl. 2003;42:4742–58. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- 78.Dalle-Donne I, Rossi R, Giustarini D, et al. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radic Biol Med. 2002;32:927–37. doi: 10.1016/s0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 79.Krishnamurthy R, Manning MC. The stability factor: importance in formulation development. Curr Pharm Biotechnol. 2002;3:361–71. doi: 10.2174/1389201023378229. [DOI] [PubMed] [Google Scholar]
- 80.Berti PJ, Ekiel I, Lindahl P, Abrahamson M, Storer AC. Affinity purification and elimination of methionine oxidation in recombinant human cystatin C. Protein Expr Purif. 1997;11:111–8. doi: 10.1006/prep.1997.0763. [DOI] [PubMed] [Google Scholar]
- 81.Lam XM, Yang JY, Cleland JL. Antioxidants for prevention of methionine oxidation in recombinant monoclonal antibody HER2. J Pharm Sci. 1997;86:1250–5. doi: 10.1021/js970143s. [DOI] [PubMed] [Google Scholar]
- 82.Fransson J, Florin-Robertsson E, Axelsson K, Nyhlen C. Oxidation of human insulin-like growth factor I in formulation studies: kinetics of methionine oxidation in aqueous solution and in solid state. Pharm Res. 1996;13:1252–7. doi: 10.1023/a:1016032808039. [DOI] [PubMed] [Google Scholar]
- 83.Chen K, Thomas SR, Keaney JF., Jr Beyond LDL oxidation: ROS in vascular signal transduction. Free Radic Biol Med. 2003;35:117–32. doi: 10.1016/s0891-5849(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 84.Stadtman ER, Levine RL. Why have cells selected reactive oxygen species to regulate cell signaling events? Hum Exp Toxicol. 2002;21:83. doi: 10.1191/0960327102ht215oa. [DOI] [PubMed] [Google Scholar]
- 85.Klotz LO. Oxidant-induced signaling: effects of peroxynitrite and singlet oxygen. Biol Chem. 2002;383:443–56. doi: 10.1515/BC.2002.047. [DOI] [PubMed] [Google Scholar]
- 86.Sauer H, Wartenberg M, Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem. 2001;11:173–86. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 87.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–83. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 88.Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5:577–82. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- 89.Hoshi T, Heinemann S. Regulation of cell function by methionine oxidation and reduction. J Physiol. 2001;531:1–11. doi: 10.1111/j.1469-7793.2001.0001j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pattison DI, Davies MJ. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem Res Toxicol. 2001;14:1453–64. doi: 10.1021/tx0155451. [DOI] [PubMed] [Google Scholar]
- 91.Moskovitz J. Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim Biophys Acta. 2005;1703:213–9. doi: 10.1016/j.bbapap.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 92.Park EM, Park YM, Gwak YS. Oxidative damage in tissues of rats exposed to cigarette smoke. Free Radic Biol Med. 1998;25:79–86. doi: 10.1016/s0891-5849(98)00041-0. [DOI] [PubMed] [Google Scholar]
- 93.Carnevali S, Petruzzelli S, Longoni B, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284:L955–63. doi: 10.1152/ajplung.00466.2001. [DOI] [PubMed] [Google Scholar]
- 94.Sandhir R, Subramanian S, Koul A. Long-term smoking and ethanol exposure accentuates oxidative stress in hearts of mice. Cardiovasc Toxicol. 2003;3:135–40. doi: 10.1385/ct:3:2:135. [DOI] [PubMed] [Google Scholar]
- 95.Burke A, Fitzgerald GA. Oxidative stress and smoking-induced vascular injury. Prog Cardiovasc Dis. 2003;46:79–90. doi: 10.1016/s0033-0620(03)00076-8. [DOI] [PubMed] [Google Scholar]
- 96.Alberg A. The influence of cigarette smoking on circulating concentrations of antioxidant micronutrients. Toxicology. 2002;180:121–37. doi: 10.1016/s0300-483x(02)00386-4. [DOI] [PubMed] [Google Scholar]
- 97.Panda K, Chattopadhyay R, Chattopadhyay D, Chatterjee IB. Cigarette smoke-induced protein oxidation and proteolysis is exclusively caused by its tar phase: prevention by vitamin C. Toxicol Lett. 2001;123:21–32. doi: 10.1016/s0378-4274(01)00376-9. [DOI] [PubMed] [Google Scholar]
- 98.Howard DJ, Ota RB, Briggs LA, Hampton M, Pritsos CA. Environmental tobacco smoke in the workplace induces oxidative stress in employees, including increased production of 8-hydroxy-2'-deoxyguanosine. Cancer Epidemiol Biomarkers Prev. 1998;7:141–6. [PubMed] [Google Scholar]
- 99.Wurzel H, Yeh CC, Gairola C, Chow CK. Oxidative damage and antioxidant status in the lungs and bronchoalveolar lavage fluid of rats exposed chronically to cigarette smoke. J Biochem Toxicol. 1995;10:11–7. [PubMed] [Google Scholar]
- 100.Chow CK. Cigarette smoking and oxidative damage in the lung. Ann N Y Acad Sci. 1993;686:289–98. doi: 10.1111/j.1749-6632.1993.tb39189.x. [DOI] [PubMed] [Google Scholar]
- 101.Cosio MG, Majo J, Cosio MG. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest. 2002;121:160S–5S. doi: 10.1378/chest.121.5_suppl.160s. [DOI] [PubMed] [Google Scholar]
- 102.Jimenez Ruiz CA, Rajas O, Ruiz A, et al. Bronchoalveolar lavage in smokers: quantification of alveolar macrophages and neutrophils as markers of bronchial obstruction. In Vivo. 1998;12:427–30. [PubMed] [Google Scholar]
- 103.Thompson AB, Bohling T, Heires A, Linder J, Rennard SI. Lower respiratory tract iron burden is increased in association with cigarette smoking. J Lab Clin Med. 1991;117:493–9. [PubMed] [Google Scholar]
- 104.Mayo JJ, Kohlhepp P, Zhang D, Winzerling JJ. Effects of sham air and cigarette smoke on A549 lung cells: implications for iron-mediated oxidative damage. Am J Physiol Lung Cell Mol Physiol. 2004;286:L866–76. doi: 10.1152/ajplung.00268.2003. [DOI] [PubMed] [Google Scholar]
- 105.Swaim MW, Pizzo SV. Methionine sulfoxide and the oxidative regulation of plasma proteinase inhibitors. J Leukoc Biol. 1988;43:365–79. doi: 10.1002/jlb.43.4.365. [DOI] [PubMed] [Google Scholar]
- 106.Moraga F, Janciauskiene S. Activation of primary human monocytes by the oxidized form of alpha1- antitrypsin. J Biol Chem. 2000;275:7693–700. doi: 10.1074/jbc.275.11.7693. [DOI] [PubMed] [Google Scholar]
- 107.Shapiro SD. Evolving concepts in the pathogenesis of chronic obstructive pulmonary disease. Clin Chest Med. 2000;21:621–32. doi: 10.1016/s0272-5231(05)70172-6. [DOI] [PubMed] [Google Scholar]
- 108.Griffiths SW, Cooney CL. Relationship between protein structure and methionine oxidation in recombinant human alpha 1-antitrypsin. Biochemistry. 2002;41:6245–52. doi: 10.1021/bi025599p. [DOI] [PubMed] [Google Scholar]
- 109.Taggart C, Cervantes-Laurean D, Kim G, et al. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–65. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- 110.Pryor WA, Dooley MM, Church DF. The inactivation of alpha-1-proteinase inhibitor by gas-phase cigarette smoke: protection by antioxidants and reducing species. Chem Biol Interact. 1986;57:271–83. doi: 10.1016/0009-2797(86)90002-5. [DOI] [PubMed] [Google Scholar]
- 111.Cox DW, Billingsley GD. Oxidation of plasma alpha 1-antitrypsin in smokers and nonsmokers and by an oxidizing agent. Am Rev Respir Dis. 1984;130:594–9. doi: 10.1164/arrd.1984.130.4.594. [DOI] [PubMed] [Google Scholar]
- 112.Hubbard RC, Ogushi F, Fells GA, et al. Oxidants spontaneously released by alveolar macrophages of cigarette smokers can inactivate the active site of alpha 1-antitrypsin, rendering it ineffective as an inhibitor of neutrophil elastase. J Clin Invest. 1987;80:1289–95. doi: 10.1172/JCI113204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Churg A, Zay K, Li K. Mechanisms of mineral dust-induced emphysema. Environ Health Perspect. 1997;105 (Suppl 5):1215–8. doi: 10.1289/ehp.97105s51215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nedeljkovic ZS, Gokce N, Loscalzo J. Mechanisms of oxidative stress and vascular dysfunction. Postgrad Med J. 2003;79:195–9. doi: 10.1136/pmj.79.930.195. quiz 8–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Loscalzo J. Oxidative stress in endothelial cell dysfunction and thrombosis. Pathophysiol Haemost Thromb. 2002;32:359–60. doi: 10.1159/000073600. [DOI] [PubMed] [Google Scholar]
- 116.Viles-Gonzalez JF, Fuster V, Badimon JJ. Links between inflammation and thrombogenicity in atherosclerosis. Curr Mol Med. 2006;6:489–99. doi: 10.2174/156652406778018707. [DOI] [PubMed] [Google Scholar]
- 117.Spronk HM, van der Voort D, Ten Cate H. Blood coagulation and the risk of atherothrombosis: a complex relationship. Thromb J. 2004;2:12. doi: 10.1186/1477-9560-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109:II27–33. doi: 10.1161/01.CIR.0000129501.88485.1f. [DOI] [PubMed] [Google Scholar]
- 119.Voetsch B, Loscalzo J. Genetics of thrombophilia: impact on atherogenesis. Curr Opin Lipidol. 2004;15:129–43. doi: 10.1097/00041433-200404000-00006. [DOI] [PubMed] [Google Scholar]
- 120.Ruberg FL, Loscalzo J. Prothrombotic determinants of coronary atherothrombosis. Vasc Med. 2002;7:289–99. doi: 10.1191/1358863x02vm448ra. [DOI] [PubMed] [Google Scholar]
- 121.Christofidou-Solomidou M, Kennel S, Scherpereel A, et al. Vascular immunotargeting of glucose oxidase to the endothelial antigens induces distinct forms of oxidant acute lung injury: targeting to thrombomodulin, but not to PECAM-1, causes pulmonary thrombosis and neutrophil transmigration. Am J Pathol. 2002;160:1155–69. doi: 10.1016/S0002-9440(10)64935-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fernandez JA, Gruber A, Heeb MJ, Griffin JH. Protein C pathway impairment in nonsymptomatic cigarette smokers. Blood Cells Mol Dis. 2002;29:73–82. doi: 10.1006/bcmd.2002.0542. [DOI] [PubMed] [Google Scholar]
- 123.Espana F, Vaya A, Mira Y, et al. Low level of circulating activated protein C is a risk factor for venous thromboembolism. Thromb Haemost. 2001;86:1368–73. [PubMed] [Google Scholar]
- 124.Medina P, Navarro S, Estelles A, et al. Contribution of polymorphisms in the endothelial protein C receptor gene to soluble endothelial protein C receptor and circulating activated protein C levels, and thrombotic risk. Thromb Haemost. 2004;91:905–11. doi: 10.1160/TH03-10-0657. [DOI] [PubMed] [Google Scholar]
- 125.Griffin JH, Fernandez JA, Liu D, Cheng T, Guo H, Zlokovic BV. Activated protein C and ischemic stroke. Crit Care Med. 2004;32:S247–53. doi: 10.1097/01.ccm.0000126127.87484.2b. [DOI] [PubMed] [Google Scholar]
- 126.De Cristofaro R, Rocca B, Marchioli R, Landolfi R. Plasma protein oxidation is associated with an increase of procoagulant markers causing an imbalance between pro- and anticoagulant pathways in healthy subjects. Thromb Haemost. 2002;87:58–67. [PubMed] [Google Scholar]
- 127.Bayele HK, Murdock PJ, Perry DJ, Pasi KJ. Simple shifts in redox/thiol balance that perturb blood coagulation. FEBS Lett. 2002;510:67–70. doi: 10.1016/s0014-5793(01)03209-4. [DOI] [PubMed] [Google Scholar]
- 128.Ambrosio G, Tritto I, Golino P. Reactive oxygen metabolites and arterial thrombosis. Cardiovasc Res. 1997;34:445–52. doi: 10.1016/s0008-6363(97)00101-6. [DOI] [PubMed] [Google Scholar]
- 129.Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002;62:1524–38. doi: 10.1046/j.1523-1755.2002.00600.x. [DOI] [PubMed] [Google Scholar]
- 130.Obrosova IG. How does glucose generate oxidative stress in peripheral nerve? Int Rev Neurobiol. 2002;50:3–35. doi: 10.1016/s0074-7742(02)50071-4. [DOI] [PubMed] [Google Scholar]
- 131.Lipinski B. Pathophysiology of oxidative stress in diabetes mellitus. J Diabetes Complications. 2001;15:203–10. doi: 10.1016/s1056-8727(01)00143-x. [DOI] [PubMed] [Google Scholar]
- 132.Bonnefont-Rousselot D. Glucose and reactive oxygen species. Curr Opin Clin Nutr Metab Care. 2002;5:561–8. doi: 10.1097/00075197-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 133.Biondi-Zoccai GG, Abbate A, Liuzzo G, Biasucci LM. Atherothrombosis, inflammation, and diabetes. J Am Coll Cardiol. 2003;41:1071–7. doi: 10.1016/s0735-1097(03)00088-3. [DOI] [PubMed] [Google Scholar]
- 134.Banga JD. Coagulation and fibrinolysis in diabetes. Semin Vasc Med. 2002;2:75–86. doi: 10.1055/s-2002-23098. [DOI] [PubMed] [Google Scholar]
- 135.Candido R, Srivastava P, Cooper ME, Burrell LM. Diabetes mellitus: a cardiovascular disease. Curr Opin Investig Drugs. 2003;4:1088–94. [PubMed] [Google Scholar]
- 136.De Cristofaro R, Rocca B, Vitacolonna E, et al. Lipid and protein oxidation contribute to a prothrombotic state in patients with type 2 diabetes mellitus. J Thromb Haemost. 2003;1:250–6. doi: 10.1046/j.1538-7836.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 137.Andreotti F, Burzotta F, Manzoli A, Robinson K. Homocysteine and risk of cardiovascular disease. J Thromb Thrombolysis. 2000;9:13–21. doi: 10.1023/a:1018675624181. [DOI] [PubMed] [Google Scholar]
- 138.Cattaneo M. Hyperhomocysteinemia and thrombosis. Lipids. 2001;36 (Suppl):S13–26. doi: 10.1007/s11745-001-0677-9. [DOI] [PubMed] [Google Scholar]
- 139.Lee R, Frenkel EP. Hyperhomocysteinemia and thrombosis. Hematol Oncol Clin North Am. 2003;17:85–102. doi: 10.1016/s0889-8588(02)00090-4. [DOI] [PubMed] [Google Scholar]
- 140.Falk E, Zhou J, Moller J. Homocysteine and atherothrombosis. Lipids. 2001;36 (Suppl):S3–11. doi: 10.1007/s11745-001-0676-x. [DOI] [PubMed] [Google Scholar]
- 141.den Heijer M, Keijzer MB. Hyperhomocysteinemia as a risk factor for venous thrombosis. Clin Chem Lab Med. 2001;39:710–3. doi: 10.1515/CCLM.2001.117. [DOI] [PubMed] [Google Scholar]
- 142.Herrmann W. The importance of hyperhomocysteinemia as a risk factor for diseases: an overview. Clin Chem Lab Med. 2001;39:666–74. doi: 10.1515/CCLM.2001.110. [DOI] [PubMed] [Google Scholar]
- 143.de Jong SC, van den Berg M, Rauwerda JA, Stehouwer CD. Hyperhomocysteinemia and atherothrombotic disease. Semin Thromb Hemost. 1998;24:381–5. doi: 10.1055/s-2007-996026. [DOI] [PubMed] [Google Scholar]
- 144.Guthikonda S, Haynes WG. Homocysteine: role and implications in atherosclerosis. Curr Atheroscler Rep. 2006;8:100–6. doi: 10.1007/s11883-006-0046-4. [DOI] [PubMed] [Google Scholar]
- 145.Undas A, Brozek J, Szczeklik A. Homocysteine and thrombosis: from basic science to clinical evidence. Thromb Haemost. 2005;94:907–15. doi: 10.1160/TH05-05-0313. [DOI] [PubMed] [Google Scholar]
- 146.Olszewski AJ, McCully KS. Homocysteine metabolism and the oxidative modification of proteins and lipids. Free Radic Biol Med. 1993;14:683–93. doi: 10.1016/0891-5849(93)90151-j. [DOI] [PubMed] [Google Scholar]
- 147.Widner B, Enzinger C, Laich A, Wirleitner B, Fuchs D. Hyperhomocysteinemia, pteridines and oxidative stress. Curr Drug Metab. 2002;3:225–32. doi: 10.2174/1389200024605091. [DOI] [PubMed] [Google Scholar]
- 148.Cattaneo M, Franchi F, Zighetti ML, Martinelli I, Asti D, Mannucci PM. Plasma levels of activated protein C in healthy subjects and patients with previous venous thromboembolism: relationships with plasma homocysteine levels. Arterioscler Thromb Vasc Biol. 1998;18:1371–5. doi: 10.1161/01.atv.18.9.1371. [DOI] [PubMed] [Google Scholar]
- 149.Gerdes VE, Hovinga HA, ten Cate H, et al. Homocysteine and markers of coagulation and endothelial cell activation. J Thromb Haemost. 2004;2:445–51. doi: 10.1111/j.1538-7836.2004.00674.x. [DOI] [PubMed] [Google Scholar]
- 150.Goswami K, Nandakumar DN, Koner BC, Bobby Z, Sen SK. Oxidative changes and desialylation of serum proteins in hyperthyroidism. Clin Chim Acta. 2003;337:163–8. doi: 10.1016/j.cccn.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 151.Bednarek J, Wysocki H, Sowinski J. Oxidation products and antioxidant markers in plasma of patients with Graves' disease and toxic multinodular goiter: effect of methimazole treatment. Free Radic Res. 2004;38:659–64. doi: 10.1080/10715760410001701621. [DOI] [PubMed] [Google Scholar]
- 152.Bianchi G, Solaroli E, Zaccheroni V, et al. Oxidative stress and anti-oxidant metabolites in patients with hyperthyroidism: effect of treatment. Horm Metab Res. 1999;31:620–4. doi: 10.1055/s-2007-978808. [DOI] [PubMed] [Google Scholar]
- 153.Hofbauer LC, Heufelder AE. Coagulation disorders in thyroid diseases. Eur J Endocrinol. 1997;136:1–7. doi: 10.1530/eje.0.1360001. [DOI] [PubMed] [Google Scholar]
- 154.Verberne HJ, Fliers E, Prummel MF, Stam J, Brandjes DP, Wiersinga WM. Thyrotoxicosis as a predisposing factor for cerebral venous thrombosis. Thyroid. 2000;10:607–10. doi: 10.1089/thy.2000.10.607. [DOI] [PubMed] [Google Scholar]
- 155.Parker JL, Lawson DH. Death from thyrotoxicosis. Lancet. 1973;302:894–5. doi: 10.1016/s0140-6736(73)92019-9. [DOI] [PubMed] [Google Scholar]
- 156.Pawlak K, Naumnik B, Brzosko S, Pawlak D, Mysliwiec M. Oxidative stress - a link between endothelial injury, coagulation activation, and atherosclerosis in haemodialysis patients. Am J Nephrol. 2004;24:154–61. doi: 10.1159/000076244. [DOI] [PubMed] [Google Scholar]
- 157.Locatelli F, Canaud B, Eckardt KU, Stenvinkel P, Wanner C, Zoccali C. Oxidative stress in end-stage renal disease: an emerging threat to patient outcome. Nephrol Dial Transplant. 2003;18:1272–80. doi: 10.1093/ndt/gfg074. [DOI] [PubMed] [Google Scholar]
- 158.Casserly LF, Dember LM. Thrombosis in end-stage renal disease. Semin Dial. 2003;16:245–56. doi: 10.1046/j.1525-139x.2003.16048.x. [DOI] [PubMed] [Google Scholar]
- 159.Jikimoto T, Nishikubo Y, Koshiba M, et al. Thioredoxin as a biomarker for oxidative stress in patients with rheumatoid arthritis. Mol Immunol. 2002;38:765–72. doi: 10.1016/s0161-5890(01)00113-4. [DOI] [PubMed] [Google Scholar]
- 160.Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum. 2005;52:402–11. doi: 10.1002/art.20853. [DOI] [PubMed] [Google Scholar]
- 161.Cheras PA, Whitaker AN, Blackwell EA, Sinton TJ, Chapman MD, Peacock KA. Hypercoagulability and hypofibrinolysis in primary osteoarthritis. Clin Orthop Relat Res. 1997:57–67. [PubMed] [Google Scholar]
- 162.Jurcut C, Jurcut R, Tanasescu C. Cardiovascular risk and rheumatoid arthritis: from mechanisms of atherosclerosis to therapeutic approach. Rom J Intern Med. 2004;42:659–69. [PubMed] [Google Scholar]