

Summary
Pleiotrophin (PTN, Ptn) is an 18 kD secretory cytokine that is expressed in many human cancers, including glioblastoma. In previous experiments, interruption of the constitutive PTN signaling in human U87MG glioblastoma cells that inappropriately express endogenous Ptn reversed their rapid growth in vitro and their malignant phenotype in vivo. To seek a mechanism for the effect of the dominant negative PTN, flow cytometry was used to compare the profiles of U87MG cells and four clones of U87MG cells that express the dominant negative PTN (U87MG/PTN 1–40 cells); here, we report that the dominant negative PTN in U87MG cells induces tetraploidy and aneuploidy and arrests the tetraploid and aneuploid cells in the G1 phase of the cell cycle. The data suggest that PTN signaling may have a critical role in chromosomal segregation and cell cycle progression; the data suggest induction of tetraploidy and aneuploidy in U87MG glioblastoma cells may be an important mechanism that contributes to the loss of the malignant phenotype of U87MG cells.
Keywords: Pleiotrophin, Glioblastoma, Dominant-negative, Aneuploid, Tetraploid
Introduction
Pleiotrophin (PTN, Ptn) [1–3], also known as heparin-binding growth-associated molecule (HB-GAM)[4], is an 18 kDa secreted cytokine that shares over 50% identity in amino acid sequence and high structural homology in two heparin binding domains with midkine (MK, Mk), the only other member of the PTN/MK developmental gene family [5, 6]. High level expression of the Ptn gene has been found in many aggressive human malignancies, including human breast and prostate cancers [7, 8], neuroblastomas [9], gliomas [10], melanomas [11], colon cancers [12], pancreatic carcinomas [13], and small cell lung cancers [14, 15] and, cell lines derived from these human malignant cancers have been found to inappropriately express the endogenous Ptn [7, 8]. Different approaches to inhibit Ptn expression or constitutive PTN signaling in different malignant cells with inappropriate expression of Ptn have effectively reversed the malignant phenotype [16–19], thus supporting directly the importance of PTN-signaling in the progression and in the pathogenesis of highly malignant cancers. However, the mechanisms through which PTN stimulates a more malignant phenotype in malignant cells that inappropriately express Ptn are only beginning to be unraveled.
Glioblastomas are highly aggressive and highly vascularized tumors [20]; U87MG cells are derived from a human glioblastoma and have been found to inappropriately express high levels of Ptn [21]. In previous experiments, introduction of a dominant-negative Ptn encoding PTN amino acids 1–40 (which establishes nonfunctional PTN heterodimers during processing) [16] was introduced into human U87MG glioblastoma cells (U87MG/PTN 1–40 cells). The dominant negative PTN effectively reduced cell growth and reversed the malignant phenotype of these highly malignant cells in vivo [21], indicating a requirement of PTN-signaling in the malignant phenotype of U87MG glioblastoma cells and raising the question of the mechanism through which the dominant negative PTN reduces growth and reverses the malignant phenotype of U87MG glioblastoma cells.
To seek the (a) mechanism through which the dominant negative PTN effectively reverses the malignant phenotype of U87MG cells, we compared the profiles of U87MG mock cells with U87MG/PTN 1–40 cells using flow cytometry; it is now demonstrated that expression of the dominant negative PTN in U87MG cells induces tetraploidy and aneuploidy and furthermore, that all the tetraploid and aneuploid cells are arrested in the G1 phase of the cell cycle.
Materials and methods
Plasimid construction
The vector pcDNA3.1/PTN1-40 was constructed by inserting the cDNA fragment encoding residues −32 to 40 of human PTN protein into XbaI and BamHI of pcDNA3.1/myc-his/hygro vector (Invitrogen).
Cell culture and transfection
Human glioblastoma U87MG cells were maintained in Minimum Essential Medium Egale (ATCC) supplemented with 10% inactivated fetal bovine serum. 4 μg of pcDNA3.1/PTN1-40 or 4 μg of the empty pcDNA3.1/hygro vector were transfected into U87MG cells in 60 mm dishes using the FuGENE 6 transfection reagent (Roche, IN) according to manufacturer’s instructions. Forty-eight hours later, the cells were diluted 1:20 into fresh dishes and selected with hygromycin (Sigma) at 200 μg/ml; media were changed every 3 days until foci appeared. Cell lines were expanded from 4 different clones as noted subsequently; cells were shown to express high levels of dominant negative PTN were named (PTN1-40, clone 2, 3, 15 and 16) [21].
Flow Cytometry
To analyze cell cycle profiles, cells were digested by 0.25% typsin containing 1mM EDTA, washed with phosphate-buffered saline (PBS), and fixed in 70% methanol at 20°C for 10 min. Cells were then pelleted and resuspended in PBS with RNase A (100 □g/ml) and incubated for 30 min at 37 °C. Cells were then stained with propidium iodide (20 μg/ml) for 30 min and analyzed by flow cytometry using linear amplification. Data were collected using a FACScan flow cytometer (Becton Dickinson). For each sample, 20,000 events were collected, and aggregated cells were gated out. Data were analyzed by an auto analysis function of ModFitLT3.0 software (Verity Software, Topsham, ME).
Results
In previous studies, introduction of a dominant negative PTN into U87MG glioblastoma cells was found to reverse their malignant phenotype in vitro and in vivo [21]; U87MG glioblastoma cells that express the dominant negative PTN (U87MG/PTN 1–40 cells) grew more slowly than U87MG/vector (control) cells in monolayer culture, they formed fewer colonies in soft agar, and they grew more slowly than U87MG/vector cells as U87MG/PTN 1–40 xenografts in flanks of nude mice. U87MG cells inappropriately express Ptn; thus, the reversal of the malignant phenotype of U87MG cells with the dominant negative PTN supports the critical importance of constitutive PTN signaling in determining the malignant phenotype of U87MG glioblastoma cells. In the present studies, the goal was to uncover a mechanism potentially able to explain the reversal of the malignant phenotype of U87MG glioblastoma cells by the dominant negative PTN was sought by comparing the profiles of 4 clonal U87MG/PTN 1–40 cell lines with U87MG/vector cells using flow cytometry.
The profile of the control U87MG/vector cells depicted in Figure 1 established that these cells were 100% diploid. The relatively high level of U87MG/vector cells in G1 (Figure 1) is consistent with levels of G1 in different malignant cells characterized by a high proliferation rate. The modest increase in cells in the S phase of the cell cycle also is consistent with a rapid doubling time, a speculation supported by the high rate of growth of U87MG cells that were observed in vitro and of U87MG glioblastoma xenografts in nude mice. In contrast, each of the U87MG/PTN 1–40 clonal cell lines examined exhibited a significant fraction of cells either tetraploid or, in one case, both tetraploid and aneuploid (Figure 1); it was found that 6.38 % of U87MG/PTN 1–40-clone-2, 28.95 % of U87MG/PTN 1–40-clone-3, 5.8 % of U87MG/PTN 1–40-clone-15, and 17.01 % of U87MG/PTN 1–40-clone-16 cells were tetraploid. Furthermore, 12.75 % of U87MG/PTN 1-40-clone-16 cells also were aneuploid (summarized in Table 1). The data thus demonstrate directly that the block in U87MG/PTN 1–40 cells effectively blocks normal chromosomal segregation, leading to tetraploidy and aneuploidy. Fuethermore, the profiles of the U87MG/PTN 1–40 glioblastoma cells demonstrated that 100% of the tetraploid and aneuploid cells in each of the four clonal U87MG/PTN 1–40 cell lines were arrested in the G1 phase of the cell cycle (Figure 1), suggesting that the tetraploid and aneuploid cells arrested in G1 are likely to be targeted for apoptosis [22].
Figure 1.
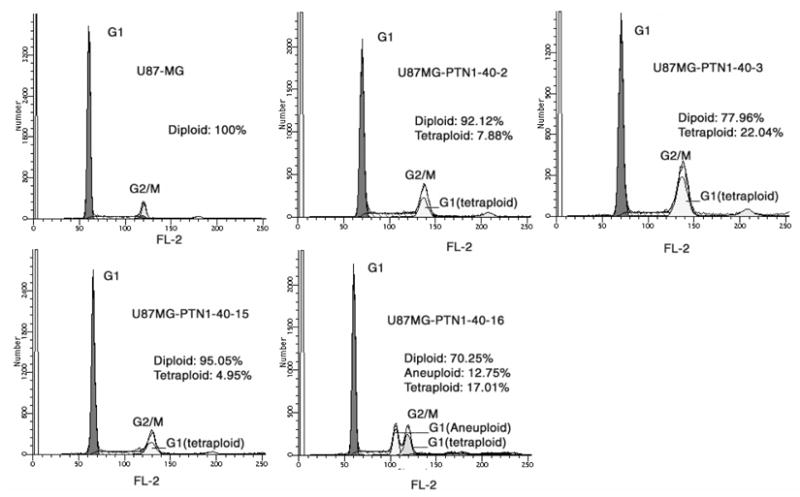
Cell cycle of U87MG glioblastoma cells expressing PTN 1–40. Representative histograms are shown. Data was obtained and analyzed as described in Materials and Methods.
Table 1.
Cell Cycle Profile of different U87MG Cell lines that express exogenous dominant negative PTN gene*.
| Cell line | Diploid (%) | Tetraploid (%) | Aneuploid (%) |
|---|---|---|---|
| U87MG/vector | 99.83 | 0.17 | 0 |
| U87MG/PTN 1-40-2 | 93.62 | 6.38 | 0 |
| U87MG/PTN 1-40-3 | 71.15 | 28.95 | 0 |
| U87MG/PTN 1-40-15 | 94.2 | 5.8 | 0 |
| U87MG/PTN 1-40-16 | 71.3 | 17.01 | 12.75 |
One representative results from three repeat experiments.
The data thus demonstrate that each of the four U87MG/PTN 1–40 cell lines contain significant numbers of cells either tetraploid or aneuploid and each of the tetraploid or aneuploid cells is arrested in the G1 phase of the cell cycle. Expression of the dominant negative PTN thus profoundly disrupts normal chromosomal separation to induce tetraploidy and aneuploidy and a G1 arrest of the tetraploid and aneuploid cells. The data suggest the possibility that induction of tetraploidy and aneuploidy and the G1 arrest of these cells may be a mechanism to slow rate of growth and reverses the malignant phenotype of U87MG glioblastoma cells and, to the best of our knowledge, these data are the first data to suggest that PTN-signaling has an important role in cell cycle progression.
Discussion
Tetraploidy can arise by exit of a cell from mitosis following a failure of spindle assembly, chromosome segregation, or cytokinesis [23]. Aneuploidy frequently follows an intermediate state of tetraploidy and is commonly found in malignant cells when tetraploid cells respond to activation of other genes through additional mutations in the cancer cells during tumor progression [24, 25]. These data in this manuscript demonstrate that expression of the dominant negative PTN in U87MG cells is associated with tetraploidy and aneuploidy and with a G1 arrest of the tetraploid and aneuploid U87MG cells; thus, interruption of constitutive PTN-signaling initiates a failure of chromosomes to segregate and a G1 arrest of the tetraploid and aneuploid cells potentially leading to apoptotic cell death.
The data suggest several conclusions; they suggest that PTN-signaling is required for a critical step in chromosomal segregation and perhaps in regulation of the cell cycle; they suggest the possibility that inappropriate Ptn expression through de-regulation of a critical step in cell cycle progression and/or perhaps separation of chromosomes during mitosis may contribute to the pathogenesis of glioblastomas; they suggest that interruption of constitutive PTN signaling in U87MG glioblastoma cells may contribute to the reversal of their highly malignant phenotype through induction of tetraploidy and aneuploidy.
The site in progression of the cell cycle at which PTN may be required is unknown; however, different studies suggest the possibility that PTN regulates the cytoskeleton reorganization required for the cell division or for spindle assembly, chromosome segregation, or cytokinesis. In different studies, PTN has been shown to increase tyrosine phosphorylation of β-catenin, to disrupt the association of N-cadherin and β-catenin, and to disrupt adherens junction complexes and homophilic cell-cell adhesion in PTN-stimulated cells [26, 27]. Pleiotrophin also has been found to activate protein kinase C (PKC) in PTN-stimulated cells; in PTN-stimulated cells, PTN stimulates the PTN-dependent PKC catalysed phosphorylation of serine residues 713 and 726 of β-adducin, which, when phosphorylated, reduces the affinity of β-adducin for actin and spectrin and disrupts β-adducin/actin/spectrin cytoskeletal complexes; the β-adducin/actin/spectrin complex is required for cytoskeleton stability and disruption of these complexes leads to distabilization of the cytoskeleton needed for cell division [28]. Perhaps the most likely site at which PTN signaling regulates cell cycle progression may be in centrioles of the mitotic spindle during mitosis; previous studies found that PTN stimulates serine phosphorylation of β-adducin at serines 713, 726 that is localized in centrioles of the mitotic spindle [28], suggesting that PTN may regulate the association of β-adducin with mitotic spindle protein complexes. The failure to disrupt these complexes in U87MG/PTN 1–40 cells through failure to activate PKC and thus to phosphorylate serines 713 and 726 of β-adducin in the U87MG/PTN 1–40 cells with a block in PTN signaling may lead to failure of chromosomal separation and tetraploidy and aneuploidy. Since the tetraploid and aneuploid cells fail to exit G1, it is postulated that the successive cell divisions will fail to progress beyond G1 and likely initiate apoptotic cell death, suggesting a potential mechanism to account for the loss of aggressive growth of U87Mg cells that express the dominant negative PTN. To the best of our knowledge, this study is the first to provide evidence that PTN signaling may be required for the orderly progression of cell cycle.
Acknowledgments
This is manuscript number 18434-MEM from the Scripps Research Institute.
This work was supported by National Institutes of Health grant RO1 CA84400 (to T.F.D.) and RO1 CA84328 (to Z.Y.W).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Milner PG, Shah D, Veile R, Donis-Keller H, Kumar BV. Cloning, nucleotide sequence, and chromosome localization of the human pleiotrophin gene. Biochemistry. 1992;31:12023–12028. doi: 10.1021/bi00163a009. [DOI] [PubMed] [Google Scholar]
- 2.Li YS, Milner PG, Chauhan AK, Watson MA, Hoffman RM, Kodner CM, Milbrandt J, Deuel TF. Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science. 1990;250:1690–1694. doi: 10.1126/science.2270483. [DOI] [PubMed] [Google Scholar]
- 3.Milner PG, Li YS, Hoffman RM, Kodner CM, Siegel NR, Deuel TF. A novel 17 kD heparin-binding growth factor (HBGF-8) in bovine uterus: purification and N-terminal amino acid sequence. Biochem Biophys Res Commun. 1989;165:1096–1103. doi: 10.1016/0006-291x(89)92715-0. [DOI] [PubMed] [Google Scholar]
- 4.Rauvala H. An 18-kd heparin-binding protein of developing brain that is distinct from fibroblast growth factors. Embo J. 1989;8:2933–2941. doi: 10.1002/j.1460-2075.1989.tb08443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohlen P, Kovesdi I. HBNF and MK, members of a novel gene family of heparin-binding proteins with potential roles in embryogenesis and brain function. Prog Growth Factor Res. 1991;3:143–157. doi: 10.1016/s0955-2235(05)80005-5. [DOI] [PubMed] [Google Scholar]
- 6.Deuel TF, Zhang N, Yeh HJ, Silos-Santiago I, Wang ZY. Pleiotrophin: a cytokine with diverse functions and a novel signaling pathway. Arch Biochem Biophys. 2002;397:162–171. doi: 10.1006/abbi.2001.2705. [DOI] [PubMed] [Google Scholar]
- 7.Vacherot F, Caruelle D, Chopin D, Gil-Diez S, Barritault D, Caruelle JP, Courty J. Involvement of heparin affin regulatory peptide in human prostate cancer. Prostate. 1999;38:126–136. doi: 10.1002/(sici)1097-0045(19990201)38:2<126::aid-pros6>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 8.Fang W, Hartmann N, Chow DT, Riegel AT, Wellstein A. Pleiotrophin stimulates fibroblasts and endothelial and epithelial cells and is expressed in human cancer. J Biol Chem. 1992;267:25889–25897. [PubMed] [Google Scholar]
- 9.Soulie P, Heroult M, Bernard-Pierrot I, Caruelle D, Oglobine J, Barritault D, Courty J. Correlation of elevated plasma levels of two structurally related growth factors, heparin affin regulatory peptide and midkine, in advanced solid tumor patients. Cancer Detect Prev. 2004;28:319–324. doi: 10.1016/j.cdp.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Mabuchi T, Satoh E, Maeda S, Nukui H, Naganuma H. Overexpression of heparin-binding growth-associated molecule in malignant glioma cells. Neurol Med Chir (Tokyo) 2004;44:637–643. 644–635. doi: 10.2176/nmc.44.637. [DOI] [PubMed] [Google Scholar]
- 11.Wu H, Barusevicius A, Babb J, Klein-Szanto A, Godwin A, Elenitsas R, Gelfand JM, Lessin S, Seykora JT. Pleiotrophin expression correlates with melanocytic tumor progression and metastatic potential. J Cutan Pathol. 2005;32:125–130. doi: 10.1111/j.0303-6987.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- 12.Souttou B, Juhl H, Hackenbruck J, Rockseisen M, Klomp HJ, Raulais D, Vigny M, Wellstein A. Relationship between serum concentrations of the growth factor pleiotrophin and pleiotrophin-positive tumors. J Natl Cancer Inst. 1998;90:1468–1473. doi: 10.1093/jnci/90.19.1468. [DOI] [PubMed] [Google Scholar]
- 13.Klomp HJ, Zernial O, Flachmann S, Wellstein A, Juhl H. Significance of the expression of the growth factor pleiotrophin in pancreatic cancer patients. Clin Cancer Res. 2002;8:823–827. [PubMed] [Google Scholar]
- 14.Jager R, List B, Knabbe C, Souttou B, Raulais D, Zeiler T, Wellstein A, Aigner A, Neubauer A, Zugmaier G. Serum levels of the angiogenic factor pleiotrophin in relation to disease stage in lung cancer patients. Br J Cancer. 2002;86:858–863. doi: 10.1038/sj.bjc.6600202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jager R, Noll K, Havemann K, Pfluger KH, Knabbe C, Rauvala H, Zugmaier G. Differential expression and biological activity of the heparin-binding growth-associated molecule (HB-GAM) in lung cancer cell lines. Int J Cancer. 1997;73:537–543. doi: 10.1002/(sici)1097-0215(19971114)73:4<537::aid-ijc14>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 16.Zhang N, Zhong R, Wang ZY, Deuel TF. Human breast cancer growth inhibited in vivo by a dominant negative pleiotrophin mutant. J Biol Chem. 1997;272:16733–16736. doi: 10.1074/jbc.272.27.16733. [DOI] [PubMed] [Google Scholar]
- 17.Czubayko F, Schulte AM, Berchem GJ, Wellstein A. Melanoma angiogenesis and metastasis modulated by ribozyme targeting of the secreted growth factor pleiotrophin. Proc Natl Acad Sci U S A. 1996;93:14753–14758. doi: 10.1073/pnas.93.25.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber D, Klomp HJ, Czubayko F, Wellstein A, Juhl H. Pleiotrophin can be rate-limiting for pancreatic cancer cell growth. Cancer Res. 2000;60:5284–5288. [PubMed] [Google Scholar]
- 19.Grzelinski M, Bader N, Czubayko F, Aigner A. Ribozyme-targeting reveals the rate-limiting role of pleiotrophin in glioblastoma. Int J Cancer. 2005 doi: 10.1002/ijc.21276. [DOI] [PubMed] [Google Scholar]
- 20.Jensen RL. Growth factor-mediated angiogenesis in the malignant progression of glial tumors: a review. Surg Neurol. 1998;49:189–195. 196. doi: 10.1016/s0090-3019(97)00218-8. [DOI] [PubMed] [Google Scholar]
- 21.Chang Y, Ezquerra L, Herradon G, Deuel TF. Dominant negative pleiotrophin gene and treatment with captopril reverse the malignant phenotype and tumor angiogenesis of U87MG glioblastoma cells in a similar manner. submitted. [Google Scholar]
- 22.Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, Pequignot MO, Casares N, Valent A, Mouhamad S, Schmitt E, Modjtahedi N, Vainchenker W, Zitvogel L, Lazar V, Garrido C, Kroemer G. Apoptosis regulation in tetraploid cancer cells. Embo J. 2006;25:2584–2595. doi: 10.1038/sj.emboj.7601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andreassen PR, Martineau SN, Margolis RL. Chemical induction of mitotic checkpoint override in mammalian cells results in aneuploidy following a transient tetraploid state. Mutat Res. 1996;372:181–194. doi: 10.1016/s0027-5107(96)00138-8. [DOI] [PubMed] [Google Scholar]
- 24.Shackney SE, Smith CA, Miller BW, Burholt DR, Murtha K, Giles HR, Ketterer DM, Pollice AA. Model for the genetic evolution of human solid tumors. Cancer Res. 1989;49:3344–3354. [PubMed] [Google Scholar]
- 25.Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS, Reid BJ. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci U S A. 1996;93:7081–7084. doi: 10.1073/pnas.93.14.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perez-Pinera P, Dimitrov T, Vega JA, 4, Deuel TF. Pleiotrophin disrupts calcium dependent homophilic cell-cell adhesion and initiates a transition to a mesenchymal-like phenotype in pleiotrophin-stimulated cells. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pariser H, Herradon G, Ezquerra L, Perez-Pinera P, Deuel TF. Pleiotrophin regulates serine phosphorylation and the cellular distribution of {beta}-adducin through activation of protein kinase C. Proc Natl Acad Sci U S A. 2005;102:12407–12412. doi: 10.1073/pnas.0505901102. [DOI] [PMC free article] [PubMed] [Google Scholar]