

Abstract
ERK5 is involved in proliferation of vascular smooth muscle cells (VSMC). The proliferative actions of insulin and angiotensin-II (A-II) in VSMC are mediated in part by ERK1/2. We hypothesized that insulin and A-II also regulate ERK5 activity in VSMC. Acute treatment (< 60 min) with insulin or A-II increased phosphorylation of ERK1/2 at 15 min and ERK5 at 5 min. Chronic treatment (≤ 8 h) with insulin increased ERK1/2 phosphorylation by 4 h and ERK5 by 8 h. A-II stimulated phosphorylation of ERK1/2 by 8 h and ERK5 by 4 h. The EC50 for insulin treatment effecting ERK1/2 and ERK5 phosphorylation was 1.5 nM and 0.1 nM, whereas the EC50 for A-II was 2 nM, each. Insulin plus A-II induced an additive effect only on ERK5 phosphorylation. Inhibition of insulin- and A-II-stimulated phosphorylation of ERK5 and ERK1/2 by PD98059 and Wortmannin exhibited differential and time-dependent effects. Taken together, these data indicate that insulin and A-II regulate the activity of ERK5, but different from that seen for ERK1/2.
Keywords: ERK5, Insulin, Angiotensin-II, MEF2C
INTRODUCTION
Mitogen-activated protein (MAP) kinases are highly conserved molecules that transduce extracellular stimuli into intracellular responses. Members of the MAP kinase family include, but are not limited to, extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 kinase. ERK MAP kinases respond to growth factors, whereas JNK and p38 MAP kinases mediate environmental stress and pro-inflammatory cytokines [1].
ERK5 has recently been identified as a new member of the MAP kinase family. ERK5 has been shown to be involved with both differentiation and proliferation in adipocytes, neurons, vascular endothelial cells, skeletal muscle cells and cancer cell lines [2–7]. More recently, ERK5 has been shown to be involved in vascular smooth muscle cell (VSMC) proliferation [8].
Insulin and angiotensin-II (A-II) stimulate proliferation of VSMC [9, 10]. Insulin mitogenic action is mediated by the Ras-ERK1/2 pathway [11]. A-II inflammatory actions appear to be mediated by the Akt and NFκB pathways [10, 12], whereas its mitogenic action has been shown to be mediated via the Ras-ERK1/2 pathway [13]. In this study, we were interested in determining whether the ERK5 pathway is part of insulin and A-II signaling in VSMC. Here we show for the first time, that insulin and A-II regulate the phosphorylation of ERK5 in rat aorta smooth muscle cells (RASMC) in a time- and dose-dependent manner. Additionally, we show that insulin and A-II appear to have an additive effect on ERK5 phosphorylation and have differential effects on ERK5 and ERK1/2 phosphorylation in the absence and presence of the MEK inhibitor, PD98059, and the phosphatidylinositol-3 (PI-3) kinase inhibitor, wortmannin.
MATERIALS AND METHODS
Materials
All basic laboratory reagents were from Sigma-Aldrich (St. Louis, MO.). Anti-phospho-ERK5, anti-ERK5, anti-phospho-MEF2C and anti-MEF2C antibodies were from Cell Signaling (Beverly, MA). SDS-polyacrylamide gels were from Pierce (Rockford, IL), PVDF and protein-gel apparatus were from Bio-Rad (Hercules, CA). Rat aorta vascular smooth muscle cells (CRL-1444) were from ATCC (Manassas, VA). Minimal Essential Medium (MEM), fetal bovine serum, phosphate buffered saline (PBS), Penicillin/Streptomycin and non-essential amino acids were from Gibco/Invitrogen (Carlsbad, CA). Fungizone was from Gemini Bio-products (Calabasas, CA).
Cell Culturing
Rat aorta smooth muscle cells (RASMC) were grown in Minimal Essential Medium (MEM) growth medium (5 mL Pencillin/Stretomycin, 5 mL Non-essential amino acids, 5 mL L-glutamine and 10% FBS in 500 mL of MEM) at 37°C and 5% CO2. Prior to treatment, all cells were placed in serum-reduced medium (0.1% FBS) for 24 hr to maintain quiescence before further treatment. For the time course experiments, RASMC were grown to sub-confluence and incubated for acute (0 to 60 min) or chronic (0 to 24 hrs) stimulus of vehicle alone, insulin (10 nM), angiotensin-II (100 nM) or designated concentrations of insulin plus A-II. For the dose response experiments, and after reaching sub-confluence, RASMC were incubated without or with designated concentrations of insulin or A-II at the optimal times as previously determined in the time-course assays, to effect changes in the magnitude of phosphorylation. For experiments that included the inhibitors, PD98059 and Wortmannin, cells were pre-incubated in the absence or presence of PD98059 (10 μM) or Wortmannin (100 nM) for 30 minutes before insulin or A-II treatment, and remained in the medium throughout the experiment.
Western blot analysis
RASMC were grown to sub-confluence in MEM growth medium. Prior to treatment with insulin and/or A-II, cells were incubated in serum-reduced medium for 24 h and then challenged with indicated concentrations of insulin or A-II for designated times and lysed in ice-cold lysis buffer (150 mM NaCl, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol 1 mM sodium vandate, 1 mM sodium phosphate, 1% Triton X-100, 0.5% SDS, 10 μg/mL aprotinin, 10 μg/mL leupetin, 50 mM HEPES, pH 7.5). Crude lysates were sonicated and centrifuged at 10,000 rpm for 5 min at 4°C. Total protein concentration from the resultant supernatant was determined by the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL) and normalized to 1mg/mL. Aliquots of whole cell lysates were dried in a Speedvac Concentrator (Savant, Holbrook, NY), denatured in Laemmli sample buffer, and stored at −20°C until use. Samples were boiled for 10 min, and 30 μg of protein per lane was resolved with SDS-polyacrylamide gel electrophoresis before transferring to polyvinylidene fluoride (PVDF) membranes. Membranes were probed with rabbit polyclonal antibodies specific for phosphoryated and total proteins then incubated with goat anti-rabbit polyclonal antibodies conjugated to horseradish peroxidase. Proteins were detected using an ECL detection kit (Amersham, Piscataway, NJ), and quantitated by densitometry, using a Bio-Rad Fluor-S MultiImager (Bio-Rad, Hercules, CA).
Statistical Analysis
All statistics were analyzed by Student’s t test, with a “P” value of < 0.05 considered significant. Results were expressed as the mean ± SEM of three or more independent experiments.
RESULTS
Insulin and A-II stimulate the phosphorylation of ERK5 and ERK1/2 with temporally different profiles
We first characterized the temporal effects of insulin and A-II on the phosphorylation of ERK5 and and ERK1/2 in rat aorta vascular smooth muscle cells (RASMC) by incubating RASMC in reduced-serum medium without or with insulin (10 nM) and in the absence or presence of A-II (100 nM) for designated times.
Acutely, insulin significantly (P < 0.05) increased the magnitude of phosphorylation of ERK5 2-fold at 5 min and ERK1/2 6-fold at 15 min (Fig 1). A-II significantly (P < 0.05) increased the amount of phosphorylation of ERK5 6-fold at 5 min and ERK1/2 10-fold at 15 min (Supplemental Fig 1) and returned to basal values by 60 min.
Figure 1. Acute treatment of insulin stimulates the phosphorylation of ERK5 and ERK1/2 at different times in RASMC.
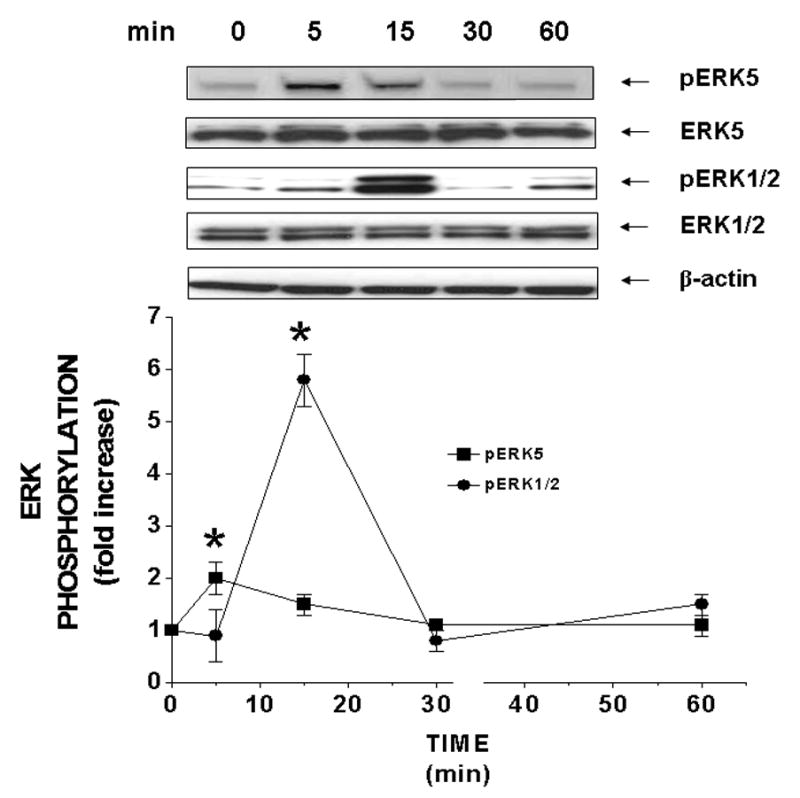
Sub-confluent cells in serum-reduced medium were treated acutely (0 – 60 min) without or with insulin (10 nM). Phosphorylated and total proteins were determined by Western blot analysis. Blots are representative of three independent experiments. Results are expressed as fold increase above controls of phosphorylated ERK5 (■) and ERK1/2 (●) and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Chronic treatment of insulin (10 nM) (Supplemental Fig 2) stimulated a 2.5-fold and 3-fold increase in ERK5 phosphorylation by 4 h and 8 h, respectively, and 3-fold and 2-fold phosphorylation of ERK1/2 at 4h and 8 h, respectively. A-II-stimulated equal increases in ERK5 and ERK1/2 phosphorylation: 2-fold at 4 h and 3-fold for 8 h, respectively (Supplemental Fig 3). Although A-II-stimulated ERK5 phosphorylation returned to basal levels by 24 hours, A-II-induced phosphorylation of ERK1/2 increased to 5-fold by 24 hours.
Insulin and A-II stimulate the phosphorylation of ERK5 and ERK1/2 differentially in a dose-dependent manner
Next, we determined the magnitude of phosphorylation of ERK5 and ERK1/2 in the presence of various concentrations of insulin and A-II at acute (< 60 m) and chronic (8 h) time points.
Acutely, increasing concentrations of insulin (Supplemental Fig 4) and A-II (Supplemental Fig 5) increased ERK5 and ERK1/2 phosphorylation in a dose-dependent manner, and both ERK5 and ERK1/2 were more sensitive (responsive) to insulin than A-II. The EC50 for insulin at acute times for ERK5 and ERK1/2 were 0.1 nM and 1.5 nM, respectively. In comparison, the EC50 for A-II was 2 nM for both ERK5 and ERK1/2.
Using the eight hour time point, we determined that the EC50 for insulin at 8 h for ERK5 and ERK1/2 phosphorylation were 0.4 nM and 0.3 nM, respectively (Supplemental Fig 6), and the EC50 for A-II had increased to 3 nM and 5 nM for ERK5 and ERK1/2, respectively (Supplemental Fig 7).
Insulin augments A-II-stimulated phosphorylation of ERK5
Because hyperinsulinemia (HI) is a chronic disorder and augments other growth factor-stimulated phosphorylation of ERK1/2 in VSMC [14, 15], we examined the effects of HI on A-II action on ERK5 phosphorylation. We incubated RASMC in serum-reduced medium in the absence and presence of insulin (0.3 nM) and without or with A-II (2 nM), and determined the amount of ERK5 and ERK1/2 phosphorylation.
Insulin and A-II increased the phosphorylation of ERK5 and ERK1/2 at acute (Supplemental Fig 8) and chronic (Fig 2) times. During acute and chronic stimulation, insulin augmented A-II-stimulated phosphorylation of ERK5, but not ERK1/2. This suggested that although phosphorylation of ERK5 and ERK1/2 does occur at acute and chronic stimulation from the actions of insulin and A-II, a more potent signal is relayed to ERK5.
Figure 2. Chronic treatment of insulin augments A-II-stimulated phosphorylation of ERK5, but not ERK1/2 in RASMC.
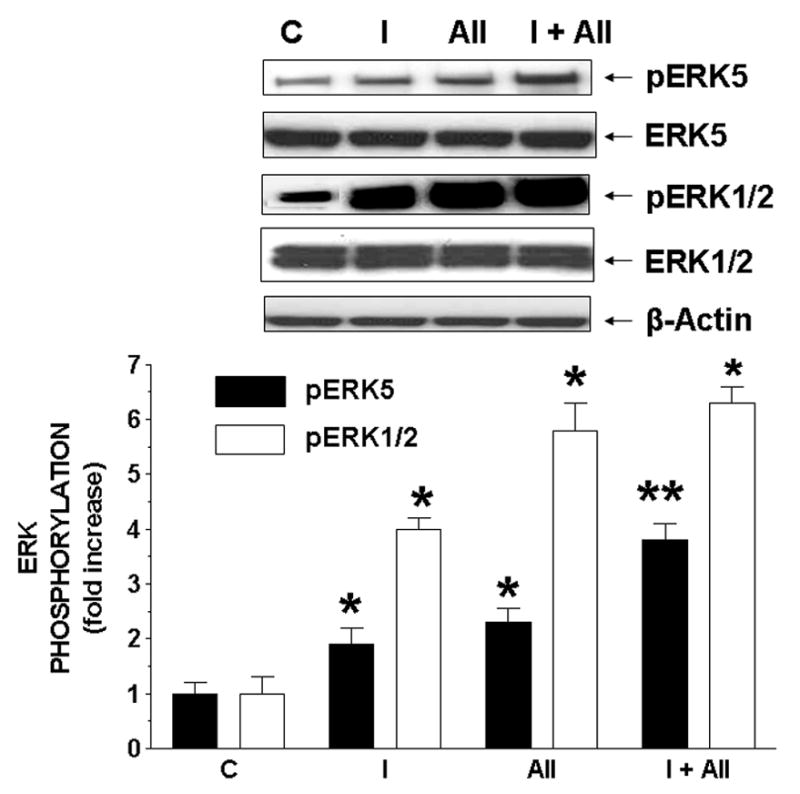
Cells were treated without or with insulin (0.3 nM) and/or A-II (2 nM) for chronic times. Results are expressed as fold increase of phosphorylated ERK5 (closed bars) and ERK1/2 (open bars) relative to controls and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls. **, P < 0.05 vs. A-II alone.
Effects of PD98059 and Wortmannin on insulin- and A-II-stimulated ERK5 and ERK1/2 phosphorylation
Although PD98059 (PD) and Wortmannin (WT) have been shown to inhibit insulin-stimulated phosphorylation of ERK1/2 and PI-3 kinase, respectively [16, 17], to our knowledge no one yet has determined the effects of acute and chronic treatments of PD and WT on insulin- and A-II-stimulated ERK5 phosphorylation in RASMC. Thus, we treated RASMC with insulin and A-II = in the absence and presence of PD (10 μM) or WT (100 nM).
At acute times (Fig. 3), both PD and WT did not have an effect on basal levels of ERK5 or ERK1/2 phosphorylation nor did they inhibit insulin-stimulated ERK5 phosphorylation. PD significantly (P < 0.05) decreased insulin-stimulated ERK1/2 phosphorylation, whereas WT had no affect on insulin-stimulated ERK1/2 phosphorylation. In comparison, PD significantly (P < 0.05) decreased A-II-stimulated ERK5 and ERK1/2 phosphorylation by 50% and 98%, respectively. WT inhibited A-II-induced ERK5 phosphorylation by 50% at acute times (Fig 3) and 25% at chronic times (Supplemental Fig 9), but had no affect on A-II-stimulated ERK1/2 phosphorylation at either time point.
Figure 3. Effect of PD98059 and Wortmannin on acute treatment of insulin- and angiotensin-II-stimulated phosphorylation of ERK5, ERK1/2 and MEF2C.
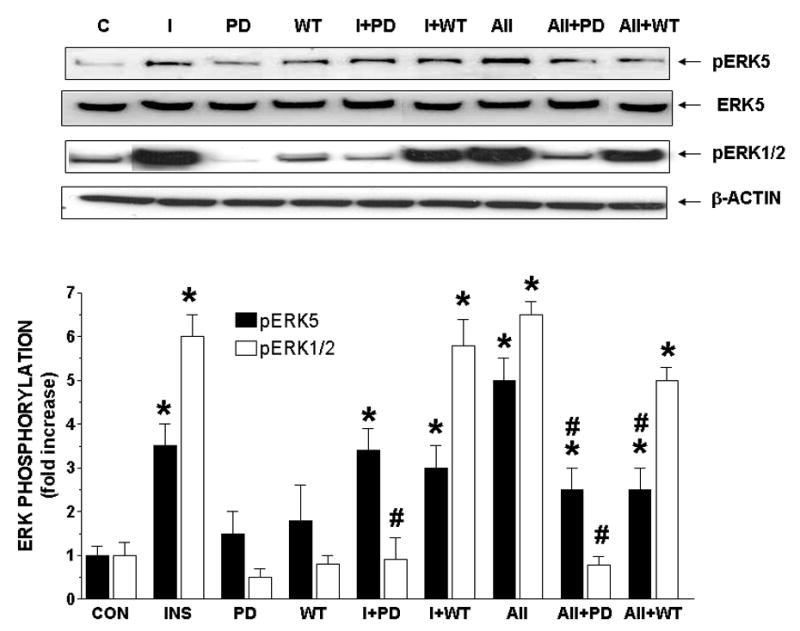
Cells were treated without or with insulin (10 nM) or A-II (100 nM) and in the absence or presence of PD98059 (10 μM) or Wortmannin (100 nM) for acute times. Results are expressed as fold increase above controls and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls. #, P < 0.05 vs. insulin or A-II alone.
At chronic times (Supplemental Fig 9), different patterns of phosphorylation existed for ERK5 and ERK1/2. First, insulin and A-II significantly (P < 0.05) stimulated increases in ERK5 phosphorylation. Second, both PD and WT alone significantly (P < 0.05) increased the phosphorylation of ERK5 above that seen for controls. However, WT alone increased ERK1/2 phosphorylation above controls. PD was without this effect on ERK1/2. Neither PD nor Wortmannin decreased insulin-stimulated ERK5 phosphorylation.
Third, PD inhibited insulin-stimulated ERK1/2 phosphorylation, whereas it had no effect on insulin-stimulated ERK5 phosphorylation. Wortmannin had no inhibitory effect on insulin-stimulated phosphorylation of ERK1/2, yet significantly augmented insulin-stimulated ERK1/2 phosphorylation, suggesting a mimicry of insulin resistance: a perturbed insulin/PI-3 kinase signaling with concomitant insulin ERK1/2 signaling. Although Wortmannin did not inhibit insulin-stimulated ERK5 phosphorylation, it did not have any additive effect on insulin-stimulated ERK5 phosphorylation either.
Fourth, PD had no effect on A-II-stimulated ERK5 phosphorylation, but reduced A-II-stimulated ERK1/2 phosphorylation by 50%. Wortmannin significantly (P < 0.05) decreased A-II-induced phosphorylation of ERK5, but only moderately and not significantly inhibited A-II-stimulated ERK1/2 phosphorylation.
Insulin- and A-II-stimulated activation of ERK5 induces MEF2C phosphorylation
Because activated ERK5 has been shown to phosphorylate myocyte enhancer factor-2C (MEF2C) [18], we examined whether insulin and A-II stimulated the phosphorylation of MEF2C.
Insulin and A-II significantly (P < 0.05) stimulated the phosphorylation of MEF2C by 10 min of treatment (Supplemental Fig 10), continuing through 15 min and returned to basal levels by 30 min. Because insulin- and A-II-stimulated phosphorylation of MEF2C occurred before 15 min (the time at which ERK1/2 exhibited insulin- and A-II-stimulated phosphorylation) suggested that insulin and A-II stimulation of MEF2C was mediated by ERK5 and not ERK1/2.
Finally, we investigated whether the inhibitors that were responsible for changes in insulin- and A-II-stimulated phosphorylation of ERK5, would also affect insulin- and A-II-stimulated MEF2C phosphorylation. We treated RASMC without or with insulin and/or A-II in the absence or presence of PD or WT for designated times. Insulin and A-II stimulated increases in MEF2C phosphorylation (Fig 4) and exhibited an additive effect of MEF2C phosphorylation. PD effected an increase in insulin-stimulated MEF2C phosphorylation as compared to insulin alone, whereas it inhibited A-II-stimulated MEF2C phosphorylation as compared to A-II alone. WT had no effect on insulin-stimulated MEF2C phosphorylation, but significantly (P < 0.05) inhibited A-II-stimulated phosphorylation of MEF2C, reflecting the same kinetics of these two inhibitors on insulin- and A-II-stimulated activation of ERK5.
Figure 4. Effect of PD98059 and Wortmannin on acute treatment of insulin- and angiotensin-II-stimulated phosphorylation of MEF2C.
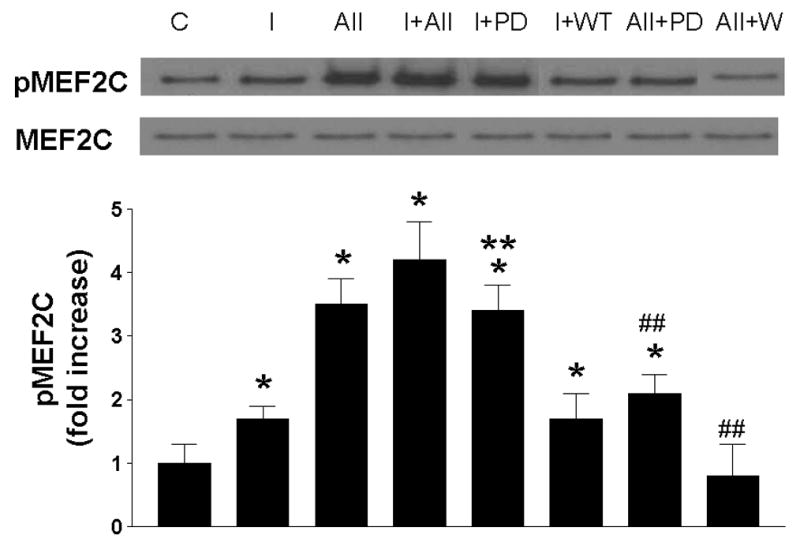
Cells were treated without or with insulin (10 nM) or A-II (100 nM) and in the absence or presence of PD98059 (10 μM) or Wortmannin (100 nM) for acute times. Results are expressed as fold increase above controls and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls. ##, P < 0.05 vs. A-II alone.
DISCUSSION
MAP kinases are highly conserved molecules in eukaryotic cells [13]. While ERK1/2 has been well characterized, ERK5 has not been fully investigated. Although ERK5 has been studied in nerve [16], skeletal muscle [7] and adipocytes [19], it has not been examined extensively in vascular smooth muscle cells.
The recently cloned ERK5 [20, 21], has been shown by many to be a critical mediator of mitogen and cytokine intracellular signaling [7, 22–24]. Although some recent studies have shown common pathways of ERK1/2 and ERK5, in many other reports different cell lines appear to have independent and distinct intracellular signaling pathways for ERK5 and ERK1/2 [24]. Even though ERK1/2 has been shown to be important in insulin-stimulated VSMC proliferation [25] and A-II-stimulated production of reactive oxygen species [26] and angiogenesis [13], it is still unclear as to the importance of ERK5 in the actions of insulin and A-II in VSMC.
In this report we have begun to tease apart the differential effects of insulin and A-II on the activation of ERK5 and ERK1/2 in RASMC. First, we showed that whether in acute or chronic presence of insulin or A-II, the time at which insulin and A-II stimulate the greatest magnitude of phosphorylation is different for ERK5 and ERK1/2. We also showed that the sensitivity of ERK1/2 and ERK5 to insulin and A-II is not only different with respect to insulin and A-II concentration, but also with respect to time. At acute times, insulin-stimulated phosphorylation of ERK5 was 15-fold more sensitive than that seen for ERK1/2. Although the sensitivity of ERK5 and ERK1/2 to A-II was equal, A-II-stimulated phosphorylation of ERK5 was 20-fold less sensitive than that observed for insulin-stimulated ERK5 phosphorylation.
We determined that insulin augmented A-II-stimulated ERK5 phosphorylation at acute and chronic times, but did not augment A-II-stimulated ERK1/2 phosphorylation at either length of treatment. Taken together, these data suggest that there are distinct upstream events that regulate insulin-augmented A-II-stimulated phosphorylation of these two MAP kinases. Furthermore, these data also indicate that because downstream events from the two receptors of insulin and A-II exhibit differential time-dependent effects on ERK5 and ERK1/2 phosphorylation and augmentation, downstream effectors of ERK5 and ERK1/2 may not only be dissimilar, but also mediate different intracellular pathways for insulin and A-II.
In order to begin characterizing the upstream events of signaling pathways of insulin- and A-II-induced phosphorylation of ERK5, we used the MEK1/2 inhibitor PD98059 and the PI-3 kinase inhibitor, Wortmannin. In this report, we demonstrated that PD inhibited only A-II-stimulated ERK5 at acute times, and had no effect on insulin- or A-II-stimulated ERK5 phosphorylation at chronic times. This suggested that (1) the insulin-stimulated Ras/ERK5 pathway is not mediated by ERK1/2 and (2) that only in acute stimulation of A-II does ERK1/2 signaling integrate with the A-II-stimulated Ras/ERK5 pathway, whereas in chronic stimulation, ERK1/2 has no influence on A-II-stimulated ERK5 activation. In comparison, PD inhibited insulin- and A-II-stimulated ERK1/2 at acute and chronic times, suggesting that ERK1/2 mediates both insulin and A-II activity in RASMC.
Interestingly, the presence of WT, which acts as a pharmacological model of insulin resistance, did not affect the basal magnitude of ERK5 or ERK1/2 phosphorylation at acute times, significantly (P < 0.05) induced increased basal levels of phosphorylated ERK5 and ERK1/2 at chronic times. These data suggested that chronic insulin resistance would upregulate both ERK1/2 and ERK5 phosphorylation, and thus lead to increased proliferative events.
Whereas WT had no influence on insulin-stimulated ERK5 phosphorylation at either acute or chronic times, WT inhibited A-II-stimulated ERK5 phosphorylation at both acute and chronic treatments as compared to A-II alone. In contrast, WT did not inhibit A-II-induced ERK1/2 phosphorylation at any time. Ushio-Fukai et al. [10] have shown that WT blocked A-II-induced stimulation of Akt/PKB signaling in VSMC, and thus, taken together, their results and ours suggest that A-II intracellular activation of ERK5 may be mediated by the PI-3 kinase/Akt pathway, whereas A-II activation of ERK1/2 is not.
Finally, we demonstrated that insulin- and A-II-stimulated phosphorylation of ERK5 resulted in its activation and subsequent phosphorylation of its substrate, MEF2C. Moreover, although both insulin and A-II induced the phosphorylation of MEF2C, the intracellular routes by which this was accomplished were different. Thus, it appears that insulin-stimulated phosphorylation of ERK5 and MEF2C is mediated via an MEK1/2-independent pathway (i.e., a MEK5-ERK5 pathway) whereas the A-II-stimulated phosphorylation and activation of ERK5 and MEF2C seem to be mediated by the PI-3 kinase pathway with some modulation from the Ras-MEK1/2 pathway.
This study demonstrates that insulin and A-II regulate the phosphorylation and activation of ERK5, resulting in the subsequent phosphorylation of MEF2C in RASMC. Moreover, the results of this study illustrate that intermediate intracellular mediators of insulin and A-II intracellular signal pathways, elicit differential effects on insulin- and A-II-stimulated ERK5 and ERK1/2 phosphorylation.
Insulin and A-II action are mediated by several kinase pathways such as ERK1/2 and PI-3 kinase. This study demonstrates that ERK5 is also a downstream mediator of insulin and A-II intracellular signaling in RASMC and is regulated by discrete kinase-dependent pathways. Because atherosclerosis is a pathology that includes the dysregulation of vascular tissue signaling concomitant with proliferation of vascular smooth muscle cells, and that insulin and A-II appear to be involved in the pathogenesis of atherosclerosis, ERK5 may be a molecular target for pharmacologic interventions and strategies for clinical and bedside amelioration of this devastating disease. Future studies are needed to determine the role of ERK5 in vascular smooth muscle cell proliferation and physiology and its possible involvement in the pathogenesis of atherosclerosis.
Supplementary Material
Supplemental Figure 1 – Acute treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 at different times in RASMC. Sub-confluent cells in serum-reduced medium were treated acutely (0 – 60 min) without or with A-II (100 nM). Phosphorylated and total proteins were determined by Western blot analysis. Blots are representative of three independent experiments. Results are expressed as fold increase above controls of phosphorylated ERK5 (■) and ERK1/2 (●) and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Supplemental Figure 2 – Chronic treatement of insulin stimulates the phosphorylation of ERK5 and ERK1/2 at different times in RASMC. Sub-confluent cells in serum-reduced medium were treated chronically (0 – 24 h) without or with insulin (10 nM). Phosphorylated and total proteins were determined by Western blot analysis. Blots are representative of three independent experiments. Results are expressed as fold increase above controls of phosphorylated ERK5 (closed bars) and ERK1/2 (open bars) and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Supplemental Figure 3 – Chronic treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 at different times in RASMC. Sub-confluent cells in serum-reduced medium were treated chronically (0 – 24 h) without or with A-II (100 nM). Results are expressed as fold increase above controls of phosphorylated ERK5 (closed bars) and ERK1/2 (open bars) relative to controls and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Supplemental Figure 4 – Acute treatment of insulin stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of insulin. Results are expressed as fold increase of above controls of phosphorylated ERK5 (■) and ERK1/2 (○) and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 5 – Acute treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of A-II. Results are expressed as fold increase of phosphorylated ERK5 (■) and ERK1/2 (○) above controls and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 6 – Chronic treatment of insulin stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of insulin. Results are expressed as fold increase of phosphorylated ERK5 (■) and ERK1/2 (○) relative to controls and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 7 – Chronic treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of A-II. Results are expressed as fold increase of phosphorylated ERK5 (■) and ERK1/2 (○) relative to controls and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 8 – Acute treatment of insulin augments A-II-stimulated phosphorylation of ERK5, but not ERK1/2 in RASMC. Cells were treated without or with insulin (0.3 nM) and/or A-II (2 nM) for acute times. Results are expressed as fold increase above controls of phosphorylated ERK5 (closed bars) and ERK1/2 (open bars) and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls. **, P < 0.05 vs. A-II alone.
Supplemental Figure 9 – Effect of PD98059 and Wortmannin on chronic treatment of insulin- and angiotensin-II-stimulated phosphorylation of ERK5, ERK1/2 and MEF2C. Cells were treated without or with insulin (10 nM) or A-II (100 nM) and in the absence or presence of PD98059 (10 μM) or Wortmannin (100 nM) for chronic times. Results are expressed as fold increase above controls and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls. #, P < 0.05 vs. insulin or A-II alone. §, P < 0.05 vs. insulin alone.
Supplemental Figure 10 – Acute treatment of insulin and angiotensin-II stimulates the phosphorylation of MEF2C in RASMC. Sub-confluent cells in serum-reduced medium were treated acutely without or with insulin (10 nM, open bars) or A-II (100 nM, closed bars). Results are expressed as fold increase above controls of phosphorylated MEF2C and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Acknowledgments
We would like to thank Hillary Akana, Smith Leser, Lisa Bolan and Katrina Smith for their time in reviewing this manuscript. This work was supported in part by the Research Service of the Department of Veterans Affairs (to M.L.G.), in which Dr. Goalstone is a recipient of a VA Career Development Award; by the American Heart Assoication (0450016Z)(M.L.G.); and the NIH (R01 DK068326)(M.L.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 2.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 3.Marinissen MJ, Chiariello M, Pallante M, Gutkind JS. A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5. Mol Cell Biol. 1999;19:4289–4301. doi: 10.1128/mcb.19.6.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, Segal RA. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci. 2001;4:981–988. doi: 10.1038/nn720. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Cavanaugh JE, Wang Y, Sakagami H, Mao Z, Xia Z. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc Natl Acad Sci U S A. 2003;100:8532–8537. doi: 10.1073/pnas.1332804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sohn SJ, Sarvis BK, Cado D, Winoto A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J Biol Chem. 2002;277:43344–43351. doi: 10.1074/jbc.M207573200. [DOI] [PubMed] [Google Scholar]
- 7.Dinev D, Jordan BW, Neufeld B, Lee JD, Lindemann D, Rapp UR, Ludwig S. Extracellular signal regulated kinase 5 (ERK5) is required for the differentiation of muscle cells. EMBO Rep. 2001;2:829–834. doi: 10.1093/embo-reports/kve177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao M, Liu Y, Bao M, Kato Y, Han J, Eaton JW. Vascular smooth muscle cell proliferation requires both p38 and BMK1 MAP kinases. Arch Bioch Biophys. 2002;400:199–207. doi: 10.1016/S0003-9861(02)00028-0. [DOI] [PubMed] [Google Scholar]
- 9.Wang CC, Gurevich I, Draznin B. Insulin affects vascular smooth muscle cell phenotype and migration via distinct signaling pathways. Diabetes. 2003;52:2562–2569. doi: 10.2337/diabetes.52.10.2562. [DOI] [PubMed] [Google Scholar]
- 10.Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- 11.Leitner JW, Kline T, Carel K, Goalstone M, Draznin B. Hyperinsulinemia potentiates activation of p21Ras by growth factors. Endocrinology. 1997;138:2211–2214. doi: 10.1210/endo.138.5.5240. [DOI] [PubMed] [Google Scholar]
- 12.Ruiz-Ortega M, Lorenzo O, Ruperez M, Konig S, Wittig B, Egido J. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: molecular mechanisms. Circ Res. 2000;86:1266–1272. doi: 10.1161/01.res.86.12.1266. [DOI] [PubMed] [Google Scholar]
- 13.Aoki H, Richmond M, Izumo S, Sadoshima J. Specific role of the extracellular signal-regulated kinase pathway in angiotensin II-induced cardiac hypertrophy in vitro. Biochem J. 2000;347(Pt 1):275–284. [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaguchi K, Higashiura K, Ura N, Murakami H, Hyakukoku M, Furuhashi M, Shimamoto K. The effect of tumor necrosis factor-alpha on tissue specificity and selectivity to insulin signaling. Hypertens Res. 2003;26:389–396. doi: 10.1291/hypres.26.389. [DOI] [PubMed] [Google Scholar]
- 15.Goalstone ML, Natarajan R, Standley PR, Walsh MF, Leitner JW, Carel K, Scott S, Nadler J, Sowers JH, Draznin B. Insulin potentiates platelet-derived growth factor action in vascular smooth muscle cells. Endocrinology. 1998;139:4067–4072. doi: 10.1210/endo.139.10.6270. [DOI] [PubMed] [Google Scholar]
- 16.Cavanaugh JE, Ham J, Hetman M, Poser S, Yan C, Xia Z. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci. 2001;21:434–443. doi: 10.1523/JNEUROSCI.21-02-00434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eringa EC, Stehouwer CD, van Nieuw Amerongen GP, Ouwehand L, Westerhof N, Sipkema P. Vasoconstrictor effects of insulin in skeletal muscle arterioles are mediated by ERK1/2 activation in endothelium. Am J Physiol Heart Circ Physiol. 2004;287:H2043–2048. doi: 10.1152/ajpheart.00067.2004. [DOI] [PubMed] [Google Scholar]
- 18.Yan C, Luo H, Lee JD, Abe J, Berk BC. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J Biol Chem. 2001;276:10870–10878. doi: 10.1074/jbc.M009286200. [DOI] [PubMed] [Google Scholar]
- 19.Sharma G, Goalstone ML. Dominant negative FTase (DNFTalpha) inhibits ERK5, MEF2C and CREB activation in adipogenesis. Mol Cell Endocrinol. 2005;245:93–104. doi: 10.1016/j.mce.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 20.Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 21.Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 22.Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999;274:26563–26571. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- 23.Mulloy R, Salinas S, Philips A, Hipskind RA. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 2003;22:5387–5398. doi: 10.1038/sj.onc.1206839. [DOI] [PubMed] [Google Scholar]
- 24.Pearson G, English JM, White MA, Cobb MH. ERK5 and ERK2 cooperate to regulate NF-kappaB and cell transformation. J Biol Chem. 2001;276:7927–7931. doi: 10.1074/jbc.M009764200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solomon CS, Goalstone ML. Dominant negative farnesyltransferase α-subunit inhibits insulin mitogenic effects. Biochem Biophys Res Comm. 2001;285:161–166. doi: 10.1006/bbrc.2001.5142. [DOI] [PubMed] [Google Scholar]
- 26.Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 – Acute treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 at different times in RASMC. Sub-confluent cells in serum-reduced medium were treated acutely (0 – 60 min) without or with A-II (100 nM). Phosphorylated and total proteins were determined by Western blot analysis. Blots are representative of three independent experiments. Results are expressed as fold increase above controls of phosphorylated ERK5 (■) and ERK1/2 (●) and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Supplemental Figure 2 – Chronic treatement of insulin stimulates the phosphorylation of ERK5 and ERK1/2 at different times in RASMC. Sub-confluent cells in serum-reduced medium were treated chronically (0 – 24 h) without or with insulin (10 nM). Phosphorylated and total proteins were determined by Western blot analysis. Blots are representative of three independent experiments. Results are expressed as fold increase above controls of phosphorylated ERK5 (closed bars) and ERK1/2 (open bars) and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Supplemental Figure 3 – Chronic treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 at different times in RASMC. Sub-confluent cells in serum-reduced medium were treated chronically (0 – 24 h) without or with A-II (100 nM). Results are expressed as fold increase above controls of phosphorylated ERK5 (closed bars) and ERK1/2 (open bars) relative to controls and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.
Supplemental Figure 4 – Acute treatment of insulin stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of insulin. Results are expressed as fold increase of above controls of phosphorylated ERK5 (■) and ERK1/2 (○) and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 5 – Acute treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of A-II. Results are expressed as fold increase of phosphorylated ERK5 (■) and ERK1/2 (○) above controls and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 6 – Chronic treatment of insulin stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of insulin. Results are expressed as fold increase of phosphorylated ERK5 (■) and ERK1/2 (○) relative to controls and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 7 – Chronic treatment of angiotensin-II stimulates the phosphorylation of ERK5 and ERK1/2 in a dose- and time-dependent manner in RASMC. Cells were treated with designated concentrations of A-II. Results are expressed as fold increase of phosphorylated ERK5 (■) and ERK1/2 (○) relative to controls and represent the mean ± SEM of 4 separate experiments.
Supplemental Figure 8 – Acute treatment of insulin augments A-II-stimulated phosphorylation of ERK5, but not ERK1/2 in RASMC. Cells were treated without or with insulin (0.3 nM) and/or A-II (2 nM) for acute times. Results are expressed as fold increase above controls of phosphorylated ERK5 (closed bars) and ERK1/2 (open bars) and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls. **, P < 0.05 vs. A-II alone.
Supplemental Figure 9 – Effect of PD98059 and Wortmannin on chronic treatment of insulin- and angiotensin-II-stimulated phosphorylation of ERK5, ERK1/2 and MEF2C. Cells were treated without or with insulin (10 nM) or A-II (100 nM) and in the absence or presence of PD98059 (10 μM) or Wortmannin (100 nM) for chronic times. Results are expressed as fold increase above controls and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls. #, P < 0.05 vs. insulin or A-II alone. §, P < 0.05 vs. insulin alone.
Supplemental Figure 10 – Acute treatment of insulin and angiotensin-II stimulates the phosphorylation of MEF2C in RASMC. Sub-confluent cells in serum-reduced medium were treated acutely without or with insulin (10 nM, open bars) or A-II (100 nM, closed bars). Results are expressed as fold increase above controls of phosphorylated MEF2C and represent the mean ± SEM of 3 separate experiments. *, P < 0.05 vs. respective controls.