

Abstract
A series of methyllycaconitine (1a, MLA) analogs was synthesized where the (S)-2-methylsuccinimidobenzoyl group in MLA was replaced with a (R)-2-methyl, 2,2-dimethyl-, 2,3-dimethyl, 2-phenyl- and 2-cyclohexylsuccinimidobenzoyl (1b–f) group. The analogs 1b–f were evaluated for their inhibition of [125I]iodo MLA binding at rat brain α7 nicotinic acetylcholine receptors (nAChR). In order to determine selectivity, MLA and the analogs 1b–f were evaluated for inhibition of binding to rat brain α, β nAChR using [3H]epibatidine. At the α7 nAChR, MLA showed a Ki value of 0.87 nM, analogs 1b–e possessed Ki values of 1.68–2.16 nM, and 1f showed a Ki value of 26.8 nM. Surprisingly, the analog 1e containing the large phenyl substituent (Ki = 1.68 nM) possessed the highest affinity. None of the compounds possessed appreciable affinity for α, β nAChRs. MLA antagonized nicotine-induced seizures with an AD50 = 2mg/kg. None of the MLA analogs were as potent as MLA in this assay. MLA and all of the MLA analogs, with the exception of 1b, antagonized nicotine’s antinociceptive effects in the tail-flick assay. Compound 1c (Ki = 1.78 nM at α7 nAChR) with an AD50 value of 1.8 mg/kg was 6.7 times more potent than MLA (AD50 = 12 mg/kg) in antagonizing nicotine’s antinociceptive effects but was 5-fold less potent than MLA in blocking nicotine-induced seizures. Since MLA has been reported to show neuroprotection against β-amyloid1–42, these new analogs which have high α7 nAChR affinity and good selectivity relative to α, β nAChRs will be useful biological tools for studying the effects of α7 nAChR antagonist and neuroprotection.
Keywords: methyllycaconitine, α7 nAChR, antagonist, and MLA analogs
1. Introduction
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels formed by the association of five subunits, leading to heteromeric and homomeric structures. The α7 receptor is the only homomeric nAChR widely distributed in the mammalian central nervous system. Binding sites in the brain that show high affinity for [125I] α-bungarotoxin ([125I] α-BTX) have been correlated with the α7 nAChR.1–4 Methyllycaconitine (1a, MLA), an alkaloid isolated from Delphinium brownii,5,6 is reported to be the most potent non-protein competitive α7 nAChR antagonist presently available.7 Recent studies have suggested that α7 nAChRs may play an important role in cognitive dysfunction, neurodegenerative diseases, vestibular function, epilepsy, and possibly smoking cessation.8 It is particularly interesting to note that MLA (1a) protects against the toxicity produced by the Alzehimer’s disease related peptide β-amyloid1–42(Aβ42).9 The availability of potent and selective α7 nAChR antagonist will enhance our ability to study these processes. Although MLA binds potently to the α7 subtype, recent evidence suggests that MLA can also interact with α4β2 nAChR and presynaptic α3/α6 nAChR.10 Clearly, there is a need for additional and selective α7 nAChR antagonists to further characterize the α7 nAChR pharmacophore and to further study its biological and pharmacological roles.
In this study, we report the syntheses and inhibition of [125I]Iodo-MLA and [3H]epibatidine binding at the α7 and α, β nAChRs for MLA (1a) and the MLA analogs 1b–f. Unfortunately, there is a lack of selective behavioral measure for α7-mediated response. Therefore, we evaluated the ability of MLA analogs to antagonize nicotine-induced seizures and antinociception in mice using both the tail-flick and hot-plate assays in mice because of MLA unique actions in these three assays. These assays were chosen because seizures are mediated at least in part through α7 receptors (sensitive to MLA), the hot-plate assay is α4β2-receptor mediated (insensitive to MLA), and the tail-flick assay is mediated through multiple nAChRs (sensitive to MLA).
1.1. Chemistry
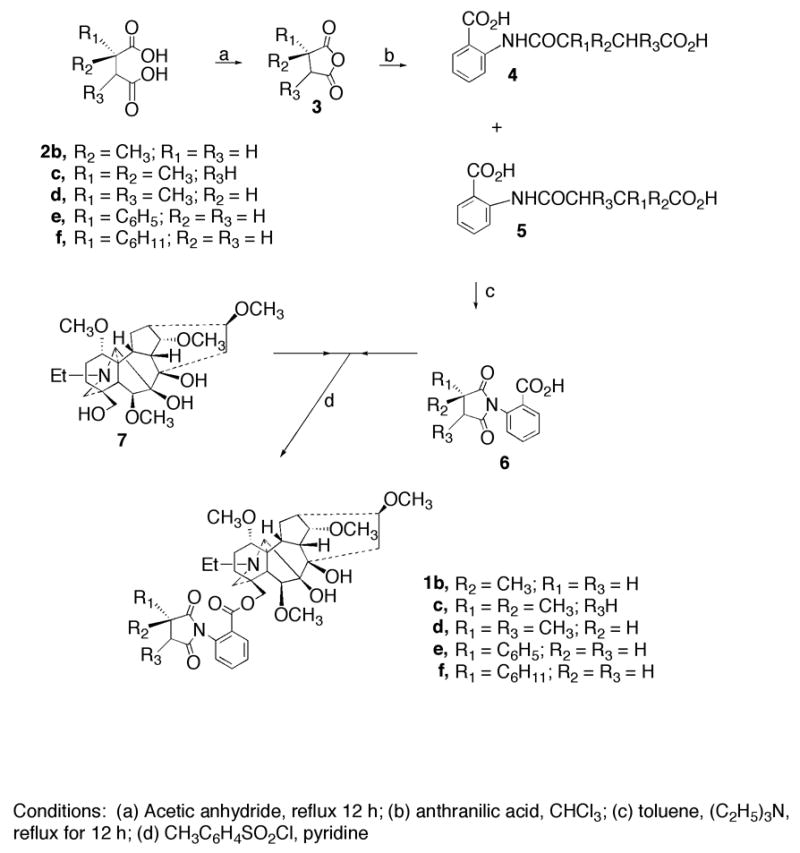
The MLA analogs 1b–f were synthesized as outlined in Scheme 1. The succinic acids 2b–f were converted to the corresponding succinic anhydrides 3 by refluxing the appropriate acid with acetic anhydride for 12 h. Treatment of each anhydride with anthranilic acid in chloroform provided the desired substituted methyllycoctonic acids, which are most likely a mixture of 4b–f and 5b–f except for the symmetrical analog (4d or 5d).11 The mixture of acids was refluxed under a Dean Stark tube in toluene containing triethylamine for 12 h to yield the appropriate 2-substituent or 2,3-disubstituted succinimidobenzoic acids 6b–f. The acids 6b–f were coupled to the primary hydroxyl group of lycoctonine (7) in the presence of p-toluenesulfonyl chloride and pyridine to give the desired MLA analogs 1b–f.12
Scheme 1.
1.2. Biology
The Ki values for the inhibition of [125I]iodo-MLA and [3H]epibatidine binding for MLA (1a) and the MLA analogs 1b–f were determined using previously reported procedures and are listed in the Table 1.13,14 MLA (1a) and the MLA analogs 1b–f were evaluated for their ability to antagonize the effects of nicotine in the tail-flick, hot-plate, and seizure test using previously reported procedures and the AD50 values are listed in Table 2.15
Table 1.
Radioligand binding data for MLA (1a) and the MLA analogs 1b–fa
| compd | RTI-7527- | R1 | R2 | R3 | α7 nAChR [125I]iodo-MLA Ki, nM (-nH) | α, β nAChR [3H]epibatidine Ki, nM (-nH) |
|---|---|---|---|---|---|---|
| 1a | MLA | CH3 | H | H | 0.87 ± 0.13 (1.09 ± 0.15) | 739 ± 43 (0.68 ± 0.02) |
| 1b | 58 | H | CH3 | H | 2.12 ± 0.47 (1.2 ± 0.34) | 352 ± 47 (0.89 ± 0.08) |
| 1c | 60 | CH3 | CH3 | H | 1.78 ± 0.15 (0.92 ± 0.05) | 602 ± 127 (0.93 ± 0.09) |
| 1d | 62 | CH3 | H | CH3 | 2.46 ± 0.87 (0.98 ± 0.18) | 321 ± 33 (0.88 ± 0.08) |
| 1e | 59 | C6H5 | H | H | 1.67 ± 0.75 (1.44 ± 0.42) | 219 ± 23 (0.75 ± 0.04) |
| 1f | 61 | C6H11 | H | H | 26.8 ± 3.62 (0.88 ± 0.21) | 1103 ± 400 (1.21 ± 0.02) |
Data represent the mean ± SE from at least three independent experiments. -nH is the Hill coefficient determined from a four-parameter logistic fit to the data.
Table 2.
Pharmacological evaluation of MLA (1a) and MLA analogs 1b–f as nicotinic antagonists. Results were expressed as AD50 (mg/kg) ± Confidence Limits (CL) or % effect at the highest dose tested.a
| compd | Seizures (AD50 mg/kg) | Tail-Flick (AD50 mg/kg) | Hot-Plate (AD50 mg/kg) |
|---|---|---|---|
| 1a | 2 (0.5–4) | 12 (2–59) | 15% @ 20 |
| 1b | 40% @ 50 | 10% @ 30 | 0% @ 30 |
| 1c | 10 (9–11) | 1.8 (0.4–9) | 13% @ 20 |
| 1d | 0% @ 50 | 7.0 (3–15) | 25% @ 20 |
| 1e | 0% @ 50 | 18.3 (10–31) | 7% @ 20 |
| 1f | 33 (10–113) | 7.0 (2.5–18) | 0% @ 20 |
Dose response curves were determined using a minimum of 4 different doses of test compound and at least eight mice were used per dose group.
2. Results and Discussion
The α7 nAChR subtype is the second most prevalent in the brain and has been implicated as playing a key role in conditions such as nicotine addiction, schizophrenia, Alzheimer’s disease, and epilepsy. Despite recent progress in the synthesis of competitive agonists selective for the α7 nAChR subtype, very few antagonists are known that bind with high affinity and selectivity at this receptor. These include the peptide toxins α-bungarotoxin and the norditerpenoid alkaloid methyllycaconitine (MLA, 1a).
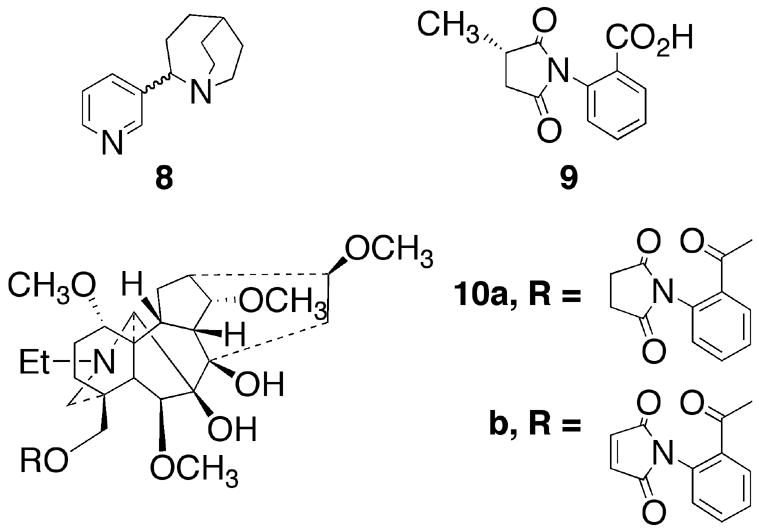
β-Amyloid1–42 binds to α7 nAChRs with high affinity.16 Functional studies have shown both α7 nAChR activation17,18 and inhibition19 with this peptide. Many studies suggest that α7 nAChRs agonist, such as TC-1698 (8),20 show neuroprotection against β-Amyloid1–42 effects on α7 nAChR function. It is well known that α7 nAChRs undergo extensive desensitization21–24 so long-term agonist treatment may not be desirable. Interestingly, Martin et al. reported that α7 nAChR antagonist MLA (1a) has neuroprotective actions against Aβ42-induced neurotoxicity and suggested that α7 nAChR antagonist might be useful pharmacotherapies in treating neurodegenerative disorders such as Alzheimer’s disease.9 Unfortunately, lack of highly selective agonists and antagonists have complicated the elucidation of the physiological function of a7 nAChRs.
MLA (1a) is an ester composed of the alcohol lycoctonine (7) and the acid, (S)-2-methylsuccinimidobenzoic acid. A number of truncated analogs of the parent MLA (1a) have been synthesized.25 None of the compounds showed appreciable activity.25,26 Reports from the literature have shown that neither the alcohol 7 nor the acid 9 possess appreciable affinity for the α7 nAChR.12,27 Thus, it appears that the (S)-2-methylsuccinimidobenzoyl group connected to the C18 oxygen of lycoctonine is essential for potent α7 nAChR binding. In addition, esters formed from the acid 9 and alcohols possessing parts of the lycoctonine structure as well as esters formed from lycoctonine with other acids were reported to possess low affinity for the α7 nAChR.27,28 In addition, the lycoctonine ester (10a), a nonditerpenoid alkaloid lacking the 2-(S)-methyl group on the succinimidobenzoyl group of MLA, and the dehydro analog (10b) are reported to show 5- and 10-fold loss in affinity relative to MLA at the α7 nAChR, which suggest that the angular-2-methyl-group on the succinimidobenzoyl group is essentially for potent α7 nAChR binding.12,29
In the present study, we found that replacement of (S)-2-methylsuccinimidobenzoyl group in MLA (1a, Ki = 0.87 nM), with the enantiomeric (R)-2-methylsuccinimidobenzoyl group to give 1b, Ki = 2.12 nM, resulted in a 2.4-fold loss in affinity for the α7 nAChR. Compounds 1c, Ki = 1.78 nM, and 1d, Ki = 2.62 nM, which possess an additional methyl group at the 2- or 3-position, showed 2.1- and 3-fold loss in affinity, respectively. Surprisingly, replacement of the 2-methyl group on the 2-methylsuccinimido group of MLA with a phenyl ring to give 1e resulted in a Ki value of 1.68 nM. Thus, the MLA pharmacophore will accommodate large flat aromatic groups in this part of the structure. In contrast, replacement of the 2-methyl group with a cyclohexyl group to give 1f resulted in a Ki value of 26.8 nM and thus a 31-fold loss of affinity. This alteration suggests that even though a phenyl ring is allowed in the 2-position of the 2-methylsuccinimido group of MLA, larger more bulky groups like cyclohexyl are not allowed. The weak ability of MLA and all the analogs 1b–f to inhibit [3H]epibatidine binding indicate that these compounds have low affinity for α, β nAChRs.
MLA analogs 1b–1e showed equipotent binding affinities toward the α7 nAChR. However, their in vivo profile in the seizures and tail-flick assays was different from each other. MLA analogs 1d–f were weak antagonists in the tail-flick assay (AD50 = 7–18.3 mg/kg). Analog 1b, which differs from MLA in only the stereochemistry of the methyl group [(R)-methyl] on the succinimidobenzoyl group, was inactive in this assay. The most potent compound, 1c, with an AD50 value of 1.8 mg/kg, was 6.7 times more potent than MLA, which has an AD50 of 12 mg/kg. The nAChR subtype or subtypes that mediate the tail-flick response are not known, but the lack of a high correlation between α7 receptor affinity and antagonistic potency in the tail-flick assay, particularly the discrepancy with enantioselectivity found in the 2-methylbenzoyl group, does not support a role for α7 receptors in mediating antinociception in this assay. It should be noted that earlier reports failed to observe a significant blockade of nicotine’s effects in the tail-flick test by MLA after s.c. injection in mice (5 mg/kg)30 and i.c.v. administration in rats (10 μg).31 However, the doses of MLA used in the present study are much higher than the reported ones. This difference in the effect observed could mean MLA lacks nAChR selectivity at higher doses or it is operating by some other mechanism. MLA and all the analogs had poor affinity for α β nAChRs as determined in the [3H]epibatidine binding assay and they were inactive at antagonizing the nicotinic effects in the hot-plate test. This is consistent with earlier observations that α4β2 receptors are required for this effect.32
MLA was also effective in blocking nicotine-induced seizures in an enantioselective manner. The seizures are mediated by α7 nAChRs and these results are consistent with an α7 mechanism. However, as with the tail-flick assay, there was a poor correlation between α7 receptor affinity and antagonistic potency. The results from the seizure study also support the view that nicotine-induced seizures and antinociception may be mediated by non-α4β2 and non-α7 receptor subtypes. Indeed, pharmacological and genetic approaches initially suggested the involvement of α7 nicotinic subtypes in nicotine-induced seizures. However, additional studies on different mouse inbred strains and transgenic mice have also implicated α4, α5, α3, and β4.33 The in vivo results show also that analogs 1b and 1e are much less potent than MLA in blocking nicotinic effects despite having good affinity for the α7 nAChR. This suggests that these two analogs have a different selectivity profile than MLA. The difference seen between in vitro binding affinity and in vivo potency in the various behavioral tests suggests that these MLA analogs have lower selectivity for the α7 receptor subtype. Ultimately, in vitro functional selectivity on various nAChRs subtypes in expressed cells would confirm this in vivo selectivity. While MLA and its derivatives antagonize both nicotine-induced antinociception and seizures, they apparently do so by acting at multiple sites or different mechanisms, since there is little correlation between antagonistic potencies in these two measures. The fact that MLA does not readily cross the blood brain barrier raises the question of whether pharmacokinetic differences may explain some of these findings. However, it seems unlikely that the pharmacokinetics would be so different among these analogs to explain their pharmacological dissimilarities.
3. Conclusions
Replacement of the (S)-2-methylsuccinimidobenzoyl group in MLA (1a) by an (R)-2-methylsuccinimidobenzoyl to give 1b caused only a small reduction in binding at the α7 nAChR, however, 1b showed no ability to antagonize seizures and antinociception induced by nicotine. Surprisingly, compound 1c which has methyl groups present in both the (2S) and (2R)-position of the succinimido group is 6.7 times more potent than MLA in antagonizing the antinociception induced by nicotine in the tail-flick test. However, 1c has essentially the same affinity for the α7 nAChR as 1b. These analogs have a pharmacological profile distinct from that of MLA despite relatively modest changes in structure. It would seem that they are acting at sites other than α7 receptors. Regardless of their mechanism of action, the (S)-2-methylsuccinimidobenzoyl group is a critical for receptor. Further development of MLA analogs may lead to even better probes for characterizing these receptor sites and to biological tools for studying the effects of α7 nAChR antagonist in several neurological disorders.
4. Experimental
Melting points were determined on a Thomas-Hoover capillary tube apparatus and are not corrected. Elemental analyses were obtained by Atlantic Microlabs, Inc. and are within ± 0.4% of the calculated values. All optical rotations were determined at the sodium D-line using a Rudolph Research Autopol III polarimeter (1-dm cell).1H NMR were determined on a Bruker Avance DPX-300 or Bruker AMX-500 spectrometer using tetramethylsilane as an internal standard. Silica gel 60 (230–400 mesh) was used for all column chromatography. All reactions were followed by thin-layer chromatrography using Whatman silica gel 60 TLC plates and were visualized by UV or by charring using 5% phosphomolybdic acid in ethanol. All solvents were reagent grade.
MLA was isolated from delphenium elatum (pacific giant) seeds (purchased from Flowers of Tomorrow, Inc., Parma, Idaho 83660) and hydrolyzed to lycoctonine (7).34,35 [3H]epibatidine (s.a. = 66.6 ci/mmol) was obtained from DuPont New England Nuclear (Boston, MA).
(R)-Methylsuccinamic Acids (4b and 5b)
A suspension of (R)-methylsuccinic acid (2b) (0.89 g, 6.75 mmol) in acetic anhydride (10 mL) was heated to reflux overnight. After removal of the excess of acetic anhydride by distillation, the residue was triturated and dried under reduced pressure to give the anhydride 3b that was dissolved in 10 mL of CHCl3 and added to a suspension of anthranilic acid (0.925 g, 6.75 mmol) in 10 mL of CHCl3. The reaction mixture was heated on a steam bath for 30 min, and the CHCl3 removed under reduced pressure. The product was purified on silica gel using hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) as eluent to give 1.56 g (92%) of succinamic acids (4b and 5b): mp 165–167 °C;1H NMR (CD3OD) δ 1.25–1.36 (2 d, 3H), 2.50–2.60 (m, 1H), 2.76–2.99 (m, 2H), 7.12–7.18 (t, 1H, aromatic), 7.53–7.56 (t, 1H, aromatic), 8.08–8.11 (d, 1H, aromatic), 8.54–8.57 (d, 1H, aromatic).
(R)-Methyllycactonic Acid (6b)
A solution of the above (R)-methylsuccinamic acids (4b and 5b) (1.5 g, 6 mmol) and triethylamine (3.05 g, 30.1 mmol) in 200 mL of toluene was heated to reflux overnight. The toluene was removed by simple distillation, and the residue was purified on silica gel using hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) as eluent to obtain 0.9 g (64%) of pure (R)-methyllycactonic acid (6b): mp 117–119 °C;1H NMR (CD3OD) δ 1.38–1.40 (d, 3H), 2.41–2.61 (m, 1H), 3.04–3.15 (m, 2H), 7.28–7.31 (d, 1H, aromatic), 7.56–7.58 (t, 1H, aromatic), 7.65–7.68 (t, 1H, aromatic), 8.11–8.14 (d, 1H, aromatic). Anal. (C12H11NO4•0.5H2O) C, H, N.
(R)-Methyllycaconitine (1b) Citrate
To a stirred solution of imido acid 6b (49.9 mg, 0.214 mmol) in 1 mL of dry pyridine was added 76.4 mg (0.428 mmol) of p-toluenesulfonyl chloride. The mixture was cooled to 0 °C, 100 mg (0.214 mmol) of lycoctonine was added, and the solution was stirred for additional 1 h at 0 °C and then placed in the refrigerator for 24 h. The yellow solution was dissolved in 10 mL of NH4OH (aq, pH 9) and extracted with 3 × 10 mL of CHCl3. Organic phases were combined and washed with 10 mL of NH4OH (aq, pH 9) and dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified on silica gel using cyclohexane:CHCl3:(Et)3N (6:4:1) as eluent to give 66 mg (45%) of pure (R)-methyllycaconitine (1b). An analytical sample was prepared as the citrate salt: mp 99 °C (dec.); [α]20D +48.3° (c, 0.64, EtOH) (free base). Anal. (C43H58N2O17•1.5H2O) C, H, N.
2,2-Dimethylsuccinamic Acids (4c and 5c)
2,2-Dimethylsuccinic anhydride (3c) (2.0 g, 15.6 mmol) was dissolved in 15 mL of CHCl3 and added to a suspension of anthranilic acid (2.14 g, 15.6 mmol) in 15 mL of CHCl3. The reaction mixture was heated on a steam bath for 30 min and CHCl3 removed under reduced pressure. The product was purified on silica gel using hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) as eluent to obtain 3.5 g (85%) of 2,2-dimethylsuccinamic acids (4c and 5c): mp 138–140 °C;1H NMR (CDCl3) δ 1.41 (s, 6H, 2CH3), 2.78 (s, 2H, -CH2), 7.07–7.20 (t, 1H, aromatic), 7.56–7.61 (t, 1H, aromatic), 8.05–8.08 (d, 1H, aromatic), 8.77–8.80 (d, 1H, aromatic). Anal. (C13H15NO5•0.5H2O) C, H, N.
2,2-Dimethyllycactonic Acid (6c)
A solution of the above 2,2-dimethylsuccinamic acids (4c and 5c) (3.5 g, 13.2 mmol) and triethylamine (6.6 g, 66 mmol) in 300 mL of toluene was heated to reflux overnight. The toluene was removed by simple distillation, and the residue was purified on silica gel using hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) as eluent to obtain 1.3 g (40%) of pure 2,2-dimethyllycactonic acid (6c): mp 134–136 °C;1H NMR (CDCl3) δ 1.39 (s, 6H, 2CH3), 2.70 (s, 2H, -CH2), 7.23–7.26 (d, 1H, aromatic), 7.47–7.52 (t, 1H, aromatic), 7.63–7.68 (t, 1H, aromatic), 8.11–8.14 (d, 1H, aromatic). Anal. (C13H13NO4•0.25H2O) C, H, N.
2,2-Dimethyllycaconitine (1c) Citrate
To a stirred solution of imido acid 6c (105.8 mg, 0.428 mmol) in 2 mL of dry pyridine was added 152.8 mg (0.856 mmol) of p-toluenesulfonyl chloride. The mixture was cooled to 0 °C, 100 mg (0.214 mmol) of lycactonine was added, and the solution was stirred for an additional 1 h at 0 °C and then placed in the refrigerator for 15 h. The deep-red solution was dissolved in 15 mL of NH4OH (aq, pH 9) and extracted with 3 × 10 mL of CHCl3. Organic phases were combined and washed with 10 mL of NH4OH (aq, pH 9) and dried over anhydrous Na2SO4. After the solvent was removed, the residue was purified on silica gel using cyclohexane:CHCl3:(Et)3N (6:4:1) as eluent to obtain 117.5 mg (79%) of the pure 2,2-dimethyllycaconitine (1c). An analytical sample was prepared as the citrate salt: mp 104 °C (dec.). Anal. (C44H60N2O17•H2O) C, H, N.
2,3-Dimethylsuccinamic Acids (4d and 5d)
A suspension of 2,3-dimethylsuccinic acid (2d) (2.0 g, 13.7 mmol) in acetic anhydride (15 mL) was heated to reflux overnight. After removal of the excess of acetic anhydride by distillation, the residue was triturated and dried under reduced pressure to give the anhydride 3d which was dissolved in 15 mL of CHCl3 and added to a suspension of anthranilic acid (1.88 g, 13.7 mmol) in 15 mL of CHCl3. The reaction mixture was heated on a steam bath for 30 min and the CHCl3 removed under reduced pressure. The product was purified on silica gel using hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) as eluent to obtain 3 g (83%) of 2,3-dimethylsuccinamic acids 4d and 5d:1H NMR (CDCl3) δ 130–138 (m, 6H), 2.75–2.85 (m, 2H), 7.07–7.12 (t, 1H, aromatic), 7.54–7.59 (t, 1H, aromatic), 8.01–8.04 (d, 1H, aromatic), 8.64–8.67 (d, 1H, aromatic).
2,3-Dimethyllycactonic Acid (6d)
A solution of the above 2,3-dimethylsuccinamic acids 4d and 5d (3 g, 11.3 mmol) and triethylamine (5.65 g, 56.5 mmol) in 300 mL of toluene was heated to reflux overnight. The toluene was removed by simple distillation, and the residue was purified on silica gel using hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) as eluent to obtain 0.8 g (29%) of pure 2,3-dimethyllycactonic acid (6d): mp 163–165 °C;1H NMR (CDCl3) δ 1.32–1.44 (2 d, 6H), 2.62–3.16 (2 m, 2H), 7.26–7.29 (d, 1H, aromatic), 7.51–7.56 (t, 1H, aromatic), 7.67–7.70 (t, 1H, aromatic), 8.17–8.20 (d, 1H, aromatic). Anal. (C13H13NO4.0.25H2O) C, H, N.
2,3-Dimethyllycaconitine (1d) Citrate
To a stirred solution of imido acid 6d (105.8 mg, 0.428 mmol) in 2 mL of dry pyridine was added 152.8 mg (0.856 mmol) of p-toluenesulfonyl chloride. The mixture was cooled to 0 °C and 100 mg (0.214 mmol) of lycactonine was added, and the solution was stirred for additional 1 h at 0 °C and was refrigerated for 18 h. The deep-red solution was taken up in 15 mL of NH4OH (aq, pH 9) and extracted with 3 × 10 mL of CHCl3. Organic phases were combined and washed with 10 mL of NH4OH (aq, pH 9) and dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified on silica gel using cyclohexane:CHCl3:(Et)3N (6:4:1) as eluent to obtain 74.6 mg (50%) of pure 2,3-dimethyllycaconitine (1d). An analytical sample was prepared as citrate salt: mp 84 °C (dec.). Anal. (C44H60N2O17•0.5H2O) C, H, N.
Phenylsuccinamic Acids (4e and 5e)
Phenylsuccinic anhydride (3e) (2.0 g, 11.36 mmol) was dissovled in 15 mL of CHCl3 and was added to a suspension of anthranilic acid (1.55 g, 11.36 mmol) in 15 mL of CHCl3. The reaction mixture was heated on a steam bath for 30 min, and then the CHCl3 solvent was evaporated to dryness under reduced pressure. The product was purified on silica gel, eluting with hexane:ethyl acetate:methanol :acetic acid (125:45:5:4) to obtain 3 g (84%) phenylsuccinamic acids (4e and 5e): mp 169–171 °C;1H NMR (DMSO) δ 2.75–2.82 (dd, 1H), 3.11–3.20 (dd, 1H), 4.02–4.10 (dd, 1H), 7.12–7.17 (m, 1H, aromatic), 7.26–7.38 (m, 5H, -C6H5), 7.55–7.60 (m, 1H, aromatic), 7.93–7.99 (m, 1H, aromatic), 8.44–8.50 (m, 1H, aromatic).
Phenyllycactonic Acid (6e)
A solution of the above phenylsuccinamic acids (4e and 5e) (3 g, 9.58 mmol) and triethylamine (4.79 g, 47.9 mmol) in 250 mL of toluene was heated to reflux overnight. The toluene solvent was removed by simple distillation, and the residue was purified on silica gel, eluting with hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) to obtain 1.1 g (39%) of pure phenyllycactonic acid (6e): mp 80–82 °C;1H NMR (CDCl3) δ 2.98–3.11 (dd, 1H), 3.36–3.45 (dd, 1H), 4.21–4.26 (dd, 1H), 7.33–7.40 (m, 6H, aromatic), 7.56–7.61 (m, 1H, aromatic), 7.72–7.76 (m, 1H, aromatic), 8.22–8.25 (m, 1H, aromatic). Anal. (C17H13NO4•0.6H2O) C, H, N.
Phenyllycaconitine (1e) Citrate
To a stirred solution of imido acid 6e (126.3 mg, 0.428 mmol) in 2 mL of dry pyridine was added 152.8 mg (0.856 mmol) of p-toluenesulfonyl chloride. The mixture was cooled to 0 °C, 100 mg (0.214 mmol) of lycactonine was added, and the solution was stirred for additional 1 h at 0 °C and was then placed in the refrigerator for 18 h. The deep-red solution was taken up in 15 mL of NH4OH aqueous solution (pH ~9) and extracted with 3 × 10 mL of CHCl3. Organic phases were combined and washed with 10 mL of NH4OH aqueous solution (pH ~9) and dried over anhydrous Na2SO4. After the solvent was removed, the residue was purified on silica gel, eluted with cyclohexane:CHCl3:(Et)3N (6:4:1) to obtain 100 mg (63%) of the pure phenyllycaconitine (1e). An analytical sample was prepared as the citrate salt: mp 105 °C (dec.). Anal. (C48H60N2O17•0.5H2O) C, H, N.
Cyclohexylsuccinamic Acids (4f and 5f)
A suspension of cyclohexylsuccinic acid (2f) (2.0 g, 10 mmol) in acetic anhydride (15 mL) was heated to reflux overnight. The excess of acetic anhydride was removed by sample distillation, the residue (3f) was triturated and dried under reduced pressure. The anhydride (3f) (1.78 g, 9.78 mmol) was dissolved in 15 mL of CHCl3 and was added to a suspension of anthranilic acid (1.34 g, 9.78 mmol) in 15 mL of CHCl3. The reaction mixture was heated on a steam bath for 30 min, and the CHCl3 solvent was evaporated to dryness under reduced pressure. The product was purified on silica gel, eluting with hexane:ethyl acetate:methanol :acetic acid (125:45:5:4) to obtain a 2.9 g (92%) of cyclohexylsuccinamic acids (4f and 5f): mp 171–173 °C,1H NMR (CDCl3) δ 1.05–2.04 (m, 11H, -C6H11), 2.61–2.68 (m, 1H), 2.76–2.88 (m, 2H), 7.08–7.13 (t, 1H, aromatic), 7.56–7.61 (t, 1H, aromatic), 8.06–8.08 (d, 1H, aromatic), 8.70–8.73 (d, 1H, aromatic).
Cyclohexyllycactonic Acid (6f)
A solution of the above cyclohexylsuccinamic acids (4f and 5f) (2.77 g, 8.68 mmol) and triethylamine (4.34 g, 43.4 mmol) in 300 mL of toluene was heated to reflux overnight. The toluene solvent was removed by simple distillation. The residue was purified on silica gel, eluting with hexane:ethyl acetate:methanol:acetic acid (125:45:5:4) to obtain 1.1 g (42%) of pure cyclohexyllycactonic acid (6f): mp 68–70 °C;1H NMR (CDCl3) δ 1.12–1.35 (m, 5H), 1.69–1.81 (m, 5H), 2.06–2.11 (m, 1H), 2.71–3.10 (m, 3H), 7.22–7.26 (d, 1H, aromatic), 7.51–7.56 (t, 1H, aromatic), 7.67–7.72 (t, 1H, aromatic), 8.16–8.19 (d, 1H, aromatic). Anal. (C17H19NO4•0.33H2O) C, H, N.
Cyclohexyllycaconitine (1f) Citrate
To a stirred solution of the imido acid (6f) (128.98 mg, 0.428 mmol) in 2 mL of dry pyridine was added 152.8 mg (0.856 mmol) of p-toluenesulfonyl chloride. The mixture was cooled to 0 °C, and 100 mg (0.214 mmol) of lycactonine was added. The solution was stirred for additional 1 h at 0 °C and was then placed in the refrigerator for 18 h. The deep-red solution was taken up in 15 mL of NH4OH aqueous solution (pH ~9) and extracted with 3 × 10 mL of CHCl3. Organic phases were combined and washed with 10 mL of NH4OH aqueous solution (pH ~9) and dried over anhydrous Na2SO4. After the solvent was removed, the residue was purified on silica gel, eluting with cyclohexane:CHCl3:(Et)3N (6:4:1) to obtain 105.7 mg (66%) of the pure cyclohexyllycaconitine (1f). An analytical sample was prepared as citrate salt: mp 72 °C (dec.). Anal. (C48H66N2O17•1.5H2O) C, H, N.
[125I]Iodo-MLA Binding Assay
Frozen male rat cerebral cortex (includes hippocampus; Pel-Freez Biologicals, Rogers, AK) was homogenized (polytron) in 39 volumes of ice-cold 50 mM Tris buffer (assay buffer; pH 7.4 @ 4 °C) containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2. The homogenate was centrifuged at 35,000 × g for 10 min at 4 °C and the supernatant discarded. The pellet was washed twice more with the original volume of buffer. After the last centrifugation, the pellet was resuspended in 1/10 the original volume of buffer and stored at −80 °C until needed. The competition binding experiments were carried out in 1.4 mL polypropylene tubes (Matrix Technologies Corporation, Hudson, NH) in a 96-well array. In a final volume of 0.5 mL triplicate samples contained approximately 3 mg of tissue (wet weight; added last), 40–50 pM [125I]iodo-MLA and 10–12 different concentrations of the test compounds. The dilutions of the test compounds were made using assay buffer containing 10% DMSO (1% final concentration). The total binding and nonspecific binding (300 uM nicotine) samples also contained a final concentration of 1% DMSO. The test compounds were pipetted using a MultiProbe IIEX (Packard Instruments, Meriden, CT) robotic liquid handling system. The samples were incubated on ice for 2 h. A MultiMate harvester (Packard) was used to separate bound radioligand from free by rapid vacuum filtration onto GF/B filters presoaked for at least 30 min in assay buffer containing 0.15% bovine serum albumin. The filters were washed with approximately 4 mL of ice cold 10 mM Tris buffer (pH 7.4 @ 4 °C) without salts and dried prior to the addition of 35 μL of Microscint 20 scintillant (Packard). The amount of radioligand remaining on each filter was determined using a Packard TopCount microplate scintillation counter (70% efficiency).
[3H]Epibatidine Binding Assay
Adult male rat cerebral cortices (Pelfreeze Biological, Rogers, AK) were homogenized in 39 volumes of ice-cold 50 mM Tris buffer (pH 7.4 at 4 °C) containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2 and centrifuged at 37,000 g for 10 min at 4 °C. The supernatant was discarded, the pellet resuspended in the original volume of buffer, and the wash procedure repeated twice more. After the last centrifugation, the pellet was resuspended in 1/10 its original homogenization volume and stored at −80 °C until needed. In a final volume of 0.5 mL, each assay tube contained 3 mg wet weight of male rat cerebral cortex homogenate (added last), 0.5 nM [3H]epibatidine (NEN Life Science Products, Wilmington, DE) and one of 10–12 different concentrations of test compound dissolved in buffer (pH 7.4 at room temperature) containing 10% DMSO resulting in a final DMSO concentration of 1%. Total and nonspecific bindings were determined in the presence of vehicle and 300 uM (–)-nicotine, respectively. After 4-h incubation at room temperature, the samples were vacuum-filtered over GF/B filter papers presoaked in 0.03% polyethylenimine using a Brandel 48-well harvester and washed with 6 mL of ice-cold buffer. The amount of radioactivity trapped on the filter was determined by standard liquid scintillation techniques in a TriCarb 2200 scintillation counter (Packard Instruments, Meriden, CT) at approximately 50% efficiency.
Data Handling
The specific binding data were fit using the nonlinear regression equations in Prism (GraphPad Prism v. 3.0; GraphPad Software, San Diego, CA). The Cheng-Prusoff equation36 [Ki = IC50/(1+([L]/Kd)] was used to calculate the Ki from the IC50. The data are reported as the mean ± S.E.M. from at least three independent experiments. Kd values for [125I]iodo-MLA and [3H]epibatidine were 1.8 and 0.02 nM, respectively. These Kd values were determined under conditions identical to their respective assays.
Tail-Flick Test
Antinociception was assessed by the tail-flick method of D’Amour and Smith.37 A control response (2–4 sec) was determined for each mouse before treatment, and a test latency was determined after drug administration. In order to minimize tissue damage, a maximum latency of 10 sec was imposed. Antinociceptive response was calculated as percent maximum possible effect (% MPE), where %MPE = [(test-control)/(10-control)] × 100. Groups of 8 to 12 animals were used for each dose and for each treatment. The mice were tested 5 min after i.t. injections of epibatidine analogs for the dose-response evaluation. Eight to 12 mice were treated per dose, and a minimum of 4 doses were performed for dose-response curve determination. Antagonism studies were carried out by pretreating the mice s.c. with either saline MLA (1a) or MLA analogs 1b–f at different times before nicotine. The animals were tested 5 min after administration of nicotine.
Hot-Plate Test
Mice were placed into a 10 cm wide glass cylinder on a hot plate (Thermojust Apparatus) maintained at 55.0 C. Two control latencies at least 10 min apart were determined for each mouse. The normal latency (reaction time) was 8 to 12 sec. Antinociceptive response was calculated as percent maximum possible effect (% MPE), where %MPE = [(test-control)/40-control) × 100]. The reaction time was scored when the animal jumped or licked its paws. Eight mice per dose were injected s.c. with epibatidine analogs and tested 5 min thereafter in order to establish a dose-response curve.
Nicotine-Induced Seizures
Male ICR (20–25 g) obtained from Harlan Laboratories (Indianapolis, IN) were used throughout the study. Following injection of nicotine, each animal was placed in a 30 × 30 cm2 Plexiglas cage and observed for 3 min. Whether a clonic seizure occurred within a 3-min time period was noted for each animal after s.c. administration of nicotine. This time was chosen because seizures occur very quickly after nicotine administration. Antagonism studies were carried out by pretreating the mice s.c. with either saline or different MLA analogs 10 min before nicotine. The percentage of animals exhibiting a seizure was calculated and dose-response curves were constructed and AD50 value determined for each of the MLA analogs.
Figure 1.
Figure 2.
Figure 3.
Acknowledgments
This research was supported by the National Institute on Drug Abuse, Grant DA12001.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albuquerque EX, Pereira EFR, Alkondon M, Eisenberg HM, Maelicke A. Neuronal nicotinic receptors and synaptic transmission in the mammalian central nervous system. Handb Exp Pharmacol. 2000;144:37–358. [Google Scholar]
- 2.Clarke PBS. Neuronal Nicotinic Receptors: Pharmacology and Therapeutic Opportunities. Wiley-Liss, Inc.: New York; 1999. Functional anatomy of nicotinic acetylcholine receptors in mammalian brain; pp. 127–139. [Google Scholar]
- 3.Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem. 2005;48:4705–4745. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- 4.Schmitt JD. Exploring the nature of molecular recognition in nicotinic acetylcholine receptors. Curr Med Chem. 2000;7:749–800. doi: 10.2174/0929867003374660. [DOI] [PubMed] [Google Scholar]
- 5.Aiyar VN, Benn MH, Hanna T, Jacyno J, Roth SH, Wilkens JL. The principal toxin of Delphinium Brownii, and its mode of action. Experientia. 1979;35:1367–1368. doi: 10.1007/BF01964013. [DOI] [PubMed] [Google Scholar]
- 6.Coates PA, Biagbrough IS, Hardick DJ, Rowan MG, Wonnacott S, Potter BVL. Rapid and efficient isolation of the nicotinic receptor antagonist methyllycaconitine from Delphinium: Assignment of the methylsuccinimide absolute stereochemistry as S. Tetrahedron Lett. 1994;35:8701–8704. [Google Scholar]
- 7.Wonnacott S, Albuquerque EX, Bertrand D. Methyllycaconitine: A new probe that discriminates between nicotinic acetylcholine receptor subclasses. Methods in Neurosciences. 1993:263–275. [Google Scholar]
- 8.Lloyd GK, Williams M. Neuronal nicotinic acetylcholine receptors as novel drug targets. J Pharmacol Exp Ther. 2000;292:461–467. [PubMed] [Google Scholar]
- 9.Martin SE, de Fiebre NE, de Fiebre CM. The alpha7 nicotinic acetylcholine receptor-selective antagonist, methyllycaconitine, partially protects against beta-amyloid1-42 toxicity in primary neuron-enriched cultures. Brain Res. 2004;1022:254–256. doi: 10.1016/j.brainres.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 10.Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther. 2002;302:197–204. doi: 10.1124/jpet.302.1.197. [DOI] [PubMed] [Google Scholar]
- 11.Stephani R, Cesare V, Sadarangani I, Lengyel I. Synthesis of New Optically Active and Racemic Phenylsuccinamic Acids. Synthesis. 2002;1:47–52. [Google Scholar]
- 12.Jacyno JM, Harwood JS, Lin N-H, Campbell JE, Sullivan JP, Holladay MW. Lycaconitine revisited: Partial synthesis and neuronal nicotinic acetylcholine receptor afinities. J Nat Prod. 1996;59:707–709. doi: 10.1021/np960352c. [DOI] [PubMed] [Google Scholar]
- 13.Navarro HA, Zhong D, Abraham P, Xu H, Carroll FI. Synthesis and pharmacological characterization of [(125)I]iodomethyllycaconitine ([(125)I]iodo-MLA). A new ligand for the α7 nicotinic acetylcholine receptor. J Med Chem. 2000;43:142–145. doi: 10.1021/jm990544f. [DOI] [PubMed] [Google Scholar]
- 14.Carroll FI, Liang F, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 2-exo-2-(2′-substituted 5′-pyridinyl)-7-azabicyclo[2.2.1]heptanes. Epibatidine analogues. J Med Chem. 2001;44:2229–2237. doi: 10.1021/jm0100178. [DOI] [PubMed] [Google Scholar]
- 15.Damaj MI, Slemmer JE, Carroll FI, Martin BR. Pharmacological characterization of nicotine’s interaction with cocaine and cocaine analogs. J Pharmacol Exp Ther. 1999;289:1229–1236. [PubMed] [Google Scholar]
- 16.Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 17.Fu W, Jhamandas JH. Beta-amyloid peptide activates non-alpha7 nicotinic acetylcholine receptors in rat basal forebrain neurons. J Neurophysiol. 2003;90:3130–3136. doi: 10.1152/jn.00616.2003. [DOI] [PubMed] [Google Scholar]
- 18.Dineley KT, Bell KA, Bui D, Sweatt JD. beta -Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem. 2002;277:25056–25061. doi: 10.1074/jbc.M200066200. [DOI] [PubMed] [Google Scholar]
- 19.Wu J, Kuo YP, George AA, Xu L, Hu J, Lukas RJ. beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem. 2004;279:37842–37851. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- 20.Breining SR, Mazurov AA, Miller CH. Neuronal nicotinic acetylcholine receptor modulators: Recent advances and therapeutic potential. In: Doherty AM, editor. Annual Reports in Medicinal Chemistry. Elsevier Academic Press; United Kingdom: 2005. pp. 3–16. [Google Scholar]
- 21.Puchacz E, Buisson B, Bertrand D, Lukas RJ. Functional expression of nicotinic acetylcholine receptors containing rat alpha 7 subunits in human SH-SY5Y neuroblastoma cells. FEBS Lett. 1994;354:155–159. doi: 10.1016/0014-5793(94)01108-7. [DOI] [PubMed] [Google Scholar]
- 22.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- 23.Seguela P, Wadiche J, Dineleymiller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain-alpha7 - a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Courturier S, Bertrand D, Matter JM, Hernandez M-C, Bertrans S, Miller N, Valera S, Barkas T, Ballivet M. A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homooligometric ion channel blocked by α-BTX. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 25.Goodall KJ, Barker D, Brimble MA. A review of advances in the synthesis of analogues of the Delphinium Alkaloid Methyllycaconitine. Synlett. 2005;12:1809–1827. [Google Scholar]
- 26.Daly JW. Nicotinic agonists, antagonists, and modulators from natural sources. Cell Mol Neurobiol. 2005;25:513–552. doi: 10.1007/s10571-005-3968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardick DJ, Blagbrough IS, Cooper G, Potter BVL, Critchley T, Wonnacott S. Nudicauline and elatine as potent norditerpenoid ligands at rat neuronal α-bungarotoxin binding sites: Importance of the 2-(methylsuccinimido)benzoyl moiety for neuronal nicotinic acetylcholine receptor binding. J Med Chem. 1996;39:4860–4866. doi: 10.1021/jm9604991. [DOI] [PubMed] [Google Scholar]
- 28.Davies ARL, Hardick DJ, Blagbrough IS, Potter BVL, Wolstenholme AJ, Wonnacott S. Characterisation of the binding of [3H]methyllycaconitine: A new radioligand for labelling α7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology. 1999;38:679–690. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- 29.Jennings KR, Starratt AN, Penaranda P, Loughton BG. Pharmacological studies of Delphinium alkaloids at the nicotinic acetylcholine receptor. R Soc Chem. 1999;232:163–174. [Google Scholar]
- 30.Damaj MI, Glassco W, Dukat M, Martin BR. Pharmacological characterization of nicotine-induced seizures in mice. J Pharmacol Exp Ther. 1999;291:1284–1291. [PubMed] [Google Scholar]
- 31.Rao TS, Correa LD, Reid RT, Lloyd GK. Evaluation of anti-nociceptive effects of neuronal nicotinic acetylcholine receptor (NAChR) ligands in the rat tail-flick assay. Neuropharmacology. 1996;35:393–405. doi: 10.1016/0028-3908(96)00013-5. [DOI] [PubMed] [Google Scholar]
- 32.Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d’Exaerde A, Huchet M, Damaj MI, Changeux JP. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- 33.Stitzel JA, Jimenez M, Marks MJ, Tritto T, Collins AC. Potential role of the alpha4 and alpha6 nicotinic receptor subunits in regulating nicotine-induced seizures. J Pharmacol Exp Ther. 2000;293:67–74. [PubMed] [Google Scholar]
- 34.Pelletier SW, Ross SA, Kulanthaivel P. New alkaloids from Delphinium elatum L. Tetrahedron. 1989;45:1887–1892. [Google Scholar]
- 35.Pelletier SW, Sawhney RS, Desai HK, Mody NV. The diterpenoid alkaloids of Consolida ambigua. J Nat Prod. 1980;43:395–406. [Google Scholar]
- 36.Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 37.D’Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]