

Abstract
Single nucleotide polymorphisms (SNPs) are hypothesized to explain the genetic predisposition to ischemic heart disease (IHD) in the general population. Lack of evidence for a role of such variation is fostering pessimism about the utility of genetic information in the practice of medicine. In this study we determined the utility of exonic and 5′ SNPs in apolipoprotein E (APOE) and lipoprotein lipase (LPL) when considered singly and in combination for predicting incidence of IHD in 8,456 individuals from the general population during 24 years of follow-up. In men, LPL D9N improved prediction of IHD (P = 0.03) beyond smoking, diabetes and hypertension. The group of men heterozygous and homozygous for the rare D9N variant had a hazard ratio (HR) of 1.69 (95% confidence interval = 1.10–2.58) relative to the most common genotype. Pairwise combinations of D9N with −219G > T in APOE and N291S and S447X in LPL significantly improved the prediction of IHD (P = 0.05 in women, P = 0.04 in men, P = 0.03 in men, respectively) beyond smoking, diabetes and hypertension, and identified subgroups of individuals (n = 6–94) with highly significant HRs of 1.92–4.35. These results were validated in a case-control study (n = 8,806). In conclusion, we present evidence that combinations of SNPs in APOE and LPL identify subgroups of individuals at substantially increased risk of IHD beyond that associated with smoking, diabetes and hypertension.
Introduction
Substantial increases in risk of ischemic heart disease (IHD) are associated with rare Mendelian disorders that explain only a minute fraction of IHD risk in the general population. In contrast, common genotypes defined by single nucleotide polymorphisms (SNPs) are assumed to account for a significant fraction of IHD risk as part of a complex adaptive system of interactions between many genes and environmental factors, each with subtle effects (Sing et al. 2003). However, evidence for this assumption is lacking, and pessimism surrounding the genetic study of complex traits has been expressed (Chakravarti and Little 2003; Pembrey 2004). Epidemiologists have argued that the genetic component of IHD is negligible compared to traditional risk factors such as smoking, diabetes and hypertension (Khot et al. 2003). We suggest that this pessimism arises from the implementation of inadequate study designs (Collins 2004; Pembrey 2004), statistical analyses based on oversimplified genetic models of disease, and the use of surrogate measures of disease risk instead of well-characterized disease endpoints (Gluckman and Hanson 2004; Pembrey 2004). We address these issues in presenting evidence that combinations of SNPs in two IHD susceptibility genes mark rare classes of genotypes that estimate increased risk of IHD beyond that associated with smoking, diabetes and hypertension in a prospective cohort study carried out by the Copenhagen City Heart Study (CCHS). We validated our findings in a case-control study of an independently ascertained sample of IHD patients from the same background population as the prospective cohort.
Apolipoprotein E (APOE) and lipoprotein lipase (LPL) are two major components of human lipid metabolism which both mediate the clearance and modulation of triglyceride-rich lipoprotein particles (Brunzell and Deeb 2001; Mahley and Rall 2001). LPL hydrolyzes triglycerides contained in the core of both chylomicrons and very low-density lipoproteins (VLDL), thus causing these particles to be transformed into chylomicron remnants and intermediate-density lipoproteins (IDL) and/or low-density lipoproteins (LDL), respectively (Brunzell and Deeb 2001; Olivecrona and Olivecrona 1995). APOE serves as a receptor-binding ligand mediating the cellular uptake of chylomicrons, VLDLs and IDLs from plasma (Mahley and Rall 2001) via members of the LDL receptor family (Gliemann 1998). Recent evidence also suggests that apoE protects against atherosclerosis, because apoE particles secreted by macrophages have local beneficial effects at lesion sites, through macrophage cholesterol efflux (Cullen et al. 1998) and anti-inflammatory mechanisms (Stannard et al. 2001). During the last three decades, the three major APOE alleles, ε2, ε3 and ε4, have been shown to influence interindividual variation in levels of cholesterol and triglycerides in populations around the world (Davignon et al. 1988; Frikke-Schmidt et al. 2000; Reilly et al. 1991). Recently, three SNPs in the 5′ end of APOE (Frikke-Schmidt et al. 2004; Stengard et al. 2002) and genetic variation in LPL (Wittrup et al. 1997, 1999a, 2002) were shown to explain variation in lipid traits. In spite of a major influence on quantitative variation in lipid traits, and in spite of involvement in severe forms of dyslipidemia such as type III hyperlipoproteinemia and the chylomicroneamia syndrome (Brunzell and Deeb 2001; Mahley and Rall 2001), the impact on IHD risk of genotypes defined by individual SNPs in APOE and LPL has been subtle, and genotypes defined by combinations of SNPs in these genes have never been evaluated in a large prospective population based study.
We present a stepwise combinatorial strategy, in which we first ask whether each of 11 exonic and 5′ SNPs in APOE and LPL predicts risk of IHD beyond smoking, diabetes and hypertension. These traditional risk factors were selected for the present analyses because they have previously been shown to be the three most important traits for predicting IHD in both genders in the cohort defined by the CCHS (Schnohr et al. 2002). Secondly, we ask whether pairwise combinations of these SNPs improve the prediction of risk of IHD beyond that offered by the three traditional risk factors. Subsequently, each pair of SNPs that predicts beyond traditional risk factors was further evaluated to test if there is improvement in prediction beyond that offered by a model that included either of the single SNPs and the traditional risk factors. Finally, we ask whether any of the significant pairwise combinations combine with a third SNP to define three-site genotypes which improve the prediction of risk of IHD beyond that offered by the pairwise combination and traditional risk factors. This analytical strategy was applied to prospectively collected data on 8,456 Danish individuals over a period of 24 years, during which 916 incident IHD events occurred. The predictive models resulting from this strategy were then validated in a case-control study of 927 independently ascertained IHD patients and 7,879 controls.
Methods
Study design and samples
Longitudinal prospective study (model building sample)
The Copenhagen City Heart Study (CCHS) is a prospective study of the Danish general population within the age span of 20 to 80+ years. The CCHS was initiated in 1976–1978 with follow-up examinations in 1981–1983 and 1991–1994 (Appleyard et al. 1989). Participants were randomly ascertained in January 1976 from the Copenhagen Population Registry of approximately 90,000 inhabitants 20 years and older living within the ten wards surrounding Rigshospitalet, the National University Hospital of Copenhagen. At the third examination (1991–1994), 16,563 individuals were invited, 10,135 participated (response rate 61%) and 9,259 gave blood for DNA extraction. Complete clinical data and genotype information on 11 SNPs in APOE and LPL were available on 8,846 individuals. Informed consent was obtained from all participants. More than 99% were white and of Danish descent. Studies were approved by Danish ethical committees: No. 100.2039/91, Copenhagen and Frederiksberg committee, and No. KA 93125, Copenhagen County committee.
Information on diagnoses of IHD (World Health Organization International Classification of Diseases, 8th edition, codes 410–414; 10th edition, I20–I25, defining myocardial infarction, angina pectoris and acute and chronic atherosclerosis related heart disease) was gathered until December 31st, 1999 from the Danish National Hospital Discharge Register, from the Danish National Register of Causes of Death and from medical records of general practitioners and hospitals. During follow-up, 999 of the 8,846 participants experienced IHD. Median follow-up time was 18 years (range 0.04–24 years), representing 156,730 person-years. Eighty-three individuals experienced a stroke (World Health Organization International Classification of Diseases, 8th edition, codes 431–438; 10th edition, I61–I69) before their diagnosis of IHD and 307 participants without IHD were recorded as having stroke. These individuals were excluded, leaving 916 cases with IHD and 7,540 participants without IHD for all further analyses.
Risk factors for IHD, i.e. smoking, diabetes and hypertension, were dichotomized and defined as ever-smokers (current smoker at any of the three examinations), ever-diabetics (self-reported disease, use of insulin, use of oral hypoglycemic drugs and/or non-fasting plasma glucose ≥11.1 mmol/l at any of the three examinations), ever-hypertensive (systolic blood pressure ≥140 mmHg and/or diastolic blood pressure ≥90 mmHg or use of anti-hypertensive drugs at any of the three examinations). Hypertension was defined according to guidelines from the European Society of Hypertension and European Society of Cardiology (European Society of Hypertension—European Society of Cardiology guidelines for the management of arterial hypertension 2003). Diabetes could not be defined according to criteria from the American Diabetes Association because fasting plasma glucose levels were not measured on the participants of the CCHS. Instead we used a level of non-fasting plasma glucose ≥11 mmol/l (Alberti and Zimmet 1998) as an additional criteria to self-reported disease, use of insulin and/or use of oral hypoglycaemic drugs.
Case-control study (validation sample)
To validate whether the SNP genotype model defined in the analysis of the prospectively collected data predicted risk of IHD in an independent sample, patients with IHD from Copenhagen University Hospital were compared with controls from the general population without IHD. A sample of 948 patients with IHD was identified in 1991–1993 among patients referred for coronary angiography to Copenhagen University Hospital because of angina pectoris. These patients were identified among 992 consecutive patients from the greater Copenhagen area (including the island of Zealand). Experienced cardiologists determined whether cases had IHD based on characteristic symptoms of stable angina pectoris [Task force of the European Society of Cardiology (1997). Management of stable angina pectoris recommendations], plus at least one of the following: ≥70% stenosis of at least one coronary artery or ≥50% stenosis of the left main coronary artery on coronary angiography, a previous myocardial infarction or a positive exercise electrocardiography test. Genotypes were assayed for the 927 (244 women) individuals included in the present study. Controls (n = 7,879; 4,509 women) were individuals from the CCHS that did not experience IHD events before December 31, 1999.
Laboratory analyses
Eleven SNPs in APOE and LPL were genotyped by PCR and restriction enzyme digestion or allele-specific amplification methods as previously described (Frikke-Schmidt et al. 2000, 2004; Hixson and Vernier 1990; Nordestgaard et al. 1997; Wittrup et al. 1997, 1999a, 2002). All assays included positive, negative and non-template controls on each gel. If discrepancies in the positive and/or negative controls occurred or if there was contamination in the non-template control, the entire group of samples was re-genotyped. The locations of the SNPs are given in Fig. 1. These candidate genetic variations were selected on the basis of numerous a priori studies that have documented their regulatory and structural properties and their relative allele frequencies in the general population. We may have underestimated the risk prediction conferred by these two genes by not genotyping all of the genetic variations that have been revealed by resequencing (Nickerson et al. 1998, 2000).
Fig. 1.
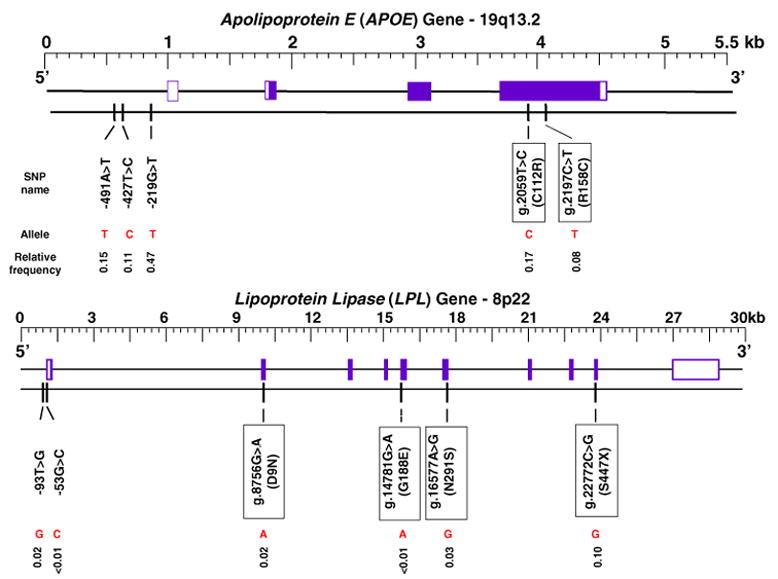
Relative frequency of least frequent allele for each SNP in APOE and LPL. Boxes indicate exonic sites. Exons in white indicate untranslated regions (UTRs)
Statistical analyses
Nominal probability values for test statistics ≤0.05 were considered statistically significant. The Cox proportional hazards model (Klein and Moeschberger 2003) was used to estimate the hazard ratios (HRs) for predictors of IHD with age as the time scale using left truncation (or delayed entry). We first constructed a reduced model that included smoking, diabetes and hypertension. The genotypes defined by a single SNP were then added to form a complete model. The χ2 = twice (ln likelihood of the complete − ln likelihood of the reduced model) with degrees of freedom = number of genotype classes −1 was used to test the hypothesis that genotype variation defined by the SNP does not contribute to the risk of IHD beyond that offered by smoking, diabetes and hypertension. For the evaluation of each pair of SNPs we added the genotypes defined by the pair to the reduced model to form a complete model. Those pairwise combinations that significantly increased risk prediction beyond the reduced model by a likelihood ratio test were further evaluated to determine if risk prediction was improved compared to a reduced model that included smoking, diabetes, hypertension and either of the SNPs that defined the pair. Finally, each SNP was added to each of the significant pairwise models to test whether genotypes defined by the resultant three-SNP-models improved prediction beyond the reduced model that included smoking, diabetes, hypertension and genotypes defined by the pairwise combination. Single, pairwise and three site analyses were also performed when the two exonic SNPs in APOE were considered as one six-genotype locus (ε22, ε32, ε42, ε33, ε43 and ε44).
Logistic regression was used to estimate odds ratios (ORs) in the case-control validation study, for single and pairwise SNP genotypes that improved the prediction of IHD in the prospective study. A likelihood ratio test statistic was used to compare a reduced model that included age in 10-year age groups and a complete model that included the genotypes defined by a single SNP and age. For the evaluation of each pair of SNPs we added the genotypes defined by the pair to the reduced model to form a complete model.
The r2 measure of linkage disequilibrium was estimated for each pair of SNPs (Pritchard and Przeworski 2001). The statistical significance of r2 was evaluated using the χ2 statistic. Homogeneity of relative allele frequencies across 20-year age groups was evaluated using the χ2 statistic.
Results
Traditional risk factors for IHD are summarized in Table 1 for incident IHD cases and for participants without IHD until time of censoring the CCHS cohort. Incident IHD cases were significantly older and smokers, diabetics and hypertensives were overrepresented in both female and male cases compared with participants without IHD.
Table 1.
Characteristics of participants in the Copenhagen City Heart Study
| Women
|
Men
|
|||
|---|---|---|---|---|
| Participants without IHD (n = 4,342) | Participants with IHD (n = 369) | Participants without IHD (n = 3,198) | Participants with IHD (n = 547) | |
| Age (years, ± SD) | 45 ± 11.5 | 55 ± 8.6* | 44 ± 11.6 | 53 ± 9.5* |
| Smoking (%) | 55 | 66* | 64 | 76* |
| Diabetes mellitus (%) | 2 | 8* | 5 | 11* |
| Hypertension (%) | 53 | 83* | 63 | 87* |
Participants without IHD were compared with participants with IHD by Pearson χ2 test or Student’s t test
Significant at 0.001 level of probability
Allele frequencies
The relative frequency of the least frequent SNP allele ranged from 0.001 to 0.47 (Fig. 1). The −53G > C and G188E LPL SNPs had minor allele frequencies <0.01, whereas the nine other SNPs had minor allele frequencies >0.01. C and T substitutions at the g.2059 and g.2197 sites in exon 4 of the APOE gene cause arginine–cysteine interchanges at amino acid residues 112 and 158 in the apoE protein, and give rise to three traditional APOE alleles, ε2, ε3 and ε4 which encode the three common apoE isoforms. The fourth possible combination of these two sites has not been observed. Hence, six traditional APOE genotypes are present in the general population: ε22, ε32, ε42, ε33, ε43, ε44 and ε32, ε33 and ε43 are the most frequent (Table 2). Except for the alleles of the −219G > T APOE SNP in females (P = 0.004), the relative frequencies of the two alleles for each of the LPL SNPs and the remaining regulatory APOE SNPs and the three traditional APOE alleles did not vary significantly among 20-year age groups in either gender. Except for the genotypes of the S447X SNP in the sample of 20- to 39-year-old individuals (P = 0.05), the relative frequencies of the three genotypes for each of the LPL SNPs and the regulatory APOE SNPs and the six traditional APOE genotypes did not deviate significantly from those predicted by the Hardy–Weinberg law in each 20-year age group (0.12 ≤ P ≤ 0.99).
Table 2.
Risk of IHD associated with single SNP genotype variation in APOE and LPL after fitting smoking, diabetes and hypertension. Each SNP is considered separately
| Women
|
Men
|
|||
|---|---|---|---|---|
| n (relative frequency) | Hazard ratioa (95% CI) | n (relative frequency) | Hazard ratioa (95% CI) | |
| Smoking | 2,642 (0.56) | 1.96* (1.58–2.44) | 2,470 (0.66) | 1.54* (1.27–1.88) |
| Diabetes mellitus | 136 (0.03) | 2.34* (1.61–3.41) | 223 (0.06) | 1.36** (1.04–1.78) |
| Hypertension | 2,382 (0.51) | 1.31** (1.02–1.68) | 2,245 (0.49) | 1.44* (1.17–1.77) |
| Genotype | ||||
| APOE −491A > T | ||||
| AA | 3,348 (0.71) | 1 | 2,710 (0.72) | 1 |
| AT | 1,240 (0.26) | 0.92 (0.73–1.16) | 953 (0.25) | 0.93 (0.77–1.14) |
| TT | 123 (0.03) | 0.78 (0.37–1.66) | 82 (0.03) | 1.13 (0.64–2.01) |
| APOE −427T > C | ||||
| TT | 3,737 (0.79) | 1 | 2,983 (0.80) | 1 |
| TC | 908 (0.19) | 0.85 (0.64–1.12) | 722 (0.19) | 1.09 (0.88–1.34) |
| CC | 66 (0.02) | 1.36 (0.61–3.05) | 40 (0.01) | 0.78 (0.25–2.44) |
| APOE −219G > T | ||||
| GG | 1,303 (0.28) | 1 | 1,059 (0.28) | 1 |
| GT | 2,336 (0.49) | 1.17 (0.92–1.50) | 1,872 (0.50) | 1.02 (0.84–1.24) |
| TT | 1,072 (0.23) | 1.20 (0.90–1.61) | 814 (0.22) | 1.01 (0.80–1.29) |
| APOE g.2059T > C (C112R) | ||||
| TT | 3,256 (0.69) | 1 | 2,625 (0.70) | 1 |
| TC | 1,318 (0.28) | 1.18 (0.94–1.47) | 1,032 (0.28) | 1.05 (0.87–1.26) |
| CC | 137 (0.03) | 1.59 (0.89–2.84) | 88 (0.02) | 1.39 (0.87–2.23) |
| APOE g.2197C > T (R158C) | ||||
| CC | 3,968 (0.84) | 1 | 3,162 (0.84) | 1 |
| CT | 716 (0.15) | 0.77 (0.56–1.04) | 565 (0.15) | 0.82 (0.64–1.06) |
| TT | 27 (<0.01) | 1.58 (0.59–4.24) | 18 (<0.01) | 1.84 (0.69–4.94) |
| APOE ε2, ε3, ε4 (C112R +R158C) | ||||
| ε22 | 27 (<0.01) | 1.69 (0.63–4.54) | 18 (<0.01) | 1.88 (0.70–5.03) |
| ε32 | 601 (0.13) | 0.83 (0.59–1.16) | 465 (0.12) | 0.81 (0.62–1.08) |
| ε42 | 115 (0.02) | 0.78 (0.37–1.65) | 100 (0.03) | 0.95 (0.55–1.66) |
| ε33 | 2,628 (0.55) | 1 | 2,142 (0.57) | 1 |
| ε43 | 1,203 (0.26) | 1.18 (0.93–1.50) | 932 (0.25) | 1.02 (0.84–1.25) |
| ε44 | 137 (0.03) | 1.54 (0.86–2.77) | 88 (0.02) | 1.35 (0.84–2.17) |
| LPL −93T > G | ||||
| TT | 4,561 (0.97) | 1 | 3,637 (0.97) | 1 |
| TG + GG | 150 (0.03) | 1.51 (0.89–2.58) | 108 (0.03) | 1.67** (1.09–2.57) |
| LPL −53G > C | ||||
| GG | 4,699 (>0.99) | 1 | 3,737 (>0.99) | 1 |
| GC | 12 (<0.01) | 1.03 (0.15–7.37) | 8 (<0.01) | 3.09 (0.99–9.64) |
| LPL g.8756G > A (D9N) | ||||
| GG | 4,568 (0.97) | 1 | 3,641 (0.97) | 1 |
| GA + AA | 143 (0.03) | 1.68 (0.98–2.87) | 104 (0.03) | 1.69** (1.10–2.58) |
| LPL g.14781G > A (G188E) | ||||
| GG | 4,709 (>0.99) | No test | 3,741 (>0.99) | No test |
| GA | 2 (<0.01) | 4 (<0.01) | ||
| LPL g.16577A > G (N291S) | ||||
| AA | 4,488 (0.95) | 1 | 3,551 (0.95) | 1 |
| AG + GG | 223 (0.05) | 1.13 (0.71–1.79) | 194 (0.05) | 0.96 (0.67–1.37) |
| LPL g.22772C > G (S447X) | ||||
| CC | 3,819 (0.81) | 1 | 3,060 (0.82) | 1 |
| CG | 836 (0.18) | 1.08 (0.82–1.42) | 643 (0.17) | 1.11 (0.89–1.37) |
| GG | 56 (0.01) | 1.12 (0.46–2.72) | 42 (0.01) | 1.03 (0.43–2.50) |
All exonic sites are named according to human mutation nomenclature (den Dunnen and Antonarakis 2001). To correspond with well-established literature names of promoter variants in APOE and LPL, nucleotide numbering is counted from transcriptional start site. For the common exonic APOE polymorphism, literature names are also given (alleles ε2, ε3, ε4; genotypes ε22, ε32, ε42, ε33, ε43, ε44). Due to low numbers of homozygotes for the minor allele, heterozygotes and homozygotes were pooled for the −93T > G and D9N SNPs in men (homozygotes absent in women) and for both genders for the N291S SNP
CI confidence interval
Significance levels of probability at 0.001
Significance levels of probability at 0.05
Adjusted for smoking, diabetes and hypertension
Linkage disequilibrium
Estimates of r2 are presented in Fig. 2 (below the diagonal) with corresponding significance levels (above the diagonal). As expected for unlinked loci, none of the five APOE SNPs were in significant linkage disequilibrium (LD) with any of the six LPL SNPs. The estimates of LD between pairs of APOE SNPs were all ≤ 0.2 and highly significant in all but one case. The − 93T > G and D9N SNPs in LPL were in almost complete LD. The estimates of LD between the remaining pairs of LPL SNPs were all ≤0.2 and statistically significant in only three cases.
Fig. 2.

Pairwise linkage disequilibrium between 11 SNPs in APOE and LPL. r2 values below diagonal and χ2 test of significance above diagonal
Single SNP genotypes and risk of IHD
As expected, from previous studies of this population, smoking, diabetes and hypertension each made a significant contribution to IHD in both women and men (Table 2). For the analyses of single SNP genotypes presented in Table 2, heterozygotes and homozygotes for the rare allele were pooled when the relative frequency of homozygotes was <0.001. No SNP predicted statistically significant variation in risk of IHD in women after fitting smoking, diabetes and hypertension. In men, the LPL −93T > G and D9N improved the prediction of IHD (P = 0.03 and P = 0.03, respectively) beyond smoking, diabetes and hypertension. The risk of IHD was increased in men heterozygous and homozygous for the G allele at the −93T > G SNP, HR = 1.67 (CI = 1.09–2.57), and heterozygous and homozygous for the A allele at the D9N SNP, HR = 1.69 (CI = 1.10–2.58) compared to the most common genotype (−93T > G: TT; D9N: GG). Because the −93T > G and D9N are in almost complete LD, all further results are reported for D9N only.
Pairwise SNP genotypes and risk of IHD
Figure 3 summarizes the HRs and cumulative incidence plots for the two SNP genotypes defined by each of three pairs of SNPs which made a significant contribution in at least one gender to predicting IHD after fitting smoking, diabetes and hypertension. All three pairs involved the D9N SNP. The additional contribution of the D9N/−219G > T pair to predicting risk was significant at the 0.05 level of probability in women while the added contribution of the D9N/N291S and D9N/S447X pairs was significant in men at the 0.04 and 0.03 level of statistical significance, respectively.
Fig. 3.
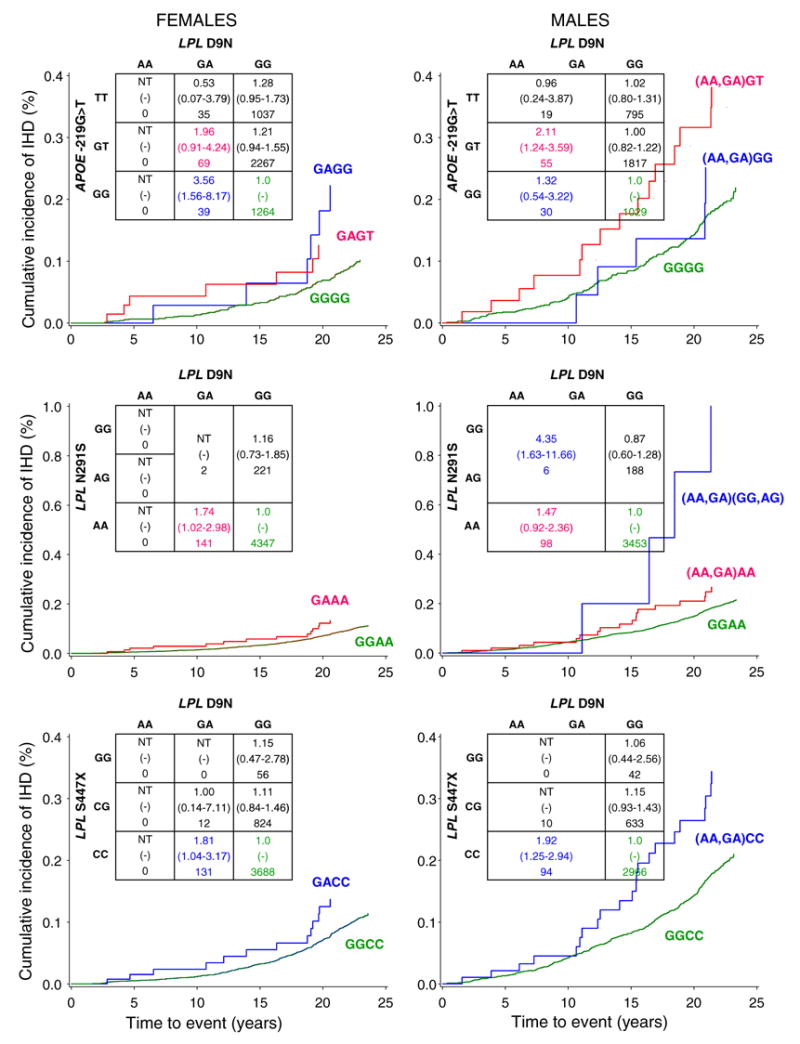
Risk of IHD associated with pairwise combinations of SNPs in APOE and LPL genes. The figure summarizes the analyses of the three pairwise combinations for which the two SNP genotypes predicted IHD in at least one gender after fitting smoking, diabetes and hypertension. All three pairs involved the D9N SNP. The additional contribution of the D9N/−219G > T pair was significant at the 0.05 level in women while the added contribution of the D9N/N291S and the D9N/S447X pairs was significant in men at the 0.04 and 0.03 level of statistical significance, respectively. The hazard ratio (95% confidence interval) and number of participants are given for each genotype. If hazard ratios for two SNP genotypes reached statistical significance in one gender, the cumulative incidence of ischemic heart disease as a function of time to event was shown for both genders (cumulative incidence plots). The color code reflects the corresponding hazard ratios and cumulative incidence plots. NT no test
Women heterozygous for LPL D9N and homozygous for APOE −219G > T (GAGG) were at greater risk of IHD, 3.56 (CI = 1.56–8.17, n = 39, P = 0.003), compared to the GGGG reference genotype (Fig. 3). Adding the −219G > T to a reduced model that included smoking, diabetes, hypertension and D9N improved the prediction of IHD in women at the 0.10 level of probability. Addition of D9N to a reduced model including the three traditional risk factors and −219G > T improved the prediction of IHD at the 0.03 level of probability. In men, the HR for IHD adjusted for the three traditional risk factors was 2.11 (CI = 1.24–3.59, n = 55, P = 0.006) for the (AA,GA)GT genotype combination (Fig. 3).
Men heterozygous or homozygous for D9N (AA,GA) and N291S (GG,AG) were at significantly greater risk of IHD, HR = 4.35 (CI = 1.63–11.66, n = 6, P = 0.003 ), compared to the GGAA reference genotype (Fig. 3). Adding the N291S to a reduced model that included smoking, diabetes, hypertension and D9N did not improve the prediction of IHD in men (P = 0.17), whereas the addition of D9N to a reduced model that included the three traditional risk factors and N291S significantly improved the prediction of IHD (P = 0.02). In women, the HR for the GAAA genotype was 1.74 (CI = 1.02–2.98, n = 141, P = 0.04).
Men heterozygous or homozygous for D9N and homozygous for S447X (AA,GA)CC were at significantly greater risk of IHD, 1.92 (CI = 1.25–2.94, n = 94, P = 0.003), compared to the GGCC reference genotype (Fig. 3). Adding the S447X to a reduced model consisting of smoking, diabetes, hypertension and D9N tended toward improvement of the prediction of IHD in men (P = 0.11), whereas the addition of D9N to a reduced model consisting of the three traditional risk factors and S447X significantly improved the prediction of IHD (P = 0.01). In women, the HR for the GACC genotype was 1.81 (CI = 1.04–3.17, n = 131, P = 0.04).
Three SNP genotypes and risk of IHD
In no case did adding a third SNP to define three SNP genotype combinations significantly improve the prediction of IHD compared to the reduced model that included smoking, diabetes, hypertension and the significant pairwise combination.
Validation of one and two-SNP genotype models
The significant contribution of the D9N SNP in LPL to the prediction of risk of IHD in men beyond smoking, diabetes and hypertension in the prospective CCHS (Table 2) was validated in the case-control study of 927 independently ascertained IHD patients and 7,879 controls. Men heterozygous or homozygous for D9N (AG + GG genotypes) were at significantly greater risk of IHD, (OR 1.93, CI = 1.23–3.02) than men with the most common GG genotype.
Two of the three models that included pairs of SNPs which predicted IHD in at least one gender in the prospective study (Fig. 3) predicted a significant increase in IHD risk in the case-control study. They were the D9N/N291S model for men (P = 0.04 in the prospective study and P = 0.06 in the case-control study) and the D9N/S447X model for men (P = 0.03 in the prospective study and P = 0.005 in the case-control study). The comparison of GAAA versus GGAA in the D9N/N291S model (Fig. 3, women, HR = 1.74, CI = 1.02–2.98), which was borderline significant in men [(AA,GA)AA versus GGAA, HR = 1.47, CI = 0.92–2.36], was validated in men in the case-control study (OR 1.91, CI = 1.21–3.02) but not in women (OR = 1.22, CI = 0.61–2.45). The statistically significant comparison of GACC or (AA,GA)CC versus GGCC in the D9N/ S447X model in both women (HR = 1.81, CI = 1.04–3.17) and men (HR = 1.92, CI = 1.25–2.94) in the prospective study was validated in men in the case-control study (OR 2.27, CI = 1.43–3.61) but not in women (OR = 1.12, CI = 0.54–2.34).
Discussion
In a prospective study of 8,456 individuals followed over 24 years we find evidence that combinations of SNPs in APOE and LPL predict increased risk of IHD beyond that associated with smoking, diabetes and hypertension. The SNPs involved do not predict, or have only small genotype effects, when considered separately. Of particular interest are the two SNPs in exon 4 of the APOE gene that define the most intensively studied genetic polymorphisms in humans (Gerdes et al. 1992; Song et al. 2004). In spite of this extensive interest and in spite of the substantial and consistent influence of both APOE and LPL on interindividual variation in quantitative lipid traits, the impact of these genes on determining risk of IHD has not been consistent among studies. Results have been dependent on context indexed by ethnicity, exposure to environmental agents and, as is the case in general for genetic effects, gender (Keavney et al. 2004; Song et al. 2004; Weiss et al. 2006; Wittrup et al. 1999b).
In the present study we were not able to confirm that genotypes defined by the common ε2, ε3, ε4 polymorphism of the APOE gene have a significant impact on risk of IHD. A recent meta-analysis of 48 studies (Song et al. 2004) of this polymorphism called to attention that many studies did not report a statistically significant result and in several of the largest studies the contribution to risk prediction did not even reach borderline statistical significance. Clearly, there is etiological heterogeneity, context dependency and subgroups of high-risk genotypes embedded within a meta-analysis that cannot be captured in the estimate of average risk for all populations. The impact of the regulatory variation in the APOE gene on prediction of risk of IHD in our study is consistent with the report by Stengard et al. (2006) which suggests the influence of both the structural variation and regulatory variation in the gene on variation in plasma measures of lipid metabolism.
Previously published case-control studies suggest that the relative frequencies of the APOE ε2, ε3 and ε4 alleles vary among age groups (Gerdes et al. 2000; Payami et al. 2005). In the present study, the relative allele frequencies differed across 20-year age groups only for the APOE −219G < T SNP in women. The observed heterogeneity may signify differential mortality, sampling variation or, for cases in case-control studies, ascertainment bias (Payami et al. 2005). However, it is important to note that such heterogeneity of allele frequencies across age is dealt with in the Cox regression analyses. The left truncation technique used in our study is the optimal choice when subjects enter a study at random with regard to their ages and are followed from this “delayed entry time” until the event occurs, or until the subjects are right-censored (Klein and Moeschberger 2003). Individuals with events are always compared to individuals without events within risk sets of similar ages on a continuous age scale. This ensures that observed effects are not influenced by relative allele frequency differences among age groups.
Because the relative frequencies of single SNP genotypes are correlated, and some two SNP genotype classes are missing, we cannot draw strong inference about the interaction of SNP effects from our study. A statistical test of non-additivity of SNP effects is confounded with the test of marginal SNP effects. We can infer substantial statistically significant increases in risk in subgroups of individuals identified by genotypes defined by combining two SNPs that each has modest genotypic effects when considered separately. This finding supports the argument that biological epistasis (Brodie 2000; de Visser et al. 2003), exemplified by non-additivity of SNP effects in the present study, may be the primary component of the genetic architecture of common disease susceptibility (Sing et al. 2003). This component is most often not given adequate consideration in the application of linear models to the analysis of single locus genetic effects.
Caution must be exercised in drawing biological inferences from statistical models of interaction (Moore and Williams 2005) because they cannot reflect the separate actions of the many participating interacting agents, most of which cannot be measured, that are involved in the causation of phenotypic variation (Cordell 2002; Rea et al. 2006). Although the biological mechanisms of epistasis cannot be directly extrapolated from the observed statistical evidence for non-additivity of SNP effects, each SNP involved in a high-risk pairwise combination has been implicated in marking biological effects in complementary studies. Lower expression of APOE (Artiga et al. 1998), lower levels of apoE in plasma and increased risk of IHD (Lambert et al. 2000) are associated with the T allele of the −219G > T SNP. The low transcriptional activity associated with the T allele has the potential to affect atherogenesis locally in the arterial wall by a decreased amount of macrophage-derived apoE, which possesses anti-atherosclerotic potential, through its cholesterol efflux properties (Cullen et al. 1998). Higher levels of triglycerides and lower levels of high-density lipoprotein (HDL) cholesterol (Gerdes et al. 1997; Wittrup et al. 1997, 1999a) are associated with the minor alleles of the −93T > G, D9N (which is in almost complete LD with −93) and N291S LPL SNPs, whereas the variant allele (G allele) of the S447X LPL SNP is associated with decreased triglyceride and increased HDL cholesterol levels (Wittrup et al. 2002).
The D9N SNP in LPL was involved in each of the three pairwise models that improved the IHD prediction beyond smoking, diabetes and hypertension. This was the only single site that had a statistically significant marginal effect. When we tested the improvement in prediction of a second site, the improvement was always significant when D9N was added as the last site, whereas the addition of the other SNP that defined the pair as the last site resulted in modest improvements. Although a statistical analysis cannot distinguish whether subgroups of individuals with combinations of D9N and −219G > T, N291S or S447X are at increased risk due to altered expression and/or function of the protein products or an interaction between them, it can provide the basis for improved risk assessment. The HR of 1.69 associated with the AA and GA D9N genotypes in men was substantially increased when combined with one of these three SNPs; to 2.11 for 55 individuals [D9N (AA,GA)/−219G > T(GT)], to 4.35 for 6 individuals [D9N (AA,GA)/N291S (GG,AG)] and to 1.92 for 94 individuals [D9N (AA,GA)/S447X (CC)]. These subgroups, with a higher risk of IHD, illustrate the importance of considering the genetic background in evaluating single SNPs as genetic predictors. In every case the risk associated with these subgroups in men exceeded the risk associated with smoking, diabetes or hypertension. This result also emphasizes the importance of considering SNPs which have no significant marginal genotypic effects in research to characterize genetically susceptible subgroups.
On the basis of the ranking of risk factors previously reported for this cohort (Schnohr et al. 2002), lipid traits were not incorporated in the primary analyses because they were weak predictors of IHD compared to smoking, diabetes and hypertension. However, because the products of the APOE and LPL genes play a major role in lipid metabolism genes, with well-established effects on total cholesterol, HDL cholesterol and triglycerides levels, we carried out a sub-analysis to determine whether the observed SNP effects on IHD persisted after adjustment for plasma lipid levels. In this sub-analysis the third examination was considered as the baseline survey because all three lipid traits were measured simultaneously at this time point. Individuals above the age of 45 years at the third examination were included. In this restricted analysis follow-up time was cut to approximately 8 years. In this analysis the recommendations of the National Cholesterol Education Program Expert Panel, National Institute of Health, USA, were used to define dyslipemic subgroups (National Cholesterol Education Program National Heart, Lung, and Blood Institute 2002). Dyslipidemia was diagnosed when total cholesterol exceeded 200 mg/ dl (5.18 mmol/l), triglycerides exceeded 150 mg/dl (1.60 mmol/l) or HDL cholesterol was less than 40 mg/ dl (1.04 mmol/l). Inclusion of the diagnosis of dyslipidemia for each of the three measures of lipids into the Cox regression model did not substantially alter the HR estimated for the GA + AA genotypes of the LPL D9N SNP in men (model without lipid traits: 1.62, CI = 0.89–2.96, P = 0.12; model with lipid traits: 1.55, CI = 0.84–2.83, P = 0.16). Addition of the lipid trait categories to the pairwise models had minor effects on the HRs for the LPL D9N and APOE −219G < T (GAGG) combination in women (model without lipid traits: 3.81, CI = 1.38–10.5, P = 0.01; model with lipid traits: 4.09, CI = 1.48–11.31, P = 0.007), for the LPL D9N (AA,GA) and LPL N291S (GG,AG) combination in men (model without lipid traits: 14.95, CI = 4.69–47.7, P < 0.0001; model with lipid traits: 16.99, CI = 5.27–54.77, P < 0.0001) or for the LPL D9N and LPL S447X (AA,GA)CC combination in men (model without lipid traits: 1.93, CI = 1.05–3.54, P = 0.03; model with lipid traits: 1.84, CI = 1.00–3.39, P = 0.05). In summary, the single site and pairwise SNP genotypes not only predict IHD beyond smoking, diabetes and hypertension, but also predict IHD after adjustment for the effects of total cholesterol, HDL cholesterol and triglyceride levels.
A major research challenge will be to design appropriate studies to distinguish between context-dependent effects and Type I errors (false positives) (Sing et al. 2003). Ideally, the impact of genetic variation on risk of a common disease with a complex multifactorial etiology should be modeled in one large prospective cohort and the utility of the model tested in a large prospective cohort of similar size that has the same genetic and environmental history. As we do not have access to a second large prospective cohort, we validated the significant results from the prospective study in a large case-control study including 927 independently ascertained and well-characterized IHD patients with severe stenosis on coronary angiography who were recruited from the same area of Denmark as the prospectively collected sample. Using this validation strategy, we were able to replicate the single SNP result and two of the three significant likelihood ratio tests (one borderline significant) and three of the four significant point estimates for pairwise combinations of SNPs in men. The replicated point estimates also had the most convincing cumulative incidence plots. The failure to replicate the pairwise SNP results in women was most likely the result of a Type II error (false negative result), due to limited statistical power for women in the validation sample (women in prospective study with IHD, n = 369; women in case-control study with IHD, n = 244) (Clark et al. 2005).
In the present paper we combined a priori in vivo and in vitro knowledge of genetic variants with a stepwise model fitting procedure to estimate the contribution of genetic information to the prediction of IHD. A stringent statistical correction for multiple comparisons to estimate an experiment-wise error for the observed outcomes of this strategy is complicated by the difficulty in assigning probabilities to the a priori knowledge. Our strategy for dealing with false positive results was to validate the results in a second sample. One major strength of this strategy is that it tests the predictive utility of the positive results obtained from a sample of the general population in a case-control study of independently ascertained IHD patients from the same population, avoiding the Type I errors that can be generated by the selection bias inherent in case-control studies. Furthermore, the biological relevance and medical utility of a validation approach is superior to a statistical adjustment for multiple comparisons when there is a priori information about model specification that is only arbitrarily quantifiable.
The aim of the present study was not to develop risk scores for primary care as done in previous studies (Anderson et al. 1990; Conroy et al. 2003). The primary aim was to determine whether genetic information increases the ability to predict IHD risk beyond traditional risk factors. In a previous description of the CCHS cohort, smoking, diabetes and hypertension were the three most important risk factors, conferring HRs above 1.4 in both women and men (Schnohr et al. 2002). Hence, the definition of risk factors deviates from the Framingham and SCORE algorithms (Anderson et al. 1990; Conroy et al. 2003). Efforts were devoted to refining these major traditional risk factors based on extensive composite information obtained from all three examinations of the CCHS cohort (see Methods above). By using such information on hypertension we obtained a more precise risk factor definition than would have been obtained by including systolic blood pressure alone (as in the Framingham and SCORE algorithms), because only one blood pressure was measured at each of the three examinations. Furthermore, inclusion of total cholesterol, HDL cholesterol and triglycerides in a sub-analysis did not alter inferences about the role of genotypes in predicting risk of IHD. Because our primary aim was to determine whether genetic information increases the ability to predict IHD risk beyond traditional risk factors, we also included diabetes mellitus in our models, the number one risk factor in both genders in the CCHS cohort (Schnohr et al. 2002), in contrast to the SCORE algorithm.
In conclusion, our study reveals that genetic variation predicts risk of IHD beyond that associated with smoking, diabetes and hypertension, and that multiple rare classes of genotypes created by different combinations of SNPs, each having small genotype effects on risk, may identify a significant fraction of those at elevated risk of IHD in the population at large. Instead of treating individuals according to information obtained from the average effect of a single risk factor, as in the common situation in the practice of medicine, our study suggests that treatment may be best targeted towards those subgroups that harbor combinations of genetic factors that are at greatest risk. Such information can also be expected to be of value in the design of more effective and efficient preventive health programs.
Acknowledgments
We thank Kenneth G. Weiss for his persistent attention to the data analyses. Mette Refstrup, Pia T. Petersen and Hanne Dam are greatly thanked for their excellent technical assistance. This study was supported by Chief Physician Johan Boserup and Lise Boserup’s Fund, The Danish Heart Foundation, The Danish Medical Research Council, Ingeborg and Leo Dannin’s Grant, The Research Fund at Rigshospitalet, Copenhagen University Hospital, National Institute of Health HL039107 and National Institute of General Medical Science grant GM065509.
Contributor Information
Ruth Frikke-Schmidt, Department of Clinical Biochemistry KB3011, Section for Molecular Genetics, Rigshospitalet, Copenhagen University Hospital, Blegdamsvej 9, 2100 Copenhagen Ø, Denmark.
Charles F. Sing, Department of Human Genetics, University of Michigan, Ann Arbor, MI, USA
Børge G. Nordestgaard, Department of Clinical Biochemistry, Herlev University Hospital, Herlev, Denmark, The Copenhagen City Heart Study, Bispebjerg University Hospital, Copenhagen, Denmark
Rolf Steffensen, Department of Cardiology, Hillerød Hospital, Hillerød, Denmark.
Anne Tybjærg-Hansen, Department of Clinical Biochemistry KB3011, Section for Molecular Genetics, Rigshospitalet, Copenhagen University Hospital, Blegdamsvej 9, 2100 Copenhagen Ø, Denmark, e-mail: at-h@rh.dk, The Copenhagen City Heart Study, Bispebjerg University Hospital, Copenhagen, Denmark.
References
- Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Anderson KM, Odell PM, Wilson PWF, Kannel WB. Cardiovascular disease risk profiles. Am Heart J. 1990;121:293–298. doi: 10.1016/0002-8703(91)90861-b. [DOI] [PubMed] [Google Scholar]
- Appleyard M, Hansen AT, Jensen G, Schnohr P, Nyboe J. The Copenhagen City Heart Study. Østerbroundersøgelsen. A book of tables with data from the first examination (1976–78) and a five year follow-up (1981–83). The Copenhagen City Heart Study Group. Scand J Soc Med Suppl. 1989;41:1–160. [PubMed] [Google Scholar]
- Artiga MJ, Bullido MJ, Sastre I, Recuero M, Garcia MA, Aldudo J, Vazquez J, Valdivieso F. Allelic polymorphisms in the transcriptional regulatory region of apolipoprotein E gene. FEBS Lett. 1998;421:105–108. doi: 10.1016/s0014-5793(97)01543-3. [DOI] [PubMed] [Google Scholar]
- Brodie ED., III . Why evolutionary genetics does not always add up. In: Wolf JB, Brodie ED III, Wade MJ, editors. Epistasis and the evolutionary process. 1. Oxford University Press; New York: 2000. pp. 3–19. [Google Scholar]
- Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apoC-II deficiency, and hepatic lipase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and molecular bases of inherited disease. 8. McGraw-Hill; New York: 2001. pp. 2789–2816. [Google Scholar]
- Chakravarti A, Little P. Nature, nurture and human disease. Nature. 2003;421:412–414. doi: 10.1038/nature01401. [DOI] [PubMed] [Google Scholar]
- Clark AG, Boerwinkle E, Hixson JE, Sing CF. Determinants of the success of whole-genome association testing. Genome Res. 2005;15:1463–1467. doi: 10.1101/gr.4244005. [DOI] [PubMed] [Google Scholar]
- Collins FS. The case for a US prospective cohort study of genes and environment. Nature. 2004;429:475–477. doi: 10.1038/nature02628. [DOI] [PubMed] [Google Scholar]
- Conroy RM, Pyörälä K, Fitzgerald AP, Sans S, Menotti A, De Backer G, De Bacquer D, Ducimetiere P, Jousilahti P, Keil U, Njølstad I, Oganov RG, Thomsen T, Tunstall-Pedoe H, Tverdal A, Wedel H, Whincup P, Wilhelmsen L, Graham IM. Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J. 2003;24:987–1003. doi: 10.1016/s0195-668x(03)00114-3. [DOI] [PubMed] [Google Scholar]
- Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- Cullen P, Cignarella A, Brennhausen B, Mohr S, Assmann G, von Eckardstein A. Phenotype-dependent differences in apolipoprotein E metabolism and in cholesterol homeostasis in human monocyte-derived macrophages. J Clin Invest. 1998;101:1670–1677. doi: 10.1172/JCI119887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- de Visser JA, Hermisson J, Wagner GP, Ancel ML, Bagheri-Chaichian H, Blanchard JL, Chao L, Cheverud JM, Elena SF, Fontana W, Gibson G, Hansen TF, Krakauer D, Lewontin RC, Ofria C, Rice SH, von Dassow G, Wagner A, Whitlock MC. Perspective: evolution and detection of genetic robustness. Evol Int J Org Evol. 2003;57:1959–1972. doi: 10.1111/j.0014-3820.2003.tb00377.x. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001;109:121–124. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- European Society of Hypertension. European Society of Cardiology guidelines for the management of arterial hypertension. 2003. Eur Heart J. 21:1011–1053. doi: 10.1097/00004872-200306000-00001. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt R, Nordestgaard BG, Agerholm-Larsen B, Schnohr P, Tybjærg-Hansen A. Context dependent and invariant associations between lipids, lipoproteins, and apolipoproteins, and apolipoprotein E genotype. A study of 9,060 women and men from the population at large. J Lipid Res. 2000;41:1812–1822. [PubMed] [Google Scholar]
- Frikke-Schmidt R, Sing CF, Nordestgaard BG, Tybjærg-Hansen A. Gender- and age-specific contributions of additional DNA sequence variation in the 5′ regulatory region of the APOE gene to prediction of measures of lipid metabolism. Hum Genet. 2004;115:331–345. doi: 10.1007/s00439-004-1165-z. [DOI] [PubMed] [Google Scholar]
- Gerdes LU, Klausen IC, Sihm I, Færgeman O. Apolipoprotein E polymorphism in a Danish population compared to findings in 45 other study populations around the world. Genet Epidemiol. 1992;9:155–167. doi: 10.1002/gepi.1370090302. [DOI] [PubMed] [Google Scholar]
- Gerdes C, Fisher RM, Nicaud V, Boer J, Humphries SE, Talmud PJ, Færgeman O. Lipoprotein lipase variants D9N and N291S are associated with increased plasma triglyceride and lower high-density lipoprotein cholesterol concentrations: studies in the fasting and postprandial states: The European Atherosclerosis Research Studies. Circulation. 1997;96:733–740. doi: 10.1161/01.cir.96.3.733. [DOI] [PubMed] [Google Scholar]
- Gerdes LU, Jeune B, Ranberg KA, Nybo H, Vaupel JW. Estimation of apolipoprotein E genotype-specific relative mortality risks from the distribution of genotypes in centenarians and middle-aged men: Apolipoprotein E gene is a “frailty gene,” not a “longevity gene”. Genet Epidemiol. 2000;19:202–210. doi: 10.1002/1098-2272(200010)19:3<202::AID-GEPI2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Gliemann J. Receptors of the low density lipoprotein (LDL) receptor family in man. Multiple functions of the large family members via interaction with complex ligands. Biol Chem. 1998;379:951–964. [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- Keavney B, Palmer A, Parish S, Clark S, Youngman L, Danesh J, McKenzie C, Delepine M, Lathrop M, Peto R, Collins R. Lipid-related genes and myocardial infarction in 4685 cases and 3460 controls: discrepancies between genotype, blood lipid concentrations, and coronary disease risk. Int J Epidemiol. 2004;33:1002–1013. doi: 10.1093/ije/dyh275. [DOI] [PubMed] [Google Scholar]
- Khot UN, Khot MB, Bajzer CT, Sapp SK, Ohman EM, Brener SJ, Ellis SG, Lincoff AM, Topol EJ. Prevalence of conventional risk factors in patients with coronary heart disease. JAMA. 2003;290:898–904. doi: 10.1001/jama.290.7.898. [DOI] [PubMed] [Google Scholar]
- Klein JP, Moeschberger ML. Techniques for censored and truncated data. 2. Springer; Berlin Heidelberg New York: 2003. Refinements of the semiparametric proportional hazards model. Survival analysis; pp. 295–328. [Google Scholar]
- Lambert JC, Brousseau T, Defosse V, Evans A, Arveiler D, Ruidavets JB, Haas B, Cambou JP, Luc G, Ducimetiere P, Cambien F, Chartier-Harlin MC, Amouyel P. Independent association of an APOE gene promoter polymorphism with increased risk of myocardial infarction and decreased apoE plasma concentrations—the ECTIM Study. Hum Mol Genet. 2000;9:57–61. doi: 10.1093/hmg/9.1.57. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Rall SC., Jr . Type III hyperlipoproteinemia (dysbetalipoproteinemia): the role of apolipoprotein E in normal and abnormal lipoprotein metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. McGraw-Hill; New York: 2001. pp. 2835–2862. [Google Scholar]
- Moore JH, Williams SM. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays. 2005;27:637–646. doi: 10.1002/bies.20236. [DOI] [PubMed] [Google Scholar]
- National Cholesterol Education Program National Heart, Lung, and Blood Institute (2002) Third Report of the National Cholesterol Education Program (NCEP) Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. NIH Publication No. 02–5215, September
- Nickerson DA, Taylor SL, Weiss KM, Clark AG, Hutchinson RG, Stengard J, Salomaa V, Vartiainen E, Boerwinkle E, Sing CF. DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nat Genet. 1998;19:233–240. doi: 10.1038/907. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Taylor SL, Fullerton SM, Weiss KM, Clark AG, Stengard JH, Salomaa V, Boerwinkle E, Sing CF. Sequence diversity and large-scale typing of SNPs in the human apolipoprotein E gene. Genome Res. 2000;10:1532–1545. doi: 10.1101/gr.146900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordestgaard BG, Abildgaard S, Wittrup HH, Steffensen R, Jensen G, Tybjærg-Hansen A. Heterozygous lipoprotein lipase deficiency. Frequency in the general population, effect on plasma lipid levels, and risk of ischemic heart disease. Circulation. 1997;96:1737–1744. doi: 10.1161/01.cir.96.6.1737. [DOI] [PubMed] [Google Scholar]
- Olivecrona G, Olivecrona T. Triglyceride lipases and atherosclerosis. Curr Opin Lipidol. 1995;6:291–305. doi: 10.1097/00041433-199510000-00009. [DOI] [PubMed] [Google Scholar]
- Payami H, Zhu M, Montimurro J, Keefe R, McCulloch CC, Moses L. One step closer to fixing association studies: evidence for age-and gender-specific allele frequency variations and deviations from Hardy–Weinberg expectations in controls. Hum Genet. 2005;118:322–330. doi: 10.1007/s00439-005-0057-1. [DOI] [PubMed] [Google Scholar]
- Pembrey M. Genetic epidemiology: some special contributions of birth cohorts. Paediatr Perinat Epidemiol. 2004;18:3–7. doi: 10.1111/j.1365-3016.2004.00530.x. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Przeworski M. Linkage disequilibrium in humans: models and data. Am J Hum Genet. 2001;69:1–14. doi: 10.1086/321275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea T, Brown CM, Sing CF. Complex adaptive system models and the genetic analysis of plasma HDL-cholesterol levels. Perspect Biol Med. 2006 doi: 10.1353/pbm.2006.0063. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SL, Ferrell RE, Kottke BA, Kamboh MI, Sing CF. The gender-specific apolipoprotein E genotype influence on the distribution of lipids and apolipoproteins in the population of Rochester, MN. I. Pleiotropic effects on means and variances. Am J Hum Genet. 1991;49:1155–1166. [PMC free article] [PubMed] [Google Scholar]
- Schnohr P, Jensen JS, Scharling H, Nordestgaard BG. Coronary heart disease risk factors ranked by importance for the individual and community. A 21 year follow-up of 12000 men and women from The Copenhagen City Heart Study. Eur Heart J. 2002;23:620–626. doi: 10.1053/euhj.2001.2842. [DOI] [PubMed] [Google Scholar]
- Sing CF, Stengard JH, Kardia SL. Genes, Environment, and Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2003;23:1190–1196. doi: 10.1161/01.ATV.0000075081.51227.86. [DOI] [PubMed] [Google Scholar]
- Song Y, Stampfer MJ, Liu S. Meta-analysis: Apolipoprotein E genotypes and risk for coronary heart disease. Ann Intern Med. 2004;141:137–147. doi: 10.7326/0003-4819-141-2-200407200-00013. [DOI] [PubMed] [Google Scholar]
- Stannard AK, Riddell DR, Sacre SM, Tagalakis AD, Langer C, von Eckardstein A, Cullen P, Athanasopoulos T, Dickson G, Owen JS. Cell-derived apolipoprotein E (apoE) particles inhibit vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. J Biol Chem. 2001;276:46011–46016. doi: 10.1074/jbc.M104812200. [DOI] [PubMed] [Google Scholar]
- Stengard JH, Clark AG, Weiss KM, Kardia S, Nickerson DA, Salomaa V, Ehnholm C, Boerwinkle E, Sing CF. Contributions of 18 additional DNA sequence variations in the gene encoding apolipoprotein E to explaining variation in quantitative measures of lipid metabolism. Am J Hum Genet. 2002;71:501–517. doi: 10.1086/342217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengard JH, Kardia SL, Hamon SC, Frikke-Schmidt R, Tybjærg-Hansen A, Salomaa V, Boerwinkle E, Sing CF. Contribution of regulatory and structural variations in APOE to predicting dyslipidemia. J Lipid Res. 2006;47:318–328. doi: 10.1194/jlr.M500491-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Task force of the European Society of Cardiology . Management of stable angina pectoris recommendations. Eur Heart J. 1997;18:394–413. doi: 10.1093/oxfordjournals.eurheartj.a015259. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Pan L, Abney M, Ober C. The sex-specific genetic architecture of quantitative traits in humans. Nat Genet. 2006;38:218–222. doi: 10.1038/ng1726. [DOI] [PubMed] [Google Scholar]
- Wittrup HH, Tybjærg-Hansen A, Abildgaard S, Steffensen R, Schnohr P, Nordestgaard BG. A common substitution (asn291ser) in lipoprotein lipase is associated with increased risk of ischemic heart disease. J Clin Invest. 1997;99:1606–1613. doi: 10.1172/JCI119323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrup HH, Tybjærg-Hansen A, Steffensen R, Deeb SS, Brunzell JD, Jensen G, Nordestgaard BG. Mutations in the lipoprotein lipase gene associated with ischemic heart disease in men: The Copenhagen City Heart Study. Arterioscler Thromb Vasc Biol. 1999a;19:1535–1540. doi: 10.1161/01.atv.19.6.1535. [DOI] [PubMed] [Google Scholar]
- Wittrup HH, Tybjærg-Hansen A, Nordestgaard BG. Lipoprotein lipase mutations, plasma lipids and lipoproteins, and risk of ischemic heart disease: a meta-analysis. Circulation. 1999b;99:2901–2907. doi: 10.1161/01.cir.99.22.2901. [DOI] [PubMed] [Google Scholar]
- Wittrup HH, Nordestgaard BG, Steffensen R, Jensen G, Tybjærg-Hansen A. Effect of gender on phenotypic expression of the S447X mutation in LPL: the Copenhagen City Heart Study. Atherosclerosis. 2002;165:119–126. doi: 10.1016/s0021-9150(02)00183-1. [DOI] [PubMed] [Google Scholar]