

Adenosine deaminases that act on RNA (ADARs) are RNA editing enzymes that convert adenosine (A) to inosine (I) within double-stranded RNA (dsRNA) [reviewed in (1)]. ADARs are found in metazoa where they are highly expressed in neuronal tissues. Drosophila melanogaster and Caenorhabditis elegans strains lacking all ADAR activity are viable but exhibit behavioral defects (2, 3), whereas mice lacking ADARs die embryonically or shortly after birth (4, 5).
ADARs target messenger RNAs (mRNAs) in coding regions, as well as in noncoding sequences such as 5′ and 35′ untranslated regions (UTRs). A to I changes in coding sequences often alters codon meaning. For example, A to I editing in mammalian glutamate receptor B mRNA changes a glutamine (Q) codon to an arginine (R) codon at a position critical for determining ion permeability of the glutamate-gated channel; at least some of the defects of mice lacking ADARs derive from the absence of Q-R site editing (5).
The purpose of inosines within noncoding regions of an mRNA is unclear. This type of editing has been observed in mRNA of C. elegans and human brain and may be more common than editing within codons (6, 7). The double-stranded structures that mediate editing in UTRs are remarkable in their length and stability, sometimes consisting of several hundred nearly contiguous base pairs (6, 7). Given this, the question arises as to why these structures do not trigger RNA interference (RNAi). One possibility, raised by recent results (8), is that RNA editing prohibits dsRNA from triggering RNAi. A to I changes would destroy the complementarity between a dsRNA and mRNA that is required for RNAi. Further, because ADARs convert AU basepairs to IU mismatches, the UTR structures would become more single-stranded and less suited for binding dsRNA binding proteins such as Dicer. According to this scenario, the phenotypes of ADAR mutants would result because mRNAs that were normally “protected” by editing would be degraded by the RNAi pathway.
To explore this idea, we crossed C. elegans strains lacking ADARs with RNAi defective strains. The RNAi defective strains we chose contain mutations in the rde-1 or rde-4 gene, and are incapable of eliciting an RNAi response but otherwise appear normal (9–11). Previously, we observed that deletions in each or both of the C. elegans adr genes (adr-1 and adr-2) produce animals with defects in chemotaxis (2), a measure of the efficacy of a worm’s olfactory system. As shown (Fig. 1A), wild-type animals exposed to chemicals that smell like food move toward the chemoattractant in a concentration-dependent manner. The chemotaxis index reflects the number of animals reaching the chemoattractant, and adr-1(gv6);adr-2(gv42) double mutants exhibit significantly lower chemotaxis indices. We observed that two rde-1 alleles and an rde-4 allele rescued the chemotaxis defects of the adr-1(gv6);adr-2(gv42) animals. Animals containing only a mutation in the rde-1 or rde-4 gene showed normal chemotaxis behavior (Fig. 1B). Rescue was specific for the chemotaxis defects caused by mutations in the adr genes, because crossing rde-1 alleles into other chemotaxis-deficient strains did not lead to rescue (Fig. 1C).
Fig. 1.
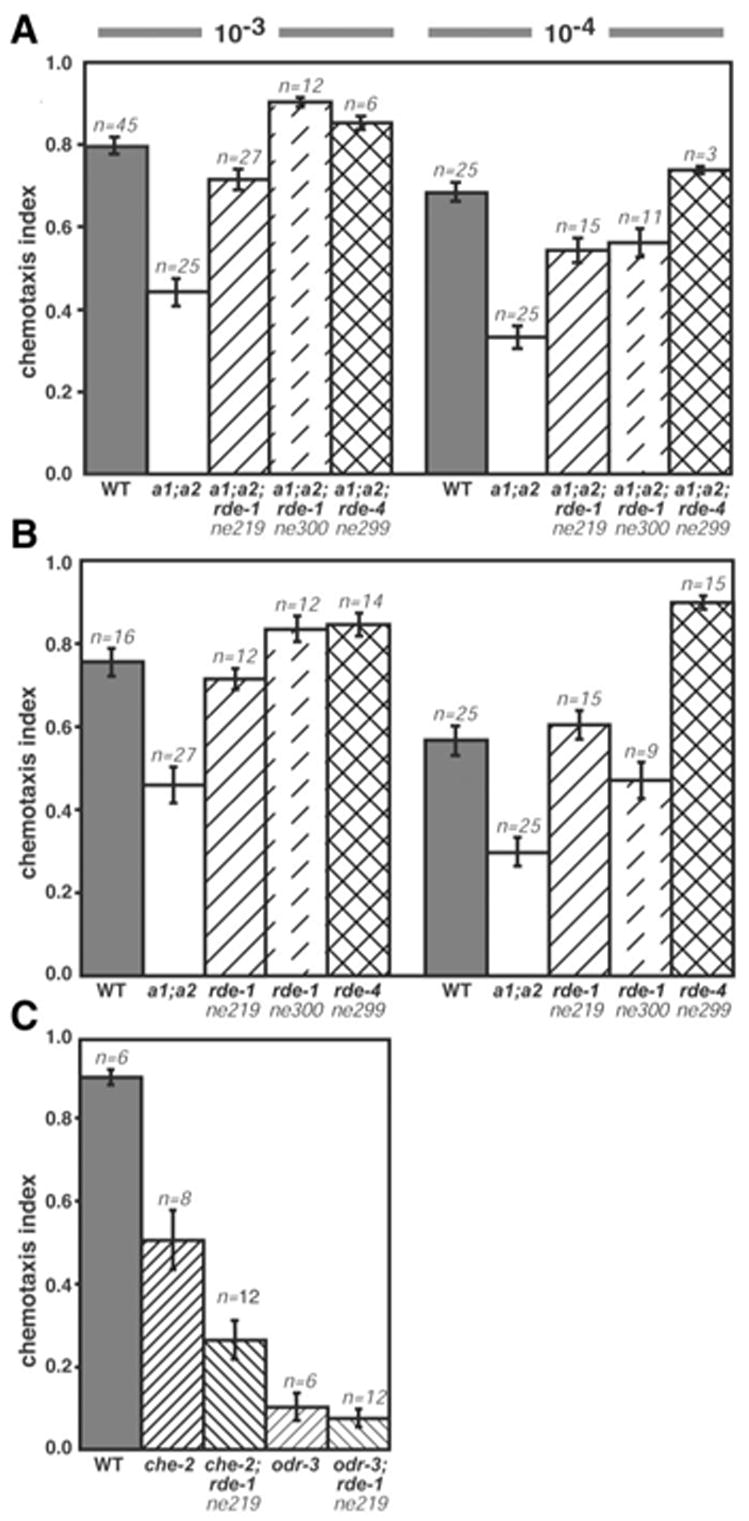
Chemotaxis assays were performed as described, except animals were cultured on plates (2). Graphs show mean chemotaxis indices for assays with 2-butanone at 10−3 and 10−4 dilutions. The number of trials is noted above error bars (SEM). Wild-type (WT) and adr-1(gv6);adr-2(gv42) double-mutants (a1;a2) were compared with (A) two rde-1 rescue strains and an rde-4 rescue strain or (B) with rde-1 and rde-4 strains. Values for rescue strains were significantly different (P ≤ 0.0001) from a1;a2 strains, but not from WT. Similar results were observed with diacetyl (fig. S1). (C) Defects of che-2(e1033) and odr-3(n2150) were not rescued by rde-1 alleles. P values are from a Student’s t test (two-tailed, unequal variance).
Our results have several implications. First, they suggest that, in addition to changing codon meaning, ADARs play a role in regulating whether a dsRNA enters the RNAi pathway. This function could be quite far-reaching: not only could ADARs regulate silencing triggered by intramolecular structures in mRNA, but also that caused by spurious antisense transcription throughout the genome (12). Finally, we speculate that dsRNA-mediated gene silencing might play a special role in the nervous system.
Supplementary Material
Footnotes
Supporting Online Material
References and Notes
- 1.Bass BL. Annu Rev Biochem. 2002;71:817. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonkin LA, et al. EMBO J. 2002;21:6025. doi: 10.1093/emboj/cdf607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. Cell. 2000;102:437. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Khillan J, Gadue P, Nishikura K. Science. 2000;290:1765. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 5.Higuchi M, et al. Nature. 2000;406:78. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 6.Morse DP, Bass BL. Proc Natl Acad Sci USA. 1999;96:6048. doi: 10.1073/pnas.96.11.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morse DP, Aruscavage PJ, Bass BL. Proc Natl Acad Sci USA. 2002;99:7906. doi: 10.1073/pnas.112704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knight SW, Bass BL. Mol Cell. 2002;10:809. doi: 10.1016/s1097-2765(02)00649-4. [DOI] [PubMed] [Google Scholar]
- 9.Tabara H, Yigit E, Siomi H, Mello CC. Cell. 2002;109:861. doi: 10.1016/s0092-8674(02)00793-6. [DOI] [PubMed] [Google Scholar]
- 10.Tabara H, et al. Cell. 1999;99:123. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- 11.Parrish S, Fire A. RNA. 2001;7:1397. [PMC free article] [PubMed] [Google Scholar]
- 12.Yelin R, et al. Nature Biotechnol. 2003;21:379. doi: 10.1038/nbt808. [DOI] [PubMed] [Google Scholar]
- 13.We thank S. Knight and E. Herrington for advice and assistance and C. Mello for rde strains. che-2(e1033) and odr-3(n2150) were provided by Caenorhabditis Genetics Center, funded by NIH National Center for Research Resources. This work was supported by funds to B.L.B. from National Institute of General Medical Sciences (GM44073). B.L.B. is a Howard Hughes Medical Institute Investigator.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.