

Abstract
The enormous complexity, wide dynamic range of relative protein abundances of interest (over 10 orders of magnitude), and tremendous heterogeneity (due to post-translational modifications, such as glycosylation) of the human blood plasma proteome severely challenge the capabilities of existing analytical methodologies. Here, we describe an approach for broad analysis of human plasma N-glycoproteins using a combination of immunoaffinity subtraction and glycoprotein capture to reduce both the protein concentration range and overall sample complexity. Six high-abundance plasma proteins were simultaneously removed using a pre-packed, immobilized antibody column. N-linked glycoproteins were then captured from the depleted plasma using hydrazide resin and enzymatically digested, and the bound N-linked glycopeptides were released using peptide-N-glycosidase F (PNGase F). Following strong cation exchange (SCX) fractionation, the deglycosylated peptides were analyzed by reversed-phase capillary liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Using stringent criteria, a total of 2053 different N-glycopeptides were confidently identified, covering 303 non-redundant N-glycoproteins. This enrichment strategy significantly improved detection and enabled identification of a number of low-abundance proteins, exemplified by interleukin-1 receptor antagonist protein (~200 pg/mL), cathepsin L (~1 ng/mL), and transforming growth factor beta 1 (~2 ng/mL). A total of 639 N-glycosylation sites were identified, and the overall high accuracy of these glycosylation site assignments as assessed by accurate mass measurement using high resolution liquid chromatography coupled to Fourier transform ion cyclotron resonance mass spectrometry (LC-FTICR) is initially demonstrated.
Keywords: human plasma, mass spectrometry, proteomics, N-glycosylation, immunoaffinity subtraction
Introduction
Human blood plasma possesses significant potential for disease diagnosis and therapeutic monitoring. For example, protein abundance changes in plasma may provide direct information on physiological and metabolic states of disease and drug response. As a result, the potential discovery of novel candidate protein biomarkers from plasma using high-throughput proteomic technologies has fostered a “gold-rush” enthusiasm in the biomedical research community1–4. However, characterization of the blood plasma proteome is analytically challenging for a number of reasons.
One of the analytical challenges of characterizing the plasma proteome stems from the wide range of concentrations among constituent proteins. For example, many of the cytokines and tissue leakage proteins that could be important indicators of changes in physiological states are present at <1 pg/mL concentrations, while serum albumin, the major carrier and transport protein in plasma, is present at a concentration of ~45 mg/mL. Moreover, 99% of the plasma protein mass is distributed across only 22 proteins1, 5.
Global proteome profiling of human plasma using either two-dimensional gel electrophoresis (2DE) or single-stage liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) has proven to be challenging because of the dynamic range of detection of these techniques. This detection range has been estimated to be in the range of 4 to 6 orders of magnitude, and allows identification of only the relatively abundant plasma proteins. A variety of depletion strategies for removing high-abundance plasma proteins6–8, as well as advances in high resolution, multidimensional nanoscale LC have been demonstrated to improve the overall dynamic range of detection. Reportedly, the use of a high efficiency two-dimensional (2-D) nanoscale LC system allowed more than 800 plasma proteins to be identified without depletion9.
Another characteristic feature of plasma that hampers proteomic analyses is its tremendous complexity; plasma contains not only “classic” plasma proteins, but also cellular “leakage” proteins that can potentially originate from virtually any cell or tissue type in the body1. In addition, the presence of an extremely large number of different immunoglobulins with highly variable regions makes it challenging to distinguish among specific antibodies on the basis of peptide sequences alone. Thus, with the limited dynamic range of detection for existing proteomic technologies, it often becomes necessary to reduce sample complexity to effectively measure the less-abundant proteins in plasma. Pre-fractionation techniques that can reduce plasma complexity prior to 2DE or 2-D LC-MS/MS analyses include depletion of immunoglobulins7, ultrafiltration (to prepare the low molecular weight protein fraction)10, size exclusion chromatography5, ion exchange chromatography5, liquid-phase isoelectric focusing11, 12, and the enrichment of specific subsets of peptides, e.g., cysteinyl peptides13–15 and glycopeptides16, 17.
The enrichment of N-glycopeptides is of particular interest for characterizing the plasma proteome because the majority of plasma proteins are believed to be glycosylated. The changes in abundance and the alternations in glycan composition of plasma proteins and cell surface proteins have been shown to correlate with cancer and other disease states. In fact, numerous clinical biomarkers and therapeutic targets are glycosylated proteins, such as the prostate-specific antigen for prostate cancer, and CA125 for ovarian cancer. N-glycosylation (the carbohydrate moiety is attached to the peptide backbone via asparagine residues) is particularly prevalent in proteins that are secreted and located on the extracellular side of the plasma membrane, and are contained in various body fluids (e.g., blood plasma)18. More importantly, because the N-glycosylation sites generally fall into a consensus NXS/T sequence motif in which X represents any amino acid residue except proline19, this motif can be used as a sequence tag prerequisite to aid in confident validation of N-glycopeptide identifications.
Recently, Zhang et al.16 developed an approach for specific enrichment of N-linked glycopeptides using hydrazide chemistry. In this study, we build on this approach by coupling multi-component immunoaffinity subtraction with N-glycopeptide enrichment for comprehensive 2-D LC-MS/MS analysis of the human plasma N-glycoproteome. A conservatively estimated dynamic range of detection of >108 was achieved by effectively reducing the protein concentration range and overall sample complexity. This overall dynamic range of detection enabled confident identification of 303 non-redundant N-glycoproteins, many of which represented low abundance secreted and extracellular proteins. The accurate mass measurements provided by Fourier transform ion cyclotron resonance mass spectrometry (FTICR) for LC-MS were used to confirm the number of N-glycosylation site(s) in glycopeptides.
Materials and Methods
Immunoaffinity Subtraction Using Multiple Affinity Removal System (MARS)
The human blood plasma sample was supplied by Stanford University School of Medicine (Palo Alto, CA); an initial protein concentration of 65 mg/mL of plasma was determined by BCA Protein Assay (Pierce, Rockford, IL). Approval for the conduct of this programmatic research was obtained from the Institutional Review Boards of the Stanford University School of Medicine and the Pacific Northwest National Laboratory in accordance with federal regulations. Six high-abundant plasma proteins – albumin, IgG, antitrypsin, IgA, transferrin, and haptoglobin – that constitute approximately 85% of the total protein mass of human plasma were removed in a single step by using the MARS affinity column (Agilent, Palo Alto, CA) on an Agilent 1100 series HPLC system (Agilent) per the manufacturer’s instruction. A total of 800 μL plasma was subjected to MARS-depletion. The flow-through fractions were pooled and desalted using BioMax centrifugal filter devices with 5 kDa molecular weight cutoffs (Millipore, Billerica, MA), and the total protein amount was determined to be 7.5 mg by Coomassie Protein Assay (Pierce).
Enrichment of Formerly N-linked glycopeptides
Hydrazide resin (Bio-Rad, Hercules, CA) was used to capture glycoproteins, using a method similar to that previously reported16. The concentrated MARS flow-through fraction was diluted 10-fold in coupling buffer (100 mM sodium acetate and 150 mM NaCl, pH 5.5) and oxidized in 15 mM sodium periodate at room temperature for 1 h in the dark, with constant shaking. The sodium periodate was subsequently removed by using a pre-packed PD-10 column (Amersham Biosciences, Piscataway, NJ) equilibrated with coupling buffer. The hydrazide resin (1 mL of 50% slurry per 100 μL of plasma) was washed five times with coupling buffer; the oxidized protein sample was then added and incubated with the resin overnight at room temperature. Non-glycoproteins were removed by washing the resin briefly three times with 100% methanol and then three times with 8 M urea in 0.4 M NH4HCO3. The glycoproteins were denatured and reduced in 8 M urea and 10 mM dithiothreitol (DTT) at 37 °C for 1 h. Protein cysteinyl residues were alkylated with 20 mM iodoacetamide for 90 min at room temperature. After washing with 8 M urea and 50 mM NH4HCO3, respectively, the resin was resuspended as 20% slurry in 50 mM NH4HCO3 and sequencing grade trypsin (Promega, Madison, WI ) was added at a 1:100 (w:w) trypsin-to-protein ratio (based on the initial plasma protein concentration of 65 mg/mL), and the sample was digested on-resin overnight at 37° C. The trypsin-released peptides were removed by washing the resin extensively with three separate solutions: 2 M NaCl, 100% methanol, and 50 mM NH4HCO3. The resin was resuspended as 50% slurry in 50 mM NH4HCO3 and the N-glycopeptides were released by incubating the resin with PNGase F (New England Biolabs, Beverly, MA) for 4 h at 37° C, using a ratio of 1 μL of PNGase F per 100 μL of plasma. The released deglycosylated peptides were then cleaned using a SPE C18 column (Supelco, Bellefonte, PA) per the manufacturer’s instructions and lyophilized under vacuum.
Strong Cation Exchange (SCX) Peptide Fractionation
Enriched deglycosylated peptides were reconstituted with 300 μL of 10 mM ammonium formate (pH 3.0)/25% acetonitrile and fractionated by strong cation exchange (SCX) chromatography on a Polysulfoethyl A 200 mm×2.1 mm column (PolyLC, Columbia, MD) that was preceded by a 10 mm×2.1 mm guard column. The separations were performed at a flow rate of 0.2 mL/min using an Agilent 1100 series HPLC system (Agilent) and mobile phases consisting of 10 mM ammonium formate (pH 3.0)/25% acetonitrile (A), and 500 mM ammonium formate (pH 6.8)/25% acetonitrile (B). After loading 300 μL of sample onto the column, the gradient was maintained at 100% A for 10 min. Peptides were then separated by using a gradient from 0–50% B over 40 min, followed by a gradient of 50–100% B over 10 min. The gradient was then held at 100% B for 10 min. A total of 30 fractions were collected, and each fraction was dried under vacuum. The fractions were dissolved in 30 μL of 25 mM NH4HCO3 and 10 μL of each fraction was analyzed by capillary LC-MS/MS.
Reversed-phase Capillary LC-MS/MS Analyses
Peptide samples were analyzed using a custom-built high pressure capillary LC system20 coupled online to either a three-dimensional ion trap mass spectrometer (LCQ; ThermoElectron, San Jose, CA) or a linear ion trap mass spectrometer (LTQ; ThermoElectron) by way of an in-house-manufactured electrospray ionization (ESI) interface. The reversed-phase capillary column was prepared by slurry packing 3-μm Jupiter C18 bonded particles (Phenomenex, Torrence, CA) into a 65-cm-long, 150 μm-i.d. × 360 μm-o.d. fused silica capillary (Polymicro Technologies, Phoenix, AZ) that incorporated a 2-μm retaining stainless steel screen in an HPLC union (Valco Instruments Co., Houston, TX). The mobile phase consisted of 0.2% acetic acid and 0.05% TFA in water (A) and 0.1% TFA in 90% acetonitrile/10% water (B). Mobile phases were degassed on-line using a vacuum degasser (Jones Chromatography Inc., Lakewood, CO). After loading 10 μL of peptides onto the column, the mobile phase was held at 100% A for 20 min. Exponential gradient elution was performed by increasing the mobile-phase composition from 0–70% B over 150 min, using a stainless steel mixing chamber. To identify the eluting peptides, the linear ion trap mass spectrometer was operated in a data-dependent MS/MS mode (m/z 400–2000), in which a full MS scan was followed by five MS/MS scans. The five most intensive precursor ions were dynamically selected in the order of highest intensity to lowest intensity and subjected to collision-induced dissociation, using a normalized collision energy setting of 35%. A dynamic exclusion duration of 1 min was used. The temperature of the heated capillary and the ESI voltage were 200 °C and 2.2 kV, respectively.
MS/MS Data Analysis and Protein Categorization
All MS/MS spectra were searched independently against the human International Protein Index (IPI) database (version 2.29 consisting of 41,216 protein entries; available online at http://www.ebi.ac.uk/IPI) and the reversed human IPI protein database using SEQUEST (ThermoFinnigan)21. The reversed human protein database was created as previously reported22 by reversing the order of the amino acid sequences for each protein. The following dynamic modifications were used: carboxamidomethylation of cysteine, oxidation of methionine, and a PNGase F-catalyzed conversion of asparagine to aspartic acid at the site of carbohydrate attachment. The false positive rates of the N-glycopeptide identifications were estimated as previously described22 by dividing the number of NXS/T-motif containing peptides from the reversed database search by the number of motif containing peptides from the normal database search. The percentages of the NXS/T-motif-containing peptides in all in silico tryptic peptides from both the normal and reversed databases were determined to be at similar level (~10%); thus, the number of false positives arising from random hits should be similar from both databases. There is a very small fraction of the peptide identifications (~0.1%) that overlap in both database searching results, but the effect of these peptides on the overall estimation of false positive rates is insignificant. Several sets of Xcorr and ΔCn cutoffs obtained from this probability-based evaluation (with an overall confidence of over 95%) were used to filter the raw peptide identifications. For example, when ΔCn ≥0.1 for the 1+ charge state, then Xcorr ≥ 1.5 for fully tryptic peptides and Xcorr ≥ 2.1 for partially tryptic peptides were used; for the 2+ charge state, Xcorr ≥ 1.8 for fully tryptic peptides and Xcorr ≥ 3.3 for partially tryptic peptides; and for the 3+ charge state, Xcorr ≥ 2.6 for fully tryptic peptides and Xcorr ≥ 4.2 for partially tryptic peptides. The presence of at least one NXS/T motif was required for all peptides. In an attempt to remove redundant protein entries in the reported results, the software ProteinProphetTM was used as a clustering tool to group similar or related protein entries into a “protein group”23. All peptides that passed the filtering criteria were given an identical probability score of 1, and entered into the ProteinProphetTM program solely for clustering analysis to generate a final list of non-redundant proteins or protein groups. Gene Ontology (GO) component, function and process terms extracted from text-based annotation files downloaded from the European Bioinformatics Institute ftp site: ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/HUMAN were used to categorize the identified proteins.
Assessing the Accuracy of N-glycosylation Site Assignments Using the Accurate Mass and Time (AMT) Tag Approach
To access the accuracy of N-glycosylation site assignments in the MS/MS identifications, a portion of the enriched deglycosylated peptides (without SCX fractionation) were analyzed by LC-FTICR24 using the same LC conditions and the AMT tag approach25, 26. Briefly, the peptide retention times from each LC-MS/MS analysis were normalized to a range of 0–1 to provide normalized elution times (NETs)27. Both the calculated mass (based on sequences without deamidation of the asparagine residues) and NET of the identified NXS/T-motif-containing peptides from the LC-MS/MS analyses were included as AMT tags in a database. Features (i.e., peaks with both a unique mass and elution time) from the LC-FTICR analysis were identified by matching the measured accurate mass and elution time of each feature to the corresponding AMT tags in the database. A maximum mass error of 5 ppm and a maximum NET error of 5% were used for the matching. The number of N-glycosylation site(s) in each peptide was confidently determined by searching the AMT tag database using dynamic deamidation on asparagine residues (0.9840 Da increment in monoisotopic mass per site).
Results
Proteomic Analysis Strategy
A combination of multi-component immunoaffinity subtraction, N-glycopeptide enrichment, SCX fractionation, and LC-MS/MS was employed in this study to effectively improve the dynamic range of detection and increase the coverage for low-abundance proteins. The proteomic workflow of this approach is illustrated in Figure 1. First, crude plasma was subjected to high-abundant-protein depletion in a highly reproducible manner by using the MARS column and fully automated LC system (data not shown). The less-abundant proteins were enriched by pooling all the flow-through fractions from an initial 800 μL of plasma sample. The hydroxyl groups on adjacent carbon atoms of carbohydrates were converted to aldehydes using periodate oxidation, and then the glycoproteins were specifically captured on the hydrazide resin by the formation of covalent hydrazone bonds between the newly formed aldehyde groups and the hydrazide groups on the resin. After in situ tryptic digestion, all non-glycopeptides were removed by extensive washing. PNGase F was used to specifically release the N-glycosylated peptides (except those peptides carrying an α1→3 linked core fucose) from the resin, which resulted in converting the asparagine residues positioned at the glycan attachment to aspartic acid residues. The O-glycosylated peptides that have a Gal β1→3GalNAc core can also be released from the resin by this process. However, due to the lack of an enzyme comparable to PNGase F, all of the external monosaccharides must be sequentially removed until only the core carbohydrate structure remains on either serine or threonine residues, before the final release of those peptides using an O-glycosidase. In this study, we focused on the identification of N-glycoproteins for two reasons: 1) N-glycosylation is particularly prevalent in blood plasma, and 2) the overall specificity of N-glycopeptide capture-and-release using hydrazide chemistry and PNGase F has been well demonstrated28. The deglycosylated peptides released by PNGase F were further fractionated into 30 fractions using SCX chromatography, and each fraction was analyzed by LC-MS/MS. Prior to SCX fractionation, a portion of the deglycosylated peptides were analyzed using LC-FTICR and the AMT tag approach to assess the accuracy of MS/MS N-glycosylation site identifications; the PNGase F-catalyzed deglycosylation reaction converts each asparagine to aspartic acid residue at the position of glycan attachment, resulting in a monoisotopic mass increment of 0.9840 Da for each glycosylation site.
Figure 1.
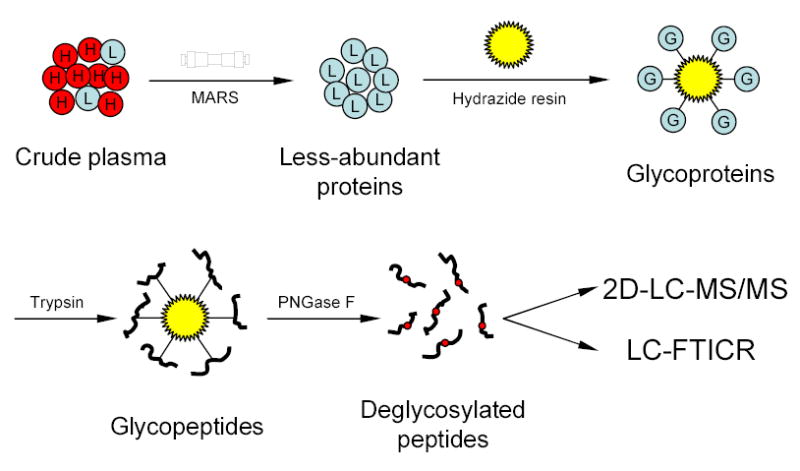
Strategy for plasma N-glycoprotein analysis using immunoaffinity subtraction, glycoprotein capture, and mass spectrometry. Crude plasma (red balls marked with “H” represent high abundance proteins; blue balls marked with “L” represent low abundance proteins) is first subjected to multi-component depletion using the pre-packed MARS column, followed by incubating the flow-through (minus the retained abundant proteins) with hydrazide resin. The captured glycoproteins (blue balls marked with “G”) are then washed and digested on-resin with trypsin, and the N-glycopeptides are specifically released from the resin by PNGase F. The resulting deglycosylated peptides can either be identified by 2-D LC-MS/MS or directly analyzed by LC-FTICR to validate the number of glycosylation sites.
Plasma N-glycoprotein Identifications
Application of the filtering criteria developed on the basis of reversed database searching provided an overall confidence level of >95%, which resulted in a total of 2053 different N-glycopeptide identifications (including partially tryptic peptides, mis-cleaved tryptic peptides, and differentially oxidized methionine-containing peptides that spanning the same glycosylation site(s)); these peptide identifications can be further collapsed to 610 representative non-redundant sequences. Consistent with previous studies, the fraction of partially tryptic peptide identifications was much greater for plasma than that observed for either cell lysates or tissue homogenates7, 10, 29. This result is most likely due to the presence of various endogenous proteases and peptidases in plasma, as well as to either the appearance of different truncated proteins from cellular and tissue “leakage” or the removal of signal peptides17, 22. A total of 303 non-redundant N-glycoprotein identifications were obtained with the majority of them being extracellular or secreted proteins. Among these non redundant identifications, 136 proteins had more than two N-glycopeptide identifications. The subcellular location and N-glycosylation information of these proteins, the representative non-redundant peptide sequences, the numbers of different peptide identifications spanning the same N-glycosylation site(s), and N-glycosylation sites, are available online as Supplementary Table 1.
A recent study of N-glycoproteins from mouse serum using hydrazide chemistry28 yielded a total of 93 N-glycoprotein identifications, while another study identified 47 N-glycoproteins from human serum using lectin affinity capture17. Both studies used single dimension LC-MS/MS analyses with three-dimensional ion trap (LCQ) mass spectrometers. The present human plasma N-glycoprotein analysis using hydrazide chemistry yielded a substantially larger set of N-glycoprotein identifications via the combined application of MARS depletion, a 2-D LC separation, and a new linear ion trap (LTQ) MS instrumentation. Experiments were conducted to further evaluate the efficiency of each of the three new components that contribute to the overall analysis improvements (Table 1). The results indicate that the 2-D LC separation made the greatest contribution (3.1-fold improvement). However, the use of new LTQ instrumentation also made 1.2-fold improvement, presumably due to its higher sensitivity (and to a lesser extent, its faster scan rate). The MARS depletion made a similar modest contribution (1.2-fold improvement), probably because the major component that was removed from the plasma during the immunosubtraction, serum albumin, is not normally glycosylated. Nonetheless, an overall 4.4-fold improvement in glycoprotein identification was achieved via the combined application of multi-component immunosubtraction, new LTQ instrumentation, and 2-D LC separation.
Table 1.
Evaluation of the efficiency of MS instrumentation, LC separation, and immunosubtraction that contribute to the overall analysis.
| MS | LC | Depletion | Unique glycoproteins | Improvement |
|---|---|---|---|---|
| LCQ | 1D | None | 69 | 1.2 |
| LTQ | 1D | None | 83 | |
| LTQ | 1D | None | 83 | 1.2 |
| LTQ | 1D | MARS | 99 | |
| LTQ | 1D | MARS | 99 | 3.1 |
| LTQ | 2D | MARS | 303 | |
| LCQ | 1D | None | 69 | 4.4 |
| LTQ | 2D | MARS | 303 |
Figure 2 shows the SCX chromatogram and the LC-MS/MS analysis of the deglycosylated peptides. A total of 30 fractions were collected from the SCX separation. Figure 2B shows the base peak chromatogram of the LC-MS/MS analysis of fraction 14, one of the peptide-rich fractions (marked with an arrow in Figure 2A). Instead of being dominated by a few high abundance species with broad elution profiles as in previous analyses of non-depleted plasma using 2D-LC-MS/MS29, a large number of peaks with narrower peak widths were observed from the base peak chromatogram, which reflects the effectively reduced sample complexity and peptide concentration range after MARS depletion and glycoprotein enrichment. Figure 2C and 2D show examples of MS scans at LC elution times of 39.41 min and 44.83 min, respectively, where almost no intense peaks appeared in the base peak chromatogram (Figure 2B). Fragmentation of low-abundant ions with m/z=638.3 and m/z=923.0 in these two MS spectra resulted in the confident identification of two fully tryptic, formerly N-glycosylated peptides K.YSVAN*DTGFVDIPKQEK.A (the asterisk indicates the N-glycosylation site and the consensus motif is in bold) and R.LQLEAVN*ITDLSENRK.Q that correspond to the two low-abundant proteins, cathepsin L (Figure 2E, 1 ng/mL) and interleukin-1 receptor antagonist protein (Figure 2F, 200 pg/mL), respectively.
Figure 2.
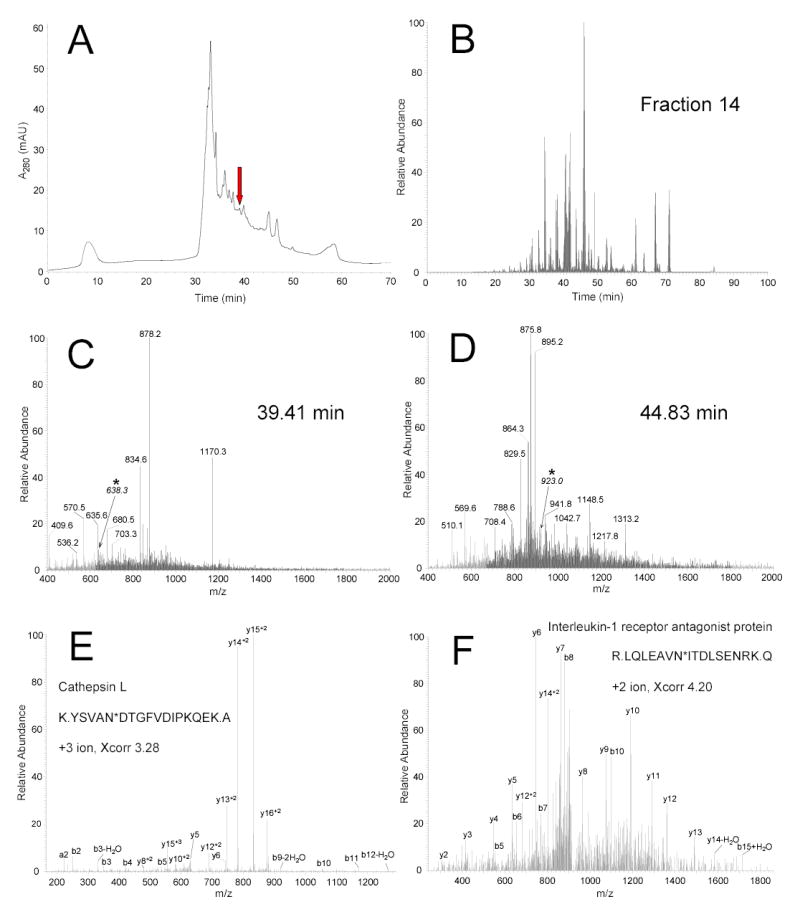
SCX fractionation and LC-MS/MS analysis. (A) The deglycosylated peptides were separated into 30 fractions using SCX chromatography. (B) Base peak chromatogram of the LC-MS/MS analysis for fraction 14 (marked with arrow in A). (C) MS spectrum for the peptides eluted at 39.41 min in the base peak chromatogram. (D) MS spectrum for the peptides eluted at 44.83 min in the same base peak chromatogram. (E) Fragmentation of low-abundance ion with m/z=638.3 (marked with asterisk in C) resulted in identification of the fully tryptic peptide K.YSVAN*DTGFVDIPKQEK.A (asterisk indicates the N-glycosylation site and the consensus motif is in bold) and originates from the low-abundance protein cathepsin L. Fragment ions that matched predicted ions are labeled as b and y ions. (F) This peptide was identified from the fragmentation of low-abundance ion with m/z=923.0 (marked with asterisk in D) as the fully tryptic peptide R.LQLEAVN*ITDLSENRK.Q and originated from the low-abundance protein interleukin-1 receptor antagonist protein.
Recently, our laboratory reported the results of several studies that involved comprehensive analysis of human plasma using various approaches, including albumin depletion, IgG depletion, and multi-dimensional and nanoscale separations9, 29, 30. 938 non-redundant plasma proteins were identified after filtering with a set of criteria developed for human plasma that provides >95% confidence level in unique peptide identifications as evaluated using reversed database searching30. In the present study, we applied the same filtering criteria and attained the same confidence level for the identification of N-glycosylated peptides, which resulted in confident identification of 303 non-redundant N-glycoproteins. Not surprisingly, the overlap of only 123 proteins between the two sets of protein identifications (ref. 30 and the work reported here) is relatively low (as shown in Figure 3), because the combination of multi-component immunoaffinity subtraction and N-glycopeptide enrichment techniques as applied in this N-glycoprotein analysis provided an enhanced dynamic range of detection that is not attainable when using either a single-component depletion step or improved LC separations alone, as in our earlier global studies. In comparing the related studies essentially all of the overlapped N-glycoprotein identifications are relatively abundant “classic” plasma proteins, e.g., α-1-acid glycoproteins, apolipoproteins, coagulation and complement factors, and protease and protease inhibitors. Table 2 lists some of the low-abundance proteins and cytokines identified in this study; a number of them have reported concentrations that range from ng/mL to pg/mL31–43. Among the low-abundance proteins are ADAM proteins, growth factors, cadherins, cathepsins, contactins, integrins, intercellular adhesion molecules, receptors for interleukins and other growth factors, neural cell adhesion molecules, and proto-oncogene tyrosine-protein kinases. In particular, the concentration of interleukin-1 receptor antagonist protein (IL-1ra) has been reported to be at the pg/mL level. The fact that numerous proteins in the low μg/mL to pg/mL range were identified suggests that the present approach can consistently detect proteins in the low ng/mL concentration range and can achieve an overall detection dynamic range of >108, which is of great significance for candidate biomarker discovery based on N-glycosylated proteins.
Figure 3.
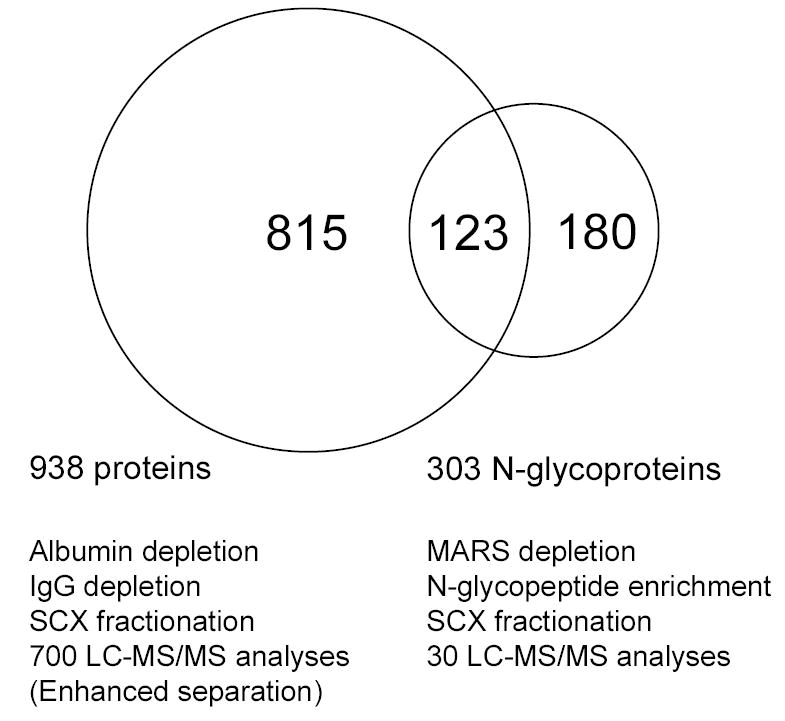
Overlap between the identified proteins from the specifically enriched N-glycopeptides and a previous study based on global tryptic peptides (Ref 30). In the previous study, the global tryptic peptides were prepared by digesting the plasma proteins with and without albumin depletion or IgG depletion, fractionated by SCX chromatography, and analyzed by LC-MS/MS with and without enhanced separation (e.g., longer gradient, smaller i.d. column), which resulted in a total of 938 non-redundant protein identifications. In contrast, 303 non-redundant N-glycoproteins were confidently identified from 30 regular LC-MS/MS analyses of the deglycosylated peptides prepared by using MARS depletion, glycoprotein capture, and SCX fractionation.
Table 2.
Selected low-abundance proteins and cytokines identified1.
| Protein ID | Description | Abbreviation | Concentration (pg/mL) |
|---|---|---|---|
| IPI00004480 | ADAM DEC1 | ADAM-Dec | |
| IPI00376784 | ADAMTS-13 protein | ADAMTS | |
| IPI00004957 | angiopoietin-related protein 3 | Angiopoietin-like 3 | |
| IPI00289346 | angiopoietin-related protein 5 | Angiopoietin-like 5 | |
| IPI00025852 | angiotensin-converting enzyme, somatic isoform | ACE | 1.20E+05 |
| IPI00032220 | angiotensinogen | Angiotensinogen | 1.00E+03 |
| IPI00005142 | basic fibroblast growth factor receptor 1 | FGFR-1 | |
| IPI00003366 | BDNF/NT-3 growth factors receptor | TRKB | |
| IPI00015916 | bone-derived growth factor | BDGF | |
| IPI00024046 | cadherin-13 | CAD-13 | |
| IPI00024035 | cadherin-6 | CAD-6 | |
| IPI00009938 | carcinoembryonic antigen-related cell adhesion molecule 1 | CD66a | 3.00E+05 |
| IPI00011229 | cathepsin D | Cathepsin D | 8.90E+03 |
| IPI00012887 | cathepsin L | Cathepsin L | 1.10E+03 |
| IPI00289819 | cation-independent mannose-6-phosphate receptor | IGF2R | |
| IPI00015102 | CD166 antigen | ALCAM | 1.60E+04 |
| IPI00024966 | contactin 2 | CNTN2 | |
| IPI00292791 | contactin 3 (plasmacytoma associated) | CNTN3 | |
| IPI00178854 | contactin 4 | CNTN4 | |
| IPI00029751 | contactin | CNTN1 | |
| IPI00029193 | hepatocyte growth factor activator | HGFA | 4.00E+05 |
| IPI00292218 | hepatocyte growth factor-like protein | HGFL | 5.90E+04 |
| IPI00018305 | insulin-like growth factor binding protein 3 | IGFBP-3 | 2.00E+04 |
| IPI00328531 | integrin alpha-1 | Integrin alpha 1 | |
| IPI00218628 | integrin alpha-IIB | Integrin alpha 2 | |
| IPI00009465 | integrin beta-1 | Integrin beta 1 | 2.10E+06 |
| IPI00291792 | integrin beta-2 | Integrin beta 2 | |
| IPI00220350 | integrin beta-3 | Integrin beta 3 | 5.50E+06 |
| IPI00027422 | integrin beta-4 | Integrin beta 4 | |
| IPI00008494 | intercellular adhesion molecule-1 | ICAM-1 | 6.30E+04 |
| IPI00009477 | intercellular adhesion molecule-2 | ICAM-2 | |
| IPI00031620 | intercellular adhesion molecule-3 | ICAM-3 (CD50) | 2.00E+05 |
| IPI00000045 | interleukin-1 receptor antagonist protein | IL-1ra | 2.36E+02 |
| IPI00297124 | interleukin-6 receptor beta chain | IL6R | 4.90E+04 |
| IPI00009243 | leptin receptor | Leptin R | 2.10E+04 |
| IPI00218795 | L-selectin | L-SELECTIN | 1.70E+04 |
| IPI00011218 | macrophage colony stimulating factor I receptor | M-CSF R | 2.60E+04 |
| IPI00009521 | macrophage receptor marco | MARCO | |
| IPI00022296 | mast/stem cell growth factor receptor | CD117 | 4.80E+03 |
| IPI00024825 | megakaryocyte stimulating factor | MSF | |
| IPI00004457 | membrane copper amine oxidase | VAP-1 | 1.20E+05 |
| IPI00032292 | metalloproteinase inhibitor 1 | TIMP1 | 1.40E+04 |
| IPI00029260 | monocyte differentiation antigen CD14 | CD14 | 1.90E+06 |
| IPI00007244 | myeloperoxidase | Mpo | 1.80E+05 |
| IPI00299059 | neural cell adhesion molecule | NCAM | |
| IPI00009476 | neural cell adhesion molecule 1, 120 kDa isoform | NCAM1 | |
| IPI00027087 | neural cell adhesion molecule L1 | NCAM | |
| IPI00299594 | neuropilin-1 | Npn | |
| IPI00006114 | pigment epithelium-derived factor | PEDF | 5.00E+06 |
| IPI00013179 | prostaglandin-H2 D-isomerase | PGDS | |
| IPI00302641 | protocadherin FAT 2 | PCAD | |
| IPI00029756 | proto-oncogene tyrosine-protein kinase MER | C-mer | |
| IPI00288965 | proto-oncogene tyrosine-protein kinase ROS | RET | |
| IPI00017480 | sonic hedgehog protein | Shh | |
| IPI00021834 | tissue factor pathway inhibitor | TFPI-1 | 7.80E+04 |
| IPI00022462 | transferrin receptor protein 1 (CD71 antigen) | TfR | 5.80E+05 |
| IPI00000075 | transforming growth factor beta 1 | TGF-beta | 2.30E+03 |
| IPI00018136 | vascular cell adhesion protein 1 | VCAM-1 | 9.40E+04 |
| IPI00293565 | vascular endothelial growth factor receptor 3 | VEGF R3 | |
| IPI00012792 | vascular endothelial-cadherin | VE-Cadherin | 3.00E+04 |
The plasma protein concentrations are approximate average values based on previous literature (31–44) and product literature from R&D Systems Quantikine immunoassay Kits (www.rndsystems.com).
Although the use of immunoaffinity approaches to remove high-abundance proteins (especially albumin) may potentially remove some other low-abundance proteins of interest due to specific or non-specific binding44, we have been able to confidently identify significantly more low-abundance proteins from the MARS-depleted plasma sample as a result of the effectively reduced protein concentration range. Immunoaffinity subtraction process using the MARS column and fully automated HPLC system is robust and reproducible chromatographically (data not shown). Moreover, in the LC-MS/MS analyses of three independently prepared samples, 66 ± 3 glycoproteins were identified from the flow-through plasma protein samples, and 26 ± 2 proteins (without glycoprotein enrichment) were identified from the bound plasma protein samples, respectively. The overlap of protein identifications in these replicated experiments is 90%, which is similar to what we normally observe in repeated analysis of less complex samples using ion trap mass spectrometers. In addition, almost all the identified MARS-bound plasma proteins are proteins targeted by the antibodies, except that there were a total of 15 different immunoglobulins identified (the peptide and protein identifications of the MARS-bound proteins are available on line in Supplementary Table 2). In a recent study on high-abundant protein depletion45, it was observed that the MARS system have no albumin, transferrin, α-1-antitrypsin, or haptoglobin present in the flow-through fraction, and the ELISA results indicated that depletion of the target proteins is typically greater than 98%. In this study, most of the target proteins except for albumin were still identified with multiple N-glycopeptides (Supplementary Table 1). This observation suggests the presence of these proteins in the sample even after >98% depletion, presumably due to the very high initial concentrations for these proteins. The overall throughput and reproducibility can be further improved by implementing automated sample processing. Thus, these processes can be readily incorporated into a quantitative proteomic strategy to enhance detection of low-abundance proteins in various biofluids for discovering candidate biomarkers. Many plasma proteins are known to be present in various post-translationally processed forms, particularly differentially glycosylated forms, which increase proteome complexity and heterogeneity. For example, in a recent large scale plasma proteome profiling reported by Pieper et al.5, using extensive pre-fractionation of the plasma proteins prior to 2DE separation, 3700 protein spots were displayed on 2D gels. However, only 325 distinct proteins were identified by MS, largely due to the presence of the different forms of the same protein that have similar molecular weights, but different isoelectric points (horizontal stripes on gels). However, since it is estimated that there is only an average of 3.6 potential N-glycopeptides per protein28 and the highly heterogeneous oligosaccharides can be removed from the enriched glycopeptides, the quantitative measurements of plasma, by either isotopic labeling16 or direct feature comparison28, will greatly benefit from the use of the enriched deglycosylated peptides due to the largely reduced sample complexity and heterogeneity.
Assessing Accuracy of N-glycosylation Site Assignments Using LC-FTICR
A total of 639 putative N-glycosylation sites were identified from the LC-MS/MS analyses. Among these sites, 225 were annotated in SWISS-PROT as known N-glycosylation sites, 300 were annotated as “probable” or “potential” N-glycosylation sites, and 114 were unknown either because the sites were not annotated or because the corresponding proteins did not have a SWISS-PROT entry (Supplementary Table 1). Twenty-six peptides had more than one putative N-glycosylation site. Two peptides were identified with 3 putative sites, and all of these sites were annotated in SWISS-PROT as known or probable N-glycosylation sites. The peptide R.ETIYPN*ASLLIQN*VTQN*DTGFYTLQVIK.S, with all 3 sites annotated as known glycosylation sites, was identified from carcinoembryonic antigen-related cell adhesion molecule 1, which has a total of 5 known sites and 15 potential sites. The other triply N-glycosylated peptide K.NN*MSFVVLVPTHFEWN*VSQVLAN*LSWDTLHPPLVWERPTK.V was identified from α-2-antiplasmin, and all 3 of the identified sites were annotated as potential sites. The ability to identify a large number of doubly or triply glycosylated peptides suggests that the glycopeptide capture-and-release method used in this study provides good coverage for abundant N-glycopeptides that originate from plasma proteins, although in situ protein digestion may be sterically hindered by the presence of large, covalently-bound carbohydrate moieties.
In LC-MS/MS analysis, the assignment of the glycosylation sites by SEQUEST was performed by searching the protein database using deamidation of asparagine as a dynamic modification (a monoisotopic mass increment of 0.9840 Da). Such a small mass difference may make the accurate assignment of glycosylation sites difficult due to the limited mass measurement accuracy of ion-trap instrumentation. This difficulty in site assignment is particularly true when the peptide has more than one NXS/T motif, since it is not necessarily always a one motif-one site scenario (e.g., one peptide that has two NXS/T motifs may have just one N-glycosylation site). Thus, to assess the LC-MS/MS glycosylation site identifications, the same deglycosylated peptide sample (without SCX fractionation) was measured using a single LC-FTICR analysis, and the results are summarized in Table 3. A total of 246 different peptides covering 95 proteins were identified using the accurate mass measurements provided by LC-FTICR; the details of these site-confirmed glycopeptide identifications are available online in Supplementary Table 3.
Table 3.
Summary of a single capillary LC-FTICR analysis of formerly N-glycosylated peptides from human plasma.
|
Number of confirmed N-glycosylation site(s) |
||||
|---|---|---|---|---|
| total | 0 site | 1 site | 2 sites | |
| Peptides containing one NXS/T1 motif | 229 | 4 | 225 | 0 |
| Peptides containing two NXS/T motifs | 17 | 0 | 4 | 13 |
NXS/T is the consensus sequence motif in which X represents any amino acid residue.
An AMT tag database was generated that contained the calculated masses (based on the unmodified peptide sequences) and NETs of all peptide identifications with at least one NXS/T motif from the LC-MS/MS analyses. Dynamic modification, corresponding to different numbers of deamidation of asparagine residues (i.e., monoisotopic mass increment of n×0.9840 Da, n=1 to 3), was applied when features were matched to this AMT tag database. Note that peptides that contain the NPS/T motif (which cannot be N-glycosylated) were also included in the AMT tag database to test the accuracy of this method. Among the 229 peptides containing one NXS/T motif, 225 peptides were determined to have only one glycosylation site, and four peptides were determined not to be glycosylated (1.3%, excluding one NPS/T motif-containing peptide included for test purposes). For the 225 one-site peptides confirmed by LC-FTICR, 169 sites were annotated as known N-glycosylation sites in SWISS-PROT and 49 sites were annotated as potential sites (Supplementary table 3). Seven additional one-NXS/T-motif-containing peptides with undescribed sites were observed by LC-FTICR with 0.9840 Da increment in mass, thereby confirming the presence of one N-glycosylation site in those peptides. Among 17 peptides that contained two NXS/T motifs, all had glycosylation sites: 13 peptides were determined to have two glycosylation sites (all sites were annotated as known sites) and the other four peptides were determined to have one glycosylation site (three known sites and one potential site). Interestingly, among all of the peptides identified that contained two NPS/T motifs, the peptide K.VVNPTQK.- from the α-1-antitrypsin-related protein was determined to be a non-glycosylated peptide based on accurate mass measurement and another one, R.LNPTVTYGN*DSFSAK.A from intercellular adhesion molecule-1, was determined to have only one site. These results indicate that accurate mass LC-FTICR and the AMT tag approach can be used to accurately determine the number of sites in formerly N-glycosylated peptides. Many “potential” or unknown sites were confirmed by this single LC-FTICR analysis. For example, five sites that were previously annotated as potential sites were validated for apolipoprotein B-100 (Supplementary Table 3). When the number of NXS/T motifs in the peptide and the number of deamidated asparagine residues confirmed by LC-FTICR are different, additional experiments may be necessary to identify the exact site of glycosylation, e.g., performing an enzymatic deglycosylation reaction in the presence of enriched 18O water to introduce a 2-Da mass increment upon deglycosylation followed by MS/MS analysis46, 47. However, the fact that only 1.3% of the one-NXS/T motif-containing peptides were shown to be non-glycosylated by LC-FTICR measurements supports the overall high accuracy of the site assignment of LC-MS/MS identifications obtained in this study.
Categorization of the Human Plasma N-glycoproteome
The 303 N-glycoproteins confidently identified in this study include common circulatory plasma proteins, coagulation and complement factors, blood transport and binding proteins, and protease and protease inhibitors that are present in moderate to high abundance, as well as other enzymes, secreted or membrane-associated proteins, and cytokines and hormones that are present in medium to low abundance in the plasma. All N-glycoproteins identified in this study were categorized on the basis of their GO component, function, and process terms.
Figure 4 shows the cellular distributions of the 303 N-glycoproteins identified in this study (A) and 938 proteins confidently identified in our previous global study30 (B). It comes as no surprise that the cellular distributions of the proteins in these two datasets are very different. Compared with the cellular components of global plasma in the previous study, significantly higher percentages of certain cellular components are identified in this study for the N-glycoproteins: extracellular (19.9% →51.1%), membrane (14.3% →34.1%), and endoplasmic reticulum, Golgi, and lysosome (2.7% →5.2%). Note that none of these N-glycoproteins are from the nuclear, mitochondria, cytoplasm, and cytoskeleton (Figure 4A). This finding suggests that most glycoproteins are either secreted, shed (from the extracellular side of the plasma membrane), or transported into the plasma. All of the N-glycoproteins identified in the single LC-FTICR analysis were also categorized using component terms, and their cellular distribution is very similar to that of the N-glycoproteins identified in the LC-MS/MS analysis (data not shown).
Figure 4.
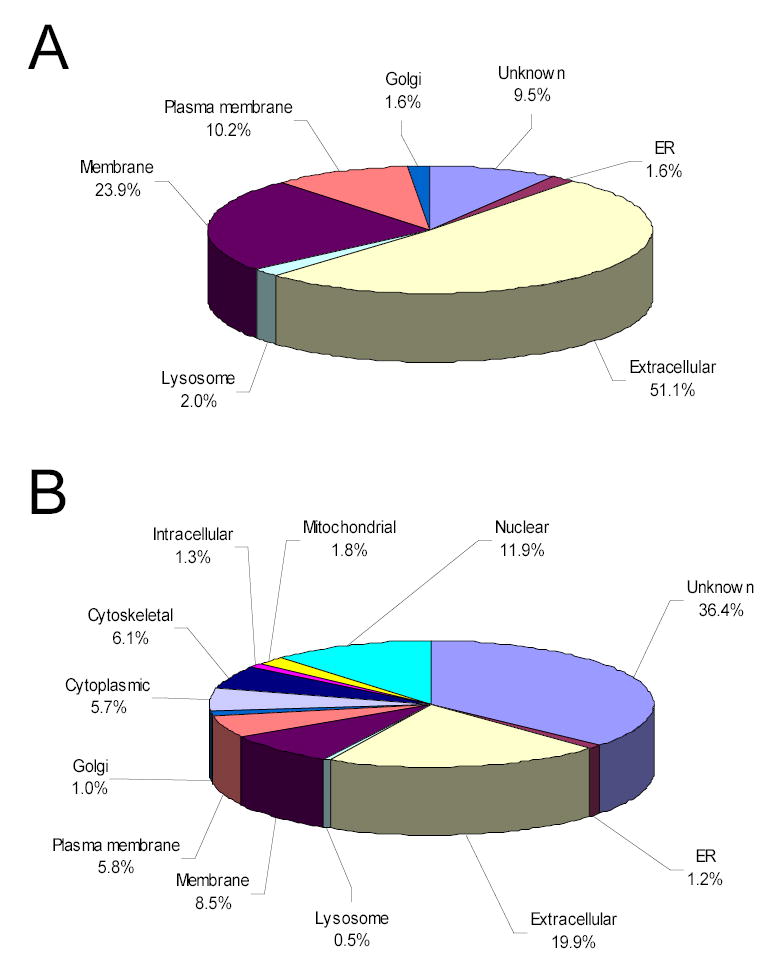
Comparison of protein categorization using GO component terms. Major component categories are shown for N-glycoprotein identifications (A) and global protein identifications (B).
In the GO function categorization, a large portion of glycoproteins possess binding activity (27.3%), while two other significant portions show receptor activity (11.8%) and transporter activity (9.2%). Protease and protease inhibitors are present at almost the same level (~10%). Glycoproteins also display activities for a variety of enzymes, e.g., kinases and phosphatases (2.0%), transferases (2.0%), and other enzymes (9.5%). Noticeably, 14.1% of the glycoproteins have cytokine and hormone activities, 3.6% of them have structural molecule activity, and 0.7% of them have transcription factor activity (Integrin β-4 and Plexin B1). The N-glycoproteins identified in this study also have been indicated to be involved in various biological processes – circulation (1.9%), coagulation and proteolysis (13.5%), immune and inflammatory responses and defensive mechanisms (19.3%), development (9.9%), signaling (12.0%), transcription (1.2%), transport (8.7%), metabolism (12.0%), and cell adhesion, death, mobility and proliferation (21.5%) – that reflect the major physiological functions of human blood, including immunity, coagulation, inflammation, small molecule transport, and lipid metabolism.
Discussion
Application of multi-component immunoaffinity subtraction and glycopeptide enrichment methods in combination with 2-D LC-MS/MS analyses significantly adds to the number of N-glycoproteins previously identified in human plasma. Using this approach to profile the human plasma N-glycoproteome resulted in confident identification of 2053 different N-glycopeptides, covering a total of 303 non-redundant proteins. Furthermore, the overall high accuracy of the LC-MS/MS N-glycosylation site assignments was assessed by LC-FTICR accurate mass measurements. An estimated dynamic range of detection >108 was achieved due largely to the greatly reduced protein concentration range and sample complexity; a series of low-abundance proteins were identified having concentrations ranging from low μg/mL to pg/mL levels (Table 2). This work provides a foundation for quantitative measurements of the human plasma proteome using either isotopic labeling or “label-free” MS-intensity measurements of the detected glycopeptides using highly sensitive LC-FTICR and the AMT tag approach. A major advantage of this quantitation strategy is that once an AMT tag database is generated from these MS/MS identifications, a large number of plasma samples derived from various disease states (e.g., clinical plasma samples) or treatments can be analyzed in a high-throughput manner using LC-MS, without the need for additional LC-MS/MS measurements25.
In addition to effective sample preparation and pre-fractionation techniques (e.g., immunoaffinity subtraction, protein/peptide separation by liquid chromatography, enrichment of subsets of peptides), sophisticated and sensitive detection technologies (e.g., ion mobility MS48, LC-FTICR) are required to overcome the large protein concentration range and sample complexity of human plasma. In particular, the use of high performance LC-FTICR together with specific peptide enrichment techniques offers significant potential for greatly accelerating the qualitative and quantitative characterization of the human plasma proteome, and more importantly, the analysis of plasma samples from clinical studies. In the case of N-glycosylation, it is still not possible to differentiate between spontaneous deamidation and enzymatic deglycosylation as the cause of asparagine to aspartic acid conversion by either LC-MS/MS or LC-FTICR. Additional sample processing, e.g., the application of an enzymatic deglycosylation reaction in the presence of enriched 18O water, can be used for the determination of the exact sites of glycosylation, since a dynamic modification of asparagine with a 2-Da mass increment can be introduced upon deglycosylation. On the other hand, the LC-FTICR measurements can be used to confidently determine the number of glycosylation sites for each peptide. In combination, these two techniques can be used to identify glycosylation sites, as well as differentiate the sites from the spontaneously deamidated asparagine residues in the peptide.
Since the majority of diagnostic and clinical markers currently used are glycosylated, the proteomic profiling of N-glycoproteins in human plasma offers significant potential for the discovery of candidate disease biomarkers and therapeutic targets. In fact, a number of glycoproteins identified in this study are known to be involved in disease processes or have potential value as tissue-specific or disease-associated biomarkers. These glycoproteins include aminopeptidase N (CD13; is used as a marker for acute myeloid leukemia and plays a role in tumor invasion), attractin (involved in the initial immune cell clustering during inflammatory response and may regulate the chemotactic activity of chemokines), carcinoembryonic antigen-related cell adhesion molecule 1 (loss of or reduced expression is a major event in colorectal carcinogenesis), cathepsin D (involved in the pathogenesis of several diseases such as breast cancer and possibly Alzheimer's disease), cathepsin L (can serve as a marker of bone resorption and bone density), CD44 antigen (expressed by cells of epithelium and highly expressed by carcinomas, and plays an important role in cell migration, tumor growth and progression), ficolin 3 (expressed in lung and is highly abundant in the serum of patients with systemic lupus erythematosus), insulin-like growth factor binding protein 3 (associated with an increased risk of endometrial cancer), lysosome-associated membrane glycoprotein 1 and 2 (implicated in tumor cell metastasis), mast/stem cell growth factor receptor (its defect is a cause of gastrointestinal stromal tumor), pregnancy zone protein (potential biomarker for ovarian cancer), and tenascin X (plays a role in supporting the growth of epithelial tumors and associates with congenital adrenal hyperplasia)49. Quantitation of the relative abundances for these specific glycoproteins in plasma may provide insight into specific disease mechanisms, as well as leads for candidate disease biomarkers. In addition to changes in relative abundances, altered glycosylation may also correlate with disease. For example, a recent study demonstrating elevations in the relative abundance of Golgi Protein 73 (GP73) in the serum of patients diagnosed with hepatocellular carcinoma further suggests that examination of the glycosylation pattern may further increase the specificity of this marker50. Targeted studies on the changes in glycan structure may also be required in addition to quantitative proteomic analyses to illustrate a specific disease mechanism in depth.
Acknowledgments
Portions of this research were supported by the National Institute of General Medical Sciences (NIGMS, Large Scale Collaborative Research Grants U54 GM-62119-02) and the NIH National Center for Research Resources (RR18522). Work was performed in the Environmental Molecular Science Laboratory, a U. S. Department of Energy (DOE) national scientific user facility located on the campus of Pacific Northwest National Laboratory (PNNL) in Richland, Washington. PNNL is a multiprogram national laboratory operated by Battelle Memorial Institute for the DOE under contract DE-AC05-76RLO-1830.
References
- 1.Anderson NL, Anderson NG. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Diamandis EP. Mol Cell Proteomics. 2004;3:367–378. doi: 10.1074/mcp.R400007-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Hanash S. Nature. 2003;422:226–232. doi: 10.1038/nature01514. [DOI] [PubMed] [Google Scholar]
- 4.Petricoin E, Wulfkuhle J, Espina V, Liotta LA. J Proteome Res. 2004;3:209–217. doi: 10.1021/pr049972m. [DOI] [PubMed] [Google Scholar]
- 5.Pieper R, Gatlin CL, Makusky AJ, Russo PS, Schatz CR, Miller SS, Su Q, McGrath AM, Estock MA, Parmar PP, Zhao M, Huang ST, Zhou J, Wang F, Esquer-Blasco R, Anderson NL, Taylor J, Steiner S. Proteomics. 2003;3:1345–1364. doi: 10.1002/pmic.200300449. [DOI] [PubMed] [Google Scholar]
- 6.Pieper R, Su Q, Gatlin CL, Huang ST, Anderson NL, Steiner S. Proteomics. 2003;3:422–32. doi: 10.1002/pmic.200390057. [DOI] [PubMed] [Google Scholar]
- 7.Adkins JN, Varnum SM, Auberry KJ, Moore RJ, Angell NH, Smith RD, Springer DL, Pounds JG. Mol Cell Proeomics. 2002;1:947–955. doi: 10.1074/mcp.m200066-mcp200. [DOI] [PubMed] [Google Scholar]
- 8.Chromy BA, Gonzales AD, Perkins J, Choi MW, Corzett MH, Chang BC, Corzett CH, McCutchen-Maloney SL. J Proteome Res. 2004;3:1120–7. doi: 10.1021/pr049921p. [DOI] [PubMed] [Google Scholar]
- 9.Shen Y, Jacobs JM, Camp DG, 2nd, Fang R, Moore RJ, Smith RD, Xiao W, Davis RW, Tompkins RG. Anal Chem. 2004;76:1134–44. doi: 10.1021/ac034869m. [DOI] [PubMed] [Google Scholar]
- 10.Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Mol Cell Proteomics. 2003;2:1096–103. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Xiao Z, Conrads TP, Lucas DA, Janini GM, Schaefer CF, Buetow KH, Issaq HJ, Veenstra TD. Electrophoresis. 2004;25:128–33. doi: 10.1002/elps.200305700. [DOI] [PubMed] [Google Scholar]
- 12.Heller M, Michel PE, Morier P, Crettaz D, Wenz C, Tissot JD, Reymond F, Rossier JS. Electrophoresis. 2005;26:1174–88. doi: 10.1002/elps.200410106. [DOI] [PubMed] [Google Scholar]
- 13.Liu T, Qian WJ, Strittmatter EF, Camp DG, 2nd, Anderson GA, Thrall BD, Smith RD. Anal Chem. 2004;76:5345–53. doi: 10.1021/ac049485q. [DOI] [PubMed] [Google Scholar]
- 14.Liu T, Qian WJ, Chen WN, Jacobs JM, Moore RJ, Anderson DJ, Gritsenko MA, Monroe ME, Thrall BD, Camp DG, 2nd, Smith RD. Proteomics. 2005;5:1263–73. doi: 10.1002/pmic.200401055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H, Li XJ, Martin DB, Aerbersold R. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 17.Bunkenborg J, Pilch BJ, Podtelejnikov AV, Wisniewski JR. Proteomics. 2004;4:454–65. doi: 10.1002/pmic.200300556. [DOI] [PubMed] [Google Scholar]
- 18.Roth J. Chem Rev. 2002;102:285–303. doi: 10.1021/cr000423j. [DOI] [PubMed] [Google Scholar]
- 19.Bause E. Biochem J. 1983;209:331–6. doi: 10.1042/bj2090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen Y, Zhao R, Belov ME, Conrads TP, Anderson GA, Tang K, Pasa-Tolic L, Veenstra TD, Lipton MS, Smith RD. Anal Chem. 2001;73:1766–1775. doi: 10.1021/ac0011336. [DOI] [PubMed] [Google Scholar]
- 21.Eng JK, McCormack AL, Yates JR., 3rd J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 22.Qian WJ, Liu T, Monroe ME, Strittmatter EF, Jacobs JM, Kangas LJ, Petritis K, Camp DG, 2nd, Smith RD. J Proteome Res. 2005;4:53–62. doi: 10.1021/pr0498638. [DOI] [PubMed] [Google Scholar]
- 23.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 24.Gorshkov MV, Pasa-Tolic L, Udseth HR, Anderson GA, Huang BM, Bruce JE, Prior DC, Hofstadler SA, Tang L, Chen L, Willett JA, Rockwood AL, Sherman MS, Smith RD. J Am Soc Mass Spectrom. 1998;9:692–700. doi: 10.1016/s1044-0305(98)00037-3. [DOI] [PubMed] [Google Scholar]
- 25.Smith RD, Anderson GA, Lipton MS, Pasa-Tolic L, Shen Y, Conrads TP, Veenstra TD, Udseth HR. Proteomics. 2002;2:513–523. doi: 10.1002/1615-9861(200205)2:5<513::AID-PROT513>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 26.Lipton MS, Pasa-Tolic L, Anderson GA, Anderson DJ, Auberry DL, Battista JR, Daly MJ, Fredrickson J, Hixson KK, Kostandarithes H, Masselon C, Markillie LM, Moore RJ, Romine MF, Shen Y, Stritmatter E, Tolic N, Udseth HR, Venkateswaran A, Wong KK, Zhao R, Smith RD. Proc Natl Acad Sci USA. 2002;99:11049–11054. doi: 10.1073/pnas.172170199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petritis K, Kangas LJ, Ferguson PL, Anderson GA, Pasa-Tolic' L, Lipton MS, Auberry KJ, Strittmatter E, Shen Y, Zhao R, Smith RD. Anal Chem. 2003;75:1039–1048. doi: 10.1021/ac0205154. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Yi EC, Li XJ, Mallick P, Kelly-Spratt KS, Masselon CD, Camp DG, 2nd, Smith RD, Kemp CJ, Aebersold R. Mol Cell Proteomics. 2005;4:144–55. doi: 10.1074/mcp.M400090-MCP200. [DOI] [PubMed] [Google Scholar]
- 29.Qian WJ, Jacobs JM, Camp DG, 2nd, Monroe ME, Moore RJ, Gritsenko MA, Calvano SE, Lowry SF, Xiao W, Moldawer LL, Davis RW, Tompkins RG, Smith RD. Proteomics. 2005;5:572–84. doi: 10.1002/pmic.200400942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian WJ, Monroe ME, Liu T, Jacobs JM, Anderson GA, Shen Y, Moore RJ, Anderson DJ, Zhang R, Calvano SE, Lowry SF, Xiao W, Moldawer LL, Davis RW, Tompkins RG, Camp DG, 2nd, Smith RD. Mol Cell Proteomics. 2005;4:700–709. doi: 10.1074/mcp.M500045-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Draberova L, Cerna H, Brodska H, Boubelik M, Watt SM, Stanners CP, Draber P. Immunology. 2000;101:279–87. doi: 10.1046/j.1365-2567.2000.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hara I, Miyake H, Yamanaka K, Hara S, Kamidono S. Oncol Rep. 2002;9:1379–83. [PubMed] [Google Scholar]
- 33.Lang TH, Willinger U, Holzer G. J Lab Clin Med. 2004;144:163–6. doi: 10.1016/j.lab.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Haab BB, Geierstanger BH, Michailidis G, Vitzthum F, Forrester S, Okon R, Saviranta P, Brinker A, Sorette M, Perlee L, Suresh S, Drwal G, Adkins JN, Omenn G. Proteomics. 2005 doi: 10.1002/pmic.200401276. In press. [DOI] [PubMed] [Google Scholar]
- 35.Shimomura T, Kondo J, Ochiai M, Naka D, Miyazawa K, Morimoto Y, Kitamura N. J Biol Chem. 1993;268:22927–32. [PubMed] [Google Scholar]
- 36.Oh JC, Wu W, Tortolero-Luna G, Broaddus R, Gershenson DM, Burke TW, Schmandt R, Lu KH. Cancer Epidemiol Biomarkers Prev. 2004;13:748–52. [PubMed] [Google Scholar]
- 37.Yamauchi M, Mizuhara Y, Maezawa Y, Toda G. Pathol Res Pract. 1994;190:984–92. doi: 10.1016/S0344-0338(11)81005-8. [DOI] [PubMed] [Google Scholar]
- 38.Martin S, Rieckmann P, Melchers I, Wagner R, Bertrams J, Voskuyl AE, Roep BO, Zielasek J, Heidenthal E, Weichselbraun Iea. J Immunol. 1995;154:1951–5. [PubMed] [Google Scholar]
- 39.Rivera M, Talens-Visconti R, Sirera R, Bertomeu V, Salvador A, Cortes R, Garcia de Burgos F, Climent V, Paya R, Martinez-Dolz L, Sancho-Tello MJ, Gonzalez-Molina A. Eur J Heart Fail. 2004;6:877–82. doi: 10.1016/j.ejheart.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 40.Jarkovska Z, Rosicka M, Krsek M, Sulkova S, Haluzik M, Justova V, Lacinova Z, Marek J. Physiol Res. 2005 In press. [PubMed] [Google Scholar]
- 41.Kamel AM, El-Sharkawy NM, Khalaf MR, Galal SH, Ghelab FM, Ghazaly T. J Egypt Natl Canc Inst. 2002;14:243–9. [Google Scholar]
- 42.Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, Simoons ML, Hamm CW, Investigators C. Circulation. 2003;108:1440–5. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 43.Petersen SV, Valnickova Z, Enghild JJ. Biochem J. 2003;374:199–206. doi: 10.1042/BJ20030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liotta LA, Ferrari M, Petricoin E. Nature. 2003;425:905. doi: 10.1038/425905a. [DOI] [PubMed] [Google Scholar]
- 45.Zolotarjova N, Martosella J, Nicol G, Bailey J, Boyes BE, Barrett WC. Proteomics. 2005;5:3304–3313. doi: 10.1002/pmic.200402021. [DOI] [PubMed] [Google Scholar]
- 46.Kuster B, Mann M. Anal Chem. 1999;71:1431–40. doi: 10.1021/ac981012u. [DOI] [PubMed] [Google Scholar]
- 47.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobe T. Nat Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- 48.Shvartsburg AA, Tang K, Smith RD. Anal Chem. 2004;76:7366–74. doi: 10.1021/ac049299k. [DOI] [PubMed] [Google Scholar]
- 49. http://us.expasy.org/sprot.
- 50.Block TM, Comunale MA, Lowman M, Steel LF, Romano PR, Fimmel C, Tennant BC, London WT, Evans AA, Blumberg BS, Dwek RA, Mattu TS, Mehta AS. Proc Natl Acad Sci USA. 2005;102:779–784. doi: 10.1073/pnas.0408928102. [DOI] [PMC free article] [PubMed] [Google Scholar]