

Abstract
In proteomics, effective methods are needed for identifying the relatively limited subset of proteins displaying significant changes in abundance between two samples. One way to accomplish this task is to target for identification by MS/MS only the “interesting” proteins based on the abundance ratio of isotopically labeled pairs of peptides. We have developed the software and hardware tools for online LC-FTICR MS/MS studies in which a set of initially unidentified peptides from a proteome analysis can be selected for identification based on their distinctive changes in abundance following a “perturbation”. We report here the validation of this method using a mixture of standard proteins combined in different ratios after isotopic labeling. We also demonstrate the application of this method to the identification of Shewanella oneidensis peptides/proteins exhibiting differential abundance in sub-oxic vs. aerobic cell cultures.
Introduction
Gel-free methods based on liquid-phase separations coupled to tandem mass spectrometry (MS/MS) now allow a large fraction of a given proteome to be observed in a single analysis. Still, methods are needed that focus on identifying the relatively limited subset of proteins displaying significant changes in abundance between two samples, in analogy with selective MS analysis of spots that show significant changes between 2-dimensional gels.
Effective determination of changes in protein abundances is important for essentially all applications of proteomics. Most approaches for this are based on stable isotopic labeling (SIL). SIL involves the mixing (at either the whole cell, protein or peptide level) and simultaneous analysis of two proteome samples distinctively labeled by stable isotopes that can be sufficiently resolved in the analysis. Examples of methodologies include metabolic labeling using 14N/15N or other stable isotope enriched or depleted media1,2 tagging of cysteine residues using isotope coded affinity tags (ICAT)3, extraction and simultaneous tagging of cysteine-containing peptides by solid-phase ICAT (SPICAT)4, and differential labeling during tryptic digestion using 16O/18O,5 to obtain precise measures of the relative abundances of proteins.
An approach for selecting peptides for MS/MS based on the relative abundance of the corresponding heavy/light ICAT pairs has been proposed by Zhang and Regnier6. This method was initially demonstrated using liquid chromatography (LC) coupled to a hybrid quadrupole time of flight (q-TOF) MS instrument.7 The experiment was performed with a fraction collection and subsequent MS/MS analysis of the fractions of interest after determination of the peptides abundance ratios (AR). Whereas this circumvents some of the problems associated with the asynchronous elution of the differentially labeled peptidesError! Bookmark not defined., the use of fraction collection raises several issues such as the choice of the collection time, and the limitation of sensitivity and dynamic range caused by the remixing of separated peptides in a single fraction. Moreover, the method would benefit from the use of high efficiency LC for separation and Fourier transform ion cyclotron resonance (FTICR) mass spectrometry, for increased resolution, sensitivity and dynamic range8.
We have developed the software and hardware tools for a targeted LC-FTICR MS/MS experiment; in which a set of initially unidentified peptides from a proteome analysis was selected for dissociation based on their distinctive changes in abundance after a “perturbation”. We report here on the validation of this method using a mixture of standard proteins combined in different ratios after isotopic labeling by the SPICAT approach. We also show an application of this method to the identification of proteins differentially abundant in Shewanella oneidensis grown under aerobic and sub-oxic conditions using a metabolic 14N/15N isotopic labeling scheme.
Experimental
Standard protein mixture
Standard proteins were purchased from Sigma (St Louis, MO) and used without further purification. The SPICAT labeled chimeric mixture samples with known AR were prepared according to previously reported procedures,4 but instead of using deuterium, we incorporated 6 (six) 13C and 1 (one) 15N into the leucine moiety of the labeling reagents. The details of SPICAT labeling are described elsewhere.9 Table 1 provides the composition of the chimeric mixture.
Bacterial Cultures
S. oneidensis MR-1 cells were grown in sub-oxic conditions and labeled with 15N according to the following procedure: A 5 mL TSB tube was started with 0.1 mL of new vial frozen stock and set on a shaker (30°C, 150 RPM) for 20.5 h. A 0.1 mL aliquot of this TSB culture was injected into 160 mL bottle with 50 mL of MR-1 medium as described in detail elsewhere10,11 with 30 mM D,L-lactate, 15N labeled NH4Cl, serine, arginine and glutamic acid, in the absence of CaCl2. After 22 h of growth on a shaker (30°C, 150 RPM), the OD600 was 0.285 and the bottle was used as an inoculum source for a fermenter. Cells were added to 2,000 mL of media in a 2,500 mL vessel connected to a New Brunswick fermenter. The culture was incubated anaerobically for 19 h at 30°C with a shaker rate of 300 RPM. Cells were harvested by centrifugation (8,000 RPM, 8 min, 4°C), resuspended in a small amount of supernatant, transferred into 3 cryogenic vials, centrifuged a second time (Minimax, 8,000 RPM, 4.5 min, 4°C), snap frozen and stored in a freezer (−80°C.)
Aerobically grown cells were cultured under similar conditions except for the presence of oxygen and the fact that NH4Cl and the amino acids used contained nitrogen in its natural isotopic abundance. All other culture and harvest procedures were identical to those described above.
Cell lysis and tryptic digestion
Cell lysis was achieved by bead beating (3 min) in a Biospec Minibeadbeater with 0.1 mm zirconia/silica beads in 0.5 mL sterile siliconized microcentrifuge tubes. Beads were immediately removed by centrifugation (16,000 x g) at 4°C for 5 min and remaining lysates placed on ice to inhibit proteolysis. The protein concentration of the lysates was determined by the Pierce BCA assay kit. Cell lysates were treated by the addition of dry urea, thiourea, and DTT to final concentrations of 7 M, 2 M, and 5 mM, respectively, and incubated at 60°C for 30 min. The sample was diluted 10 fold with 100 mM ammonium bicarbonate (pH 8.4) and CaCl2 was added to a final concentration of 1 mM. Sequencing grade, modified trypsin (Promega) was prepared by adding 20 μL of 100 mM ammonium bicarbonate (pH 8.4) to a vial containing 20 μg trypsin and then, after a 10 min incubation at 37°C, this mixture was added to the cell sample. Tryptic digestion was performed for 5 hours at 37°C using a 1:50 (w/w) trypsin-to-protein ratio. Sample cleanup was achieved using a 1 mL SPE C18 column (Supelclean SPE LC-18, Supelco). The sample was eluted using 80% acetonitrile, 20% water with 0.1% trifluoroacetic acid and concentrated via speed-vac. The final peptide concentration was determined as above. The samples were then snap frozen in liquid nitrogen and stored at −80°C until analyzed.
Liquid chromatography-Mass spectrometry
All experiments were performed using a LC-FTICR instrumentation based on a 7 tesla superconducting magnet (Oxford, UK). The instrument, which is described in detail elsewhere12,13, incorporates an electrospray ion source coupled to a cubic ICR cell through various multipoles and differentially pumped regions. Briefly, ions formed by electrospray were focused to the entrance of a collisional octopole by means of an electrodynamic ion funnel14 and transferred through a set of two selection quadrupoles before being accumulated in a 5 cm long accumulation quadrupole. After accumulation and subsequent cooling, ions were transferred to the ICR cell using a quadrupole ion guide. A high pressure LC system (10,000 psi) equipped with a dual-column (80 cm long, 50 μm I.D., 3 μm C18 particles) was coupled to the FTICR as described elsewhere.15
The experiments described here were performed using two different instrumental scripts. In a first analysis, a conventional MS acquisition was performed and the data were processed using in-house developed software (ICR-2LS) to determine the m/z and retention time of the species present in the sample as well as their AR (see results and discussions). An “attention list” was then derived based on this information. The subsequent experiment involved repeating the same MS script until a species from the attention list was detected. When this occurred, the instrument switched to MS/MS mode, selecting the species matching the attention list as the precursor ion. This was accomplished by using the Odyssey data-station to control the instrument only, while a PC running ICR-2LS handled the signal detection and processing in parallel. After every MS acquisition, ICR-2LS performed a fast Fourier transform of the signal and picked peaks in the mass spectrum having a relative abundance greater than 10%. The list of detected peaks was then compared with the attention list and if a match was found, a stored waveform inverse Fourier transform (SWIFT)16 and a sustained off-resonance irradiation (SORI)17 waveform were generated to respectively select and dissociate the species of interest during the subsequent MS/MS acquisition. The waveforms parameters were stored in a log file to allow tracking of the actual precursor species during the MS/MS experiment. After waveforms generation, the MS data was stored, and a temporary file on the Odyssey system was modified by ICR-2LS to instruct the instrument controller software to switch to MS/MS mode for the subsequent acquisition. If no match was found between the detected species and the attention list, ICR-2LS simply stored the MS data and modified the file on the Odyssey system to inform the instrument controller that the next acquisition should be MS only. The Odyssey system therefore switched from the MS only script to the MS/MS script based on the value of a string in a temporary file. This was accomplished using a Tcl/Tk script written in house and available upon request. During the MS/MS acquisition, the Odyssey instrument controller triggered the application of both SWIFT and SORI waveforms from the PC at the appropriate time during the script. The detection of the MS/MS data was accomplished in parallel on the PC and directly stored to disk without processing.
Results and discussion
LC-MS/MS is widely used for the identification of peptides. However, not all species in a complex sample are amenable to MS/MS because of time constraints (limited elution period). To circumvent this under-sampling problem to some extent, several experimental schemes have been developed including peak parking18, exclusion lists, or even multiplexed MS/MS19. Despite these efforts, many species remain unidentified, especially when the sample amount is limited, due to the impossibility to repeat the experiment many times with different precursor ions selection methods. This is especially true for low abundance species co-eluting with more abundant ones.
Many proteomics studies are aimed at gaining some insight into the differences in protein abundances between different states (e.g. normal vs. diseased). These studies would greatly benefit from an LC-MS/MS method which could target for identification the species showing significant differences between a sample and a control. This would not increase the total number of identified species, but would improve their biological relevance.
The first step in such an approach is to determine which species are the most interesting (i.e. display the most significant changes). One approach for this purpose uses on-the-fly estimates of the ARs of pairs of isotopically labeled species. However, in some cases, the different elution profiles of the two members of a pair prevent us from measuring a reliable ratio during the separationError! Bookmark not defined.. In order to circumvent this problem, we have evaluated a labeling procedure that incorporates 13C and 15N labeling instead of the 2H labels used initially.9 Although this alleviated the problem, small differences in elution behavior persist. An alternative approach is to determine the AR of peptide pairs in a separate acquisition and to include the information about their m/z and retention time in an “attention list” to be used for the selection of species to dissociate on a subsequent analysis.
Figure 1 shows the elution profile generated for a pair of SPICAT labeled peptides present in a “chimeric” protein digest mixture. The composition of this “chimeric” mixture is detailed in table 1. The peptide represented here was later identified as ETYGDMADCCEK from bovine serum albumin (BSA) with two SPICAT labeled cysteine residues. When the AR was calculated from any given spectrum, the value differed significantly from the actual ratio due to the asynchronous elution profiles of the members of the pair. In contrast, when the area under the peaks of the two elution profiles were used to calculate the ratio, the outcome was the expected value (1:6 Light:heavy SPICAT). This example illustrates the need to obtain the complete elution profiles of both members of the pair to correctly evaluate ARs, and points out a potential shortfall of using on-the-fly ratio determination for targeting the species: the correct AR can be calculated only after both versions of a peptide have eluted from the column, and are consequently unavailable for selection and dissociation except when using some kind of fraction collection method. From these considerations emerged the need to subdivide the experiment in two parts: a first LC-MS analysis allowing accurate ARs to be computed and retention times and m/z values of “interesting” peptides to be measured; and a second analysis using LC-MS/MS where those “interesting” peptides are selected and dissociated to enable their identification.
Figure 1.
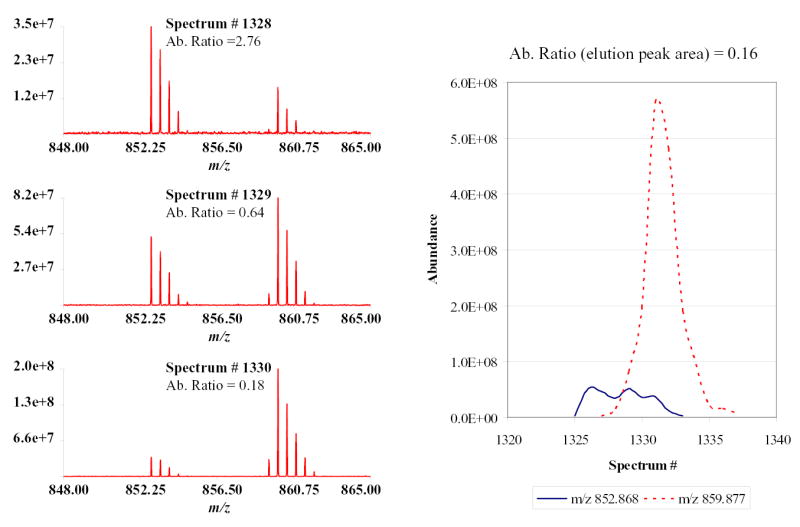
Elution profiles of two members of a differentially labeled peptide pair. The ratio calculated from 3 representative spectra were very different from the ratio estimated based on the whole elution profiles.
Table 1.
| Protein | MW (g/mol) | # Cysteine | Molar amount light- vs. heavy-SPICAT |
|---|---|---|---|
| BSA | 66,433.6 | 35 | 1 : 6 |
| Ribonuclease A | 13,690.4 | 8 | 6 : 1 |
| Lysozyme | 14,313.3 | 8 | 1 : 3 |
| Ovalbumin | 42,750.4 | 6 | 1 : 1 |
| G3P DH | 35,718.2 | 4 | 3 : 1 |
A challenge associated with this approach is to align peptide retention times in two separate analyses. In order to achieve this, we applied a retention time correction based on the retention times of two m/z markers at the beginning of the separation. Figure 2 illustrates this with the analysis of the “chimeric” mixture conducted on two different columns. The top panel (A) shows the total ion chromatogram on the first column; the bottom panel (C) represents the total ion chromatogram for the same sample when injected on a second column of same length, same internal diameter, and using the same packing material. Although both columns were identical, uncontrolled factors, such as temperature or different aging of the columns, caused different separation times. A linear correction based on the detection of two species at m/z 654.3 and m/z 664.3 that eluted before the species of interest, aligned the first separations with the second as shown in the middle panel (B). For illustrative purposes, we chose for this example an atypical case where the two elution profiles were significantly shifted relative to one another. However, even in this case, a linear correction of the retention times aligned the two analyses to within ±2 minutes over a 180 minute gradient.
Figure 2.
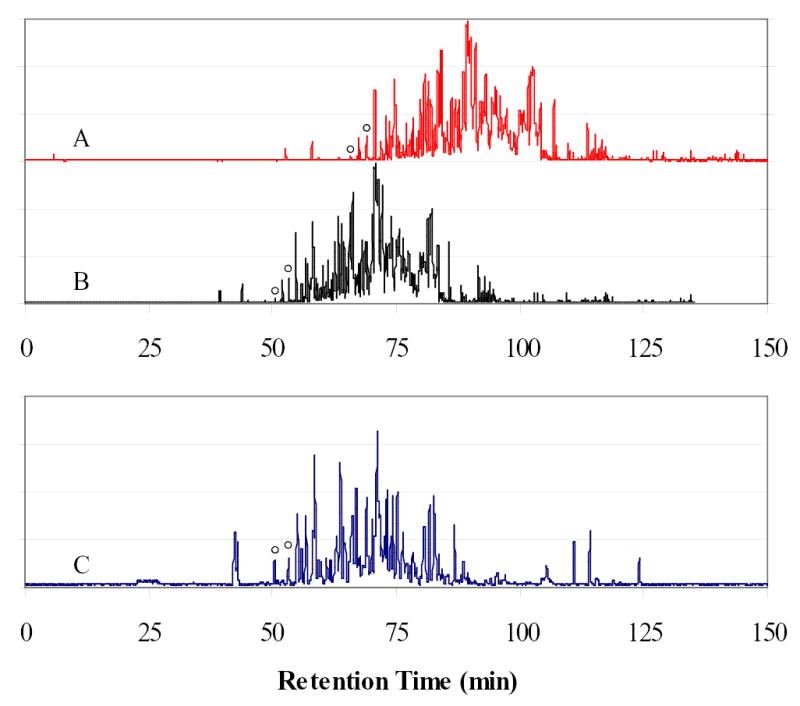
Alignment of retention times on two different columns using a linear fit. Uncorrected (A) and corrected (B) total ion chromatogram reconstructed from the LC-MS analysis of a SPICAT labeled “chimeric” mixture are compared with the reference chromatogram (C) of the same mixture analyzed using the alternative column of the dual-column system. Two target m/z values (r) were selected early in the analysis and used to correct the retention times of other species. The corrected retention times agree within 2 minutes between the two analyses.
An LC-MS experiment can be visualized as a 2 dimensional graph in which one axis represents the retention time, and the other the molecular weight of the detected species. Each point in this graph represents one detected isotopic distribution (Figure 3a). In order to extract reliable ARs from such a 2D-display, we have elaborated the concept of unique mass classes (UMC). A UMC consists of a group of measurements in a contiguous set of spectra, which can be ascribed to the same peptide detected repeatedly as it elutes from the LC column. UMCs were generated using an algorithm developed in our laboratory which calculates quasi-distances between points in the 2D-graph and links points within a pre-defined quasi-distance. Several metrics were used to evaluate this quasi-distance: the monoisotopic molecular weight; the average molecular weight; the spectrum number (equivalent to the retention time); the abundance; and the fit between the isotopic pattern and a theoretical pattern obtained from the hypothetical average protein20. An UMC was defined by the median mass of its members, the sum of their abundances, and their spectrum range (or elution time window). Once UMCs were established, the mass differences between them were computed and compared to the mass difference between the SPICAT heavy and light labels (7.02 Da). UMCs that differed by a multiple of this mass difference and for which the spectrum ranges overlapped to a predefined extent were considered paired, and their AR was calculated. For the dataset in Figure 3a, 1,938 UMCs were defined, and among them, 418 SPICAT pairs were determined. Figure 3b shows a portion of the mass vs. time display filtered to represent only the points that belonged to paired UMCs. Among those pairs, 46 had an AR between 0.05 and 0.25; and were included in the attention list. It is worth noting that the m/z and retention time (obtained by conversion of the spectrum number) of the UMC corresponding to the most intense member of each pair was incorporated in the attention list, so that the most abundant version of the peptide would be selected for dissociation.
Figure 3.
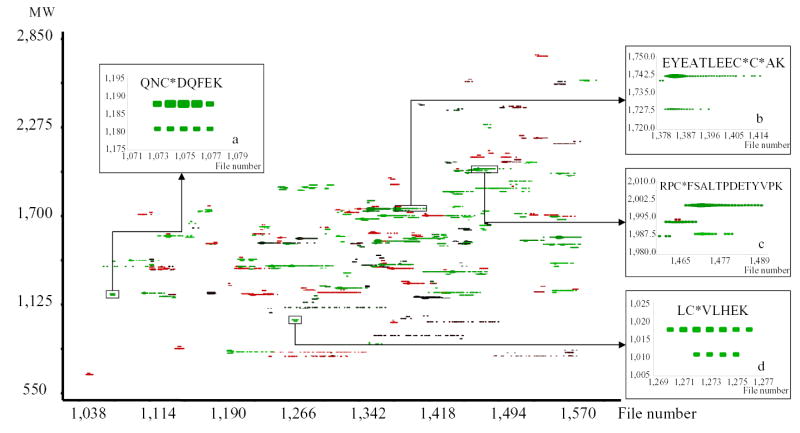
2D-display of the UMC pairs found in the initial MS dataset. Axes correspond to retention time (spectrum number) and molecular weight of the species; each point represents one detected isotopic distribution. Among the 209 established UMC pairs of this dataset, 46 pairs with 0.05≥AR≥0.25 were included in the attention list. Insets (a–d) show examples of targeted pairs and the identifications obtained during the subsequent MS/MS analysis (asterisks represent ICAT labels).
After the generation of the attention list, a second analysis was undertaken during which the instrument was set to select and dissociate only the species present in the attention list in an unattended fashion. A ± 2 minutes retention time tolerance was used and a linear correction of the attention list was applied to realign the list with the analysis at hand. This was accomplished, as described previously, by targeting two m/z values at the beginning of the separation with large retention time tolerance, and correcting the attention list based on the observed retention times of these two species.
During the second analysis, a good agreement was observed between the attention list and the species actually selected for dissociation. Nonetheless, we found some discrepancies between the experiment and the attention list; specifically, some targeted species that were not selected. We attribute this fact to the overlapping elution ranges of some peptides and the limitation of the MS/MS to one single species at a time. Dynamic exclusion of species already selected or the use of targeted multiplexed MS/MS would be beneficial to reduce this type of deficiency, and is considered for future experiments. Also, narrower elution time windows could be defined using information from closely eluting species. Among the 46 species included in the attention list, 38 were effectively selected for MS/MS. The whole dataset yielded the redundant identification of 11 peptides from bovine serum albumin; correctly revealing this protein as the one present at a 1:6 AR in the “chimeric” sample. A single peptide pointed to the protein lysozyme, indicating the possible selection of an unintended species or an error in the compilation of the attention list.
We have also used the targeted MS/MS method to identify proteins of S. oneidensis that were differentially abundant under aerobic vs. sub-oxic growth conditions. After the initial MS experiment, UMCs were generated and paired and their AR were calculated as described previously (vide supra). We subsequently incorporated in an attention list the UMCs having an AR significantly different from 1 based on the statistics of the AR distributions. This resulted in a list of 164 species which were targeted for identification in a second experiment.
Figure 4 shows a graphical display of the attention list experiment. The horizontal axis represents the retention time and the vertical axis the molecular weight since using m/z values would have yielded an even more congested graph. All spots represent targeted species, with error bars showing their elution time window, and the spot size is proportional to the abundances of the corresponding UMC as determined in the initial LC-MS experiment. The black spots correspond to species that were actually selected for dissociation during the targeted LC-MS/MS analysis, whereas the grey spot correspond to target species that have not been selected. This plot shows that the instrument in fact directed the MS/MS acquisition to the targeted species, although some were missed for the reason given above; that is, there were other species that were selected instead. This is not surprising given the complexity of the peptides mixture and density of the attention list used in this experiment. More accurately defining the retention time would help alleviate this problem, but multiplexing the MS/MS acquisition19 will be necessary to deal efficiently with co-eluting species (perhaps at the price of a somewhat lower confidence of identification). This approach is currently being evaluated.
Figure 4.
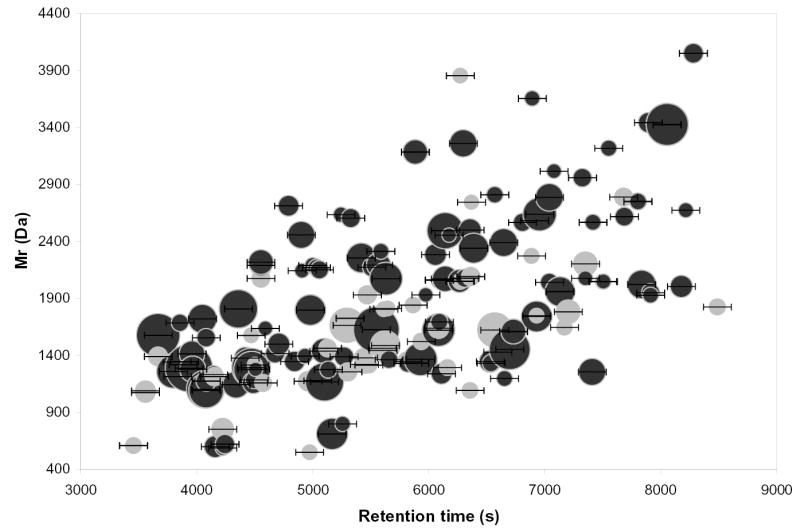
Mw vs. retention time maps of the targeted species during the targeted MS/MS acquisition of the S. oneidensis 14N/15N labeled mixture. Darkened spots correspond to the targets having effectively been selected during the experiment, while grey spots represent targeted species that haven’t been selected, mostly due to the presence of other targets overlapping their elution range.
Figure 5 shows results from a targeted LC-MS/MS study comparing S. Oneidensis in aerobic and sub-oxic conditions. In the MS dataset 9048 UMCs were determined and 1503 SPICAT pairs were revealed. The 2D-display in Figure 6 has been filtered to represent only the 3006 paired UMCs. Among those pairs, 164 exhibited AR significantly different from 1 based on the AR distribution statistics (i.e. AR<0.4 and AR>2.4) and were incorporated in the attention list. During the actual targeted MS/MS analysis, 125 species were selected for dissociation. Insets in Fig. 6 show one pair of UMCs from a targeted peptide and the corresponding MS/MS spectrum obtained in the subsequent analysis; identifying a peptide associated to the fumarate reductase flavoprotein subunit precursor (ORF #SO0970).
Figure 5.

2D-display representing UMC pairs from the initial MS dataset obtained from a mixture of 14N/15N labeled proteins from S. oneidensis grown in aerobic and sub-oxic conditions. Insets show a differentially abundant peptide pair and the corresponding MS/MS spectrum obtained in the subsequent targeted acquisition.
The database search for the MS/MS spectra was complicated by the fact that the most abundant version of the labeled peptide was targeted for any given pair, and therefore, we had to allow the peptides to be isotopically labeled. In order to account for this, we have performed two successive searches, one where only the peptides with natural abundance were considered, and the second search where all peptides were considered to be 15N-labeled. The first search against the S. Oneidensis full genome database resulted in 42 peptide hits, and the second search against the same database (but with all masses recalculated to incorporate 15N) yielded 124 peptides hits. These search results included multiple occurrences of the same identified species, and cases where several identifications were considered for the same MS/MS spectrum. We therefore filtered those search results to keep only the best hit for every MS/MS spectrum based on both the 14N and 15N identifications. We also excluded several low quality identifications. This resulted in 72 confident identifications, corresponding to 47 different peptides that could be traced to 32 non-redundant ORFs. The AR of these species were subsequently manually confirmed from the corresponding MS acquisition to assess that the selected species did indeed display differential abundances. This procedure led to the rejection of a significant number of ORFs presumably due to the identification of peptides based on erroneous estimates of their AR, and/or the unintentional selection of species with m/z values and elution time close to values in the attention list. Note that our program matches any peak with the attention list, even when this peak is not monoisotopic, therefore, overlapping isotopic patterns can potentially cause occurrences of this type. To address this issue, one would have to work with neutral masses, but the time necessary to process an MS spectrum is currently 10 to 60 seconds depending on the density of the spectrum, which is prohibitively time consuming. We are currently working on streamlining our deconvolution algorithm to enable such on-the-fly processing of spectra. After validation of ARs, a list of 19 unique peptide identifications pointing to 10 non-redundant ORFs was obtained, 6 of them showing increased abundances in sub-oxic conditions, while 4 of them had higher abundances in aerobic conditions. Fumarate reductase flavoprotein subunit precursor (ORF #SO0970) identified with 8 peptides, each of them observed 1 to 3 times, was prominently represented in the final result. The increased abundance of fumarate reductase flavoprotein subunit precursor under sub-oxic growth conditions has been previously reported21, supporting the ability of our approach to effectively target differentially abundant species. The biological interpretation of the other results will be the subject of a later publication.
Conclusion
In this work, we have demonstrated the feasibility of using “smart” instrumentation for targeted LC-MS/MS analysis based on the ARs of pairs of isotopically labeled peptides. This was accomplished using a two-phase approach, where species initially detected with “interesting” ARs, are selected for MS/MS in an automated fashion during a second analysis. To enable this approach, we have designed new software for controlling the instrumentation; we have developed the concept of UMCs for AR calculations; and introduced a correction for the attention list’s LC retention time based on the detection of retention time markers at the beginning of the targeted LC-MS/MS experiment. Using the targeted LC-MS/MS strategy, we have been able to correctly identify the protein present at the lowest AR within a “chimeric” protein mixture of standard proteins. Within a complex mixture of S. oneidensis peptides, we have been able to specifically target species corresponding to proteins differentially abundant under sub-oxic vs. aerobic growth conditions. For instance, targeted MS/MS confirmed that the amount of fumarate reductase flavoprotein subunit precursor increased by an order of magnitude under sub-oxic conditions, in agreement with previous reports. Significant improvements are expected to result from refinements of the software for ions selection and improved efficiency of dissociation through the use of RF-DC ion selection and dissociation in the external accumulation quadrupole. We believe that this method holds great potential for the analysis of clinical samples due to its focus on the most interesting features, i.e. the ones exhibiting significant changes in abundance following an insult.
Acknowledgments
The authors gratefully acknowledge Gregory Pinchuk, Jeff McLean, and Yuri Gorby for growing the S. Oneidensis cells, as well as Marina Gritsenko and Heather Mottaz for the preparation of the “chimeric” standard proteins mixture. We thank Tim Carlson for his assistance with the Tcl/Tk script for the Odyssey controller. Portions of this research were supported by the office of Biological and Environmental Research, U.S. Department of Energy, the National Cancer Institute under Grants CA81654 and CA86340, and the National Center for Research Resources (RR18522). Pacific Northwest National Laboratory is operated by Battelle Memorial Institute for the U.S. Department of Energy under contract DE-AC06-76RLO 1830.
References
- 1.Oda Y, Huang K, Cross FR, Cowburn D, Chait BT. Proc Natl Acad Sci USA. 1999;96:6591–6596. doi: 10.1073/pnas.96.12.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paša-Toliæ L, Jensen PK, Anderson GA, Lipton MS, Peden KK, Martinoviæ S, Toliæ N, Bruce JE, Smith RD. J Amer Chem Soc. 1999;121:7949–7950. [Google Scholar]
- 3.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 4.Zhou HL, Ranish JA, Watts JD, Aebersold R. Nat Biotechnol. 2002;19:512–515. doi: 10.1038/nbt0502-512. [DOI] [PubMed] [Google Scholar]
- 5.Yao XD, Freas A, Ramirez J, Demirev PA, Fenselau C. Anal Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 6.Zhang R, Regnier FE. J Proteome Res. 2002;1:139– 147. doi: 10.1021/pr015516b. [DOI] [PubMed] [Google Scholar]
- 7.Griffin TJ, Han DKM, Gygi SP, Rist B, Lee H, Aebersold R, Parker KC. J Amer Soc Mass Spectrom. 2001;12:1238–1246. doi: 10.1016/S1044-0305(01)00316-6. [DOI] [PubMed] [Google Scholar]
- 8.Shen Y, Zhao R, Belov ME, Conrads TP, Anderson GA, Tang K, Paša–Toliæ L, Veenstra TD, Lipton MS, Udseth HR, Smith RD. Anal Chem. 2001;73:1766–1775. doi: 10.1021/ac0011336. [DOI] [PubMed] [Google Scholar]
- 9.Qian WJ, Goshe MB, Camp DG, II, Yu LR, Tang K, Smith RD. Anal Chem. 2003;75:5441–5450. doi: 10.1021/ac0342774. [DOI] [PubMed] [Google Scholar]
- 10.Zachara JM, Fredrickson JK, Li S, Kennedy DW, Smith SC, Gassman PL. Am Mineral. 1998;83:1426–1443. [Google Scholar]
- 11.Myers CR, Nealson KH. J Bacteriol. 1990;172:6232–6238. doi: 10.1128/jb.172.11.6232-6238.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vilkov AN, Bogdanov B, Paša-Toliæ L, Prior DC, Anderson DA, Masselon CD, Moore RJ, Smith RD. Rapid Commun Mass Spectrom. 2004;18:1–9. doi: 10.1002/rcm.1664. [DOI] [PubMed] [Google Scholar]
- 13.Vilkov AN, Paša-Toliæ L, Bogdanov B, Ahn S, Prior DC, Anderson GA, Masselon CD, Smith RD. Proc. 51th ASMS Conf. Mass Spectrometry and Allied Topics; Montreal, Canada. June 8–12, 2003. [Google Scholar]
- 14.Belov ME, Gorshkov MV, Udseth HR, Anderson GA, Tolmachev AV, Prior DC, Harkewicz R, Smith RD. J Am Soc Mass Spectrom. 2000;11:19–23. doi: 10.1016/S1044-0305(99)00121-X. [DOI] [PubMed] [Google Scholar]
- 15.Shen Y, Zhao R, Berger SJ, Anderson GA, Rodriguez N, Smith RD. Anal Chem. 2002;74:4235–4249. doi: 10.1021/ac0202280. [DOI] [PubMed] [Google Scholar]
- 16.Grosshans PB, Marshall AG. Anal Chem. 1991;63:2057–2061. [Google Scholar]
- 17.Gauthier JW, Trautman TR, Jacobson DB. Anal Chim Acta. 1991;246:211–225. [Google Scholar]
- 18.Davis MT, Stahl DC, Hefta SA, Lee TD. Anal Chem. 1995;67:4549–4556. doi: 10.1021/ac00120a019. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Masselon CD, Anderson GA, Paša-Toliæ L, Lee S-W, Shen Y, Zhao R, Lipton MS, Conrads TP, Toliæ N, Smith RD. Anal Chem. 2001;73:3312–3322. doi: 10.1021/ac010192w. [DOI] [PubMed] [Google Scholar]
- 20.Senko MW, Beu SC, McLafferty FW. J Am Soc Mass Spectrom. 1995;6:229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 21.Giometti CS, Khare T, Tollaksen SL, Tsapin A, Zhu W, Yates JR, III, Nealson KH. Proteomics. 2003;3:777–785. doi: 10.1002/pmic.200300406. [DOI] [PubMed] [Google Scholar]