

Abstract
Acetylation of histone tails by histone acetyltransferase (HAT) enzymes is a key post-translational modification of histones associated with transcriptionally active genes. Acetylation of the physiological nucleosome substrate is performed in cells by megadalton complexes such as SAGA and NuA4. To understand how HAT enzymes specifically recognize their nucleosome and not just histone tail substrates, we have identified the catalytic SAGA and NuA4 subcomplexes sufficient to act on nucleosomes. We describe here expression and purification procedures to prepare recombinant yeast Ada2/Ada3/Gcn5 subcomplex of SAGA which acetylates histones H3 and H2B on nucleosomes, and the Piccolo NuA4 complex which acetylates histones H4 and H2A on nucleosomes. We demonstrate an unexpected benefit of using the BL21-CodonPlus strain to enhance the purity of metal affinity purified Ada2/Ada3/Gcn5 complex. We also identify E. coli EF-Tu as a contaminant that copurifies with both complexes over multiple chromatographic steps and use of hydrophobic interaction chromatography to remove the contaminant from the Piccolo NuA4 complex. The methods described here will be useful for studies into the molecular mechanism of these enzymes and for preparing the enzymes as reagents to study the interplay of nucleosome acetylation with other chromatin modification and remodeling enzymes.
Introduction
The most fundamental repeating unit of chromatin, the nucleosome core particle, contains 147 bp of DNA wrapped around a central histone octamer core of two copies each of histones H2A, H2B, H3 and H4 [1]. The nucleosome is both a packaging arrangement to compact the roughly 2 meters of DNA that must fit into a 10 μm diameter nucleus of a human cell, and also a dynamic structure involved in multiple cellular processes. For example, the nucleosome can be modified post-translationally by chromatin modification enzymes and it can be restructured by chromatin remodeling enzymes [1]. Post-translational modifications to the histones include acetylation, phosphorylation, methylation, ubiquitinylation, sumoylation and ADP-ribosylation, usually on the unstructured histone tails. Histone acetylation is perhaps the best studied modification and has long been associated with gene activation. The molecular basis for this association became apparent when it was discovered that the transcriptional coactivator Gcn5 possessed histone acetyltransferase activity and that transcriptional activity of Gcn5-dependent genes in a yeast cell correlated tightly with Gcn5's HAT activity [2–4].
Although individual HATs such as Gcn5 and Esa1 possess histone acetyltransferase activity, they do not acetylate their physiological nucleosome substrate efficiently or at all. In contrast, their SAGA and NuA4 parent complexes acetylate both histones and nucleosomes [5, 6]. Since Ada2, Ada3 and Gcn5 form a triple complex [7, 8], we hypothesized that this Ada2/Ada3/Gcn5 complex would acetylate nucleosomes. Our experiments on the Ada2/Ada3/Gcn5 complex established that this complex is sufficient for robust histone and nucleosomal HAT activity and is specific for the same histones and even the same H3 lysine residues as SAGA [9]. This suggests that the Ada2/Ada3/Gcn5 subcomplex recapitulates SAGA's HAT function of acetylating nucleosomes. However, in contrast to SAGA which is apparently directed to the promoter through its Tra1 subunit, the Ada2/Ada3/Gcn5 complex (which may be identical to the HAT-A2 complex [10, 11]) may act as a global, untargeted nucleosome acetyltransferase in the cell [12]. We have similarly identified the Piccolo NuA4 complex of Epl1, Yng2 and Esa1 as the catalytic core of the megadalton NuA4 complex [13, 14]. Interestingly, Piccolo NuA4 possesses even greater HAT activity on nucleosomes than the whole NuA4 complex [14].
To facilitate biochemical and biophysical studies of these nucleosome acetylation complexes, we have developed polycistronic expression systems which permit in vivo reconstitution of recombinant complexes in E. coli [15, 16]. We have also supplemented those primary description of the expression systems with more practical details on how to create coexpression plasmids and pitfalls to watch out for (Selleck and Tan, Methods, in press). Here we detail the expression and purification of the yeast Ada2/Ada3/Gcn5 complex which acetylates nucleosomal H3 and H2B histone tails, and the yeast Piccolo NuA4 complex which acetylates nucleosomal H4 and H2A histone tails. We highlight procedural details that we have found to affect the ease of performing the purification or the yield and purity of the preparation. Besides their use in experiments to intrinsically study how chromatin enzymes recognize and act on a nucleosome substrate, these two complexes are valuable reagents for investigating the complicated interplay between different histone modifications on a nucleosome substrate. For example, one can use these highly active HAT enzymes to acetylate individual nucleosomes or nucleosomal arrays to study how acetylation of particular histone tails affects other histone modification enzymes or chromatin remodeling complexes.
Methods
Expression and purification of recombinant yeast Ada2/Ada3/Gcn5 complex
1. Transform yeast Ada2/Ada3/Gcn5 polycistronic expression vector into E. coli
Since our polycistronic vector to express the yeast Ada2/Ada3/Gcn5 complex is based on the T7-promoter, any E. coli strain that provides an inducible T7 RNA polymerase can be used. We have compared the BL21(DE3)pLysS strain which allows tighter regulation of T7 RNA polymerase activity by low level expression of T7 lysozyme which inhibits T7 RNA polymerase [17], and the BL21-CodonPlus(DE3)-RIL strain (Stratagene) which provides extra copies of the otherwise rare tRNAs for particular Arg, Ile and Leu codons. We found that although the expression levels of the Ada3, Ada2, and Gcn5 proteins were unchanged between the two strains (data not shown), the purity of the complex after Talon metal affinity chromatography was substantially better when the proteins are expressed in BL21-CodonPlus(DE3)-RIL cells (Fig. 1). The lower molecular weight contaminants in either strain do not appear to be prematurely terminated polypeptides due to codon usage issues since polyclonal antibodies against each of the three proteins did not cross-react with these lower molecular bands in Western blots (data not shown). The exception is a truncated Gcn5 band of about 41 kDa which occurs in both BL21(DE3)pLysS and BL21-CodonPlus(DE3)-RIL cells. Thus, although the basis for the improved purity after metal affinity chromatography is unclear, use of BL21-CodonPlus(DE3)-RIL cells is preferred over BL21(DE3)pLysS cells for expression of the yeast Ada2/Ada3/Gcn5 complex.
Figure 1.
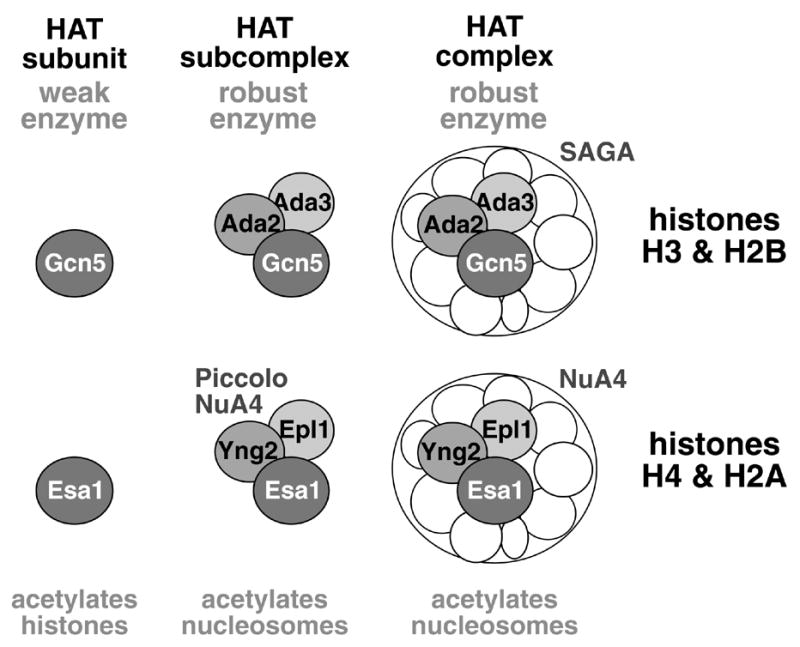
The Ada2/Ada3/Gcn5 and Piccolo NuA4 complexes are functionally sufficient for the nucleosomal HAT activity of the megadalton SAGA and NuA4 complexes. Gcn5 is the catalytic HAT subunit of SAGA, but acetylates only histone tails and weakly at that, whereas the Ada2/Ada3/Gcn5 is the sufficient subcomplex with similar robust HAT activity and histone H3 and H2B specificity for nucleosomal histones as the full megadalton SAGA complex. Similarly, Esa1 is the catalytic subunit of the NuA4 complex with weak activity on free and nucleosomal histones, but the Piccolo NuA4 subcomplex comprised of the Epl1, Yng2 and Esa1 subunits acetylates nucleosomal histones with the same preference for histones H4 and H2A as the full NuA4 complex.
2. Express yeast Ada2/Ada3/Gcn5 in E. coli
We inoculate three to five colonies from a fresh transformation plate into 100 ml 2xTY media (1.6% bacto-tryptone, 1.0% yeast extract, 0.5% NaCl) containing 50 μg/ml ampicillin and 25 μg/ml chloramphenicol, and then grow the culture at 37°C until the OD600 is between 0.1 and 0.2. For small scale expression, the culture is transferred to an 18°C incubator for continued growth until the OD600 is between 0.4 and 0.7 at which time IPTG is added to a final concentration of 0.2 mM to induce expression of the Ada2/Ada3/Gcn5 complex. For larger scale expression, 3–5 ml of the exponential phase culture is used to inoculate 12 flasks of 500 ml 2xTY + 50 μg/ml ampicillin + 25 μg/ml chloramphenicol and the culture grown at 37°C. When the OD600 of these larger cultures is between 0.1 and 0.2, the incubator temperature is lowered to 18°C and the cultures incubated at this temperature until the OD600 reaches 0.4 to 0.7 and IPTG then added to a final concentration of 0.2 mM. We harvest the induced culture after overnight incubation (10–16 hours) by centrifugation, resuspending the pellet from 100 ml cells in 20 ml P100 buffer or from 6 liters cells in 150 ml P100 buffer (P100 = 50 mM sodium phosphate pH 7.0, 100 mM NaCl, 1 mM benzamidine, 5 mM 2-mercaptoethanol) and storing the suspension at −20 or −80°C.
3. Prepare crude extract
We prepare E. coli crude extracts by freeze-thawing the cell suspension, and then sonicating the extract (2 rounds of 10 seconds, 50% full power, 50% duty cycle for smaller scale purification such as for 50 ml of cells stored in 10 ml of buffer, or 3 rounds of 10 seconds, 70% full power, 50% duty cycle for preparative purification from 6 liters of cells stored in 150 ml buffer) using a S-450D Branson cell disrupter, but other extraction methods based on detergents or high pressures should work as well. We clear the extract by centrifugation at 8–15,000 g to remove particulate matter before following chromatography steps.
4. Purify complex by Talon metal affinity chromatography
We have tagged the Ada3 subunit of the yeast Ada2/Ada3/Gcn5 complex with the hexahistidine (HIS) peptide because the HIS tag allows for efficient purification of the complex from the crude extract [18], and because tagging the Ada3 subunit allows us to separate the Ada2/Ada3/Gcn5 complex from the Ada2/Gcn5 binary complex which has similar specific HAT activity on naked histones but much less activity on nucleosomes (Ada2 can associate with either or both of Ada3 and Gcn5, but Ada3 does not associate with Gcn5 on its own [7, 8]). However, it is also possible to purify the yeast Ada2/Ada3/Gcn5 complex without using a HIS tag [9].
Ada2/Ada3/Gcn5 complexes in cell extracts prepared from 50 – 100 ml of culture can be purified by incubating the cleared crude extract with 0.5 ml of Talon resin (Clontech) pre-equilibrated in P100 buffer for 15–30 minutes at 4°C, washing the resin twice with 10 ml P100 buffer, transferring the washed resin to a Bio-Spin column (BioRad) and then eluting the complex with 4 x 0.5 ml P100 + 100 mM imidazole. If 0.5 ml fractions are collected, the majority of the eluted protein is usually found in fractions 2 and 3.
For purifying Ada2/Ada3/Gcn5 complexes from 6 liters of culture, we apply ~150 ml of cleared crude extract to 20 ml of Talon resin preequilibrated with P100 buffer, wash the column with 3–5 column volumes of P100 buffer followed by 3 column volumes of P100 + 10 mM imidazole and the complex eluted with 3 column volumes of P100 + 100 mM imidazole. Peak elution fractions as judged by SDS-PAGE are then pooled and dialyzed against T100 buffer (10 mm Tris-Cl pH 8.0, 100 mM NaCl, 5 mM 2-mercaptoethanol). It is not uncommon to observe significant precipitation during this dialysis, but the precipitant does not usually contain the Ada2/Ada3/Gcn5 complex.
The complex is usually pure enough after Talon purification, particularly in lacking contaminating protein acetylases or deacetylases, to use as an enzyme to acetylate histones or nucleosomes. Although we have not tested other metal affinity resins such as nickel-based resins, we suspect that such resins will be equally effective at purifying the HIS-tagged complex. If purer complex is desired, the sample can be fractionated further by anion- and cation-exchange chromatography as described below.
5. Purify the complex by anion-exchange chromatography
The Ada2/Ada3/Gcn5 complex is typically 50–75% pure after Talon metal affinity chromatography, with smaller molecular proteins or some degradation products contaminating the preparation. Many of the contaminating proteins can be eliminated by anion-exchange chromatography using high performance resins. We apply the Ada2/Ada3/Gcn5 Talon pool dialyzed against 20 mM Tris-Cl pH 8.0, 100 mM NaCl, 0.1 mM EDTA, 10 mM 2-mercaptoethanol to a 10 ml SourceQ column (GE Healthcare) equilibrated in the same buffer and then elute with a linear gradient from 100–500 mM NaCl. It is likely that similar loading and elution conditions can be used with other anion-exchange resins such as MonoQ (GE Healthcare).
6. Purify the complex by cation-exchange chromatography
Since the Ada2/Ada3/Gcn5 complex is stable to both anion and cation-exchange chromatography, the SourceQ pooled fractions can be purified further by SourceS cation-exchange chromatography (GE Healthcare), although with only an incremental improvement in purity. For SourceS purification, the SourceQ pool is dialyzed against 10 mM HEPES pH 7.5, 20 mM NaCl, 0.1 mM EDTA, 10 mM 2-mercaptoethanol before loading onto a 10 ml SourceS column equilibrated in the same buffer and eluted with a 20–500 mM NaCl gradient. As with the SourceQ column, other cation-exchange resins such as MonoS will likely produce similar results. The preparation is nearly pure after SourceS chromatography, with a noticeable contaminant of about 43 kDa.
To determine if the 43 kDa contaminant was a degradation product of the yeast Ada2/Ada3/Gcn5 complex or an E. coli contaminant, we used MALDI mass spectrometry to identify the polypeptide. Since a polypeptide of the same size also contaminates our Piccolo NuA4 preparations (see below), we suspected that the 43 kDa polypeptide was an E. coli contaminant. The MALDI mass spectrometry experiment confirmed this hypothesis and identified the 43 kDa polypeptide as the E. coli translation factor EF-Tu, which has a theoretical molecular weight of 43.2 kDa.
Typical yields of the yeast Ada2/Ada3/Gcn5 complex are approximately 0.5 to 0.8 mg per liter of culture.
Expression and purification of recombinant yeast Piccolo NuA4 complex
1. Transform yeast Piccolo NuA4 complex polycistronic expression vector into E. coli
Similar expression considerations for the yeast Ada2/Ada3/Gcn5 complex apply to the Piccolo NuA4 complex, although the benefit of using BL21-CodonPlus(DE3)-RIL cells are not observed for the Piccolo NuA4 complex. Therefore we use BL21(DE3)pLysS cells for expressing the Piccolo NuA4 complex in E. coli.
2. Express yeast Piccolo NuA4 complex in E. coli
We inoculate three to five colonies from a fresh transformation plate into 100 ml 2xTY media (1.6% bacto-tryptone, 1.0% yeast extract, 0.5% NaCl) containing 50 μg/ml ampicillin and 25 μg/ml chloramphenicol, grow the culture at 37°C. For small scale expression, the culture is grown at 37°C until the OD600 is between 0.5 and 0.7 at which time IPTG is added to a final concentration of 0.2 mM to induce expression of the Piccolo NuA4 complex. For larger scale expression, 3–5 ml of the exponential phase culture at OD600 ~ 0.1 is used to inoculate 12 flasks of 500 ml 2xTY + ampicillin + chloramphenicol and these larger cultures grown at 37°C. When the OD600 reaches 0.5 to 0.7, IPTG is added to a final concentration of 0.2 mM and the cultures allow to incubate at 37°C for 3–4 hours. We harvest the induced culture by centrifugation, resuspend the centrifuge pellet in P300 buffer (50 mM sodium phosphate pH 7.0, 300 mM NaCl, 1 mM benzamidine, 5 mM 2-mercaptoethanol) and store the suspended cells at −20 or −80°C.
3. Purify complex by Talon metal affinity chromatography
Our Piccolo NuA4 expression constructs are hexahistidine-tagged on one subunit, typically the Esa1 subunit. Preparation of the crude extract and purification of the HIS-tagged Piccolo NuA4 complex by Talon metal affinity chromatography is performed as described above for the yeast Ada2/Ada3/Gcn5 complex. We do use a higher salt for the metal affinity purification of Piccolo NuA4 (300 mM instead of 100 mM NaCl used for the Ada2/Ada3/Gcn5 complex) because the higher salt improves binding of the Piccolo NuA4 complex to the Talon resin. The Piccolo NuA4 complex is pure enough after the single affinity step for most enzyme assays.
4. Purify the complex by anion-exchange chromatography
Like the Ada2/Ada3/Gcn5 complex, the Piccolo NuA4 complex is typically 50–75% pure after Talon metal affinity chromatography, with smaller molecular proteins or degradation products contaminating the preparation. Furthermore, if the Esa1 subunit is HIS-tagged, it is possible that the Piccolo NuA4 prep will be contaminated with some Esa1 polypeptide that is not associated with the Epl1 and Yng2 subunits of Piccolo NuA4. This could complicate quantitative measurements of the nucleosomal HAT activity of Piccolo NuA4 relative to its naked histone HAT activity since both Esa1 and Piccolo acetylate histones but Piccolo possesses a much higher specific activity on nucleosomes than Esa1 does [13, 14]. Therefore, it may be advisable to further purify the Piccolo NuA4 complex by additional chromatography steps. Fortunately, this is relatively simple to perform since the Piccolo NuA4 complex is stable to anion- and cation-exchange chromatography as well as hydrophobic interaction chromatography.
Anion-exchange chromatography is efficient at removing many contaminants in the Talon purified Piccolo NuA4 pool. After dialyzing the Talon pool against 20 mM Tris-Cl pH 8.0, 150 mM NaCl, 0.1 mM EDTA, 10 mM 2-mercaptoethanol, the Piccolo NuA4 complex can be purified over a 10 ml SourceQ column in the same buffer, using a 100 to 500 mM NaCl gradient to elute the complex.
5. Purify the complex by cation-exchange chromatography
Further preparative purification of the Piccolo NuA4 can be achieved using a 10 ml SourceS cation-exchange column in 10 mM HEPES pH 7.6, 0.1 mM EDTA, 10 mM 2-mercaptoethanol and a linear gradient from 50 to 500 mM NaCl. The preparation is nearly pure after SourceS chromatography, except for a 43 kDa contaminant with the same electrophoretic mobility as E. coli EF-Tu that copurifies with the recombinant Ada2/Ada3/Gcn5 complex. Therefore, although we have formally identified EF-Tu as the 43 kDa contaminant for the Ada2/Ada3/Gcn5 complex, we believe the 43 kDa contaminant in the Piccolo NuA4 is also EF-Tu.
6. Purify the complex by hydrophobic interaction chromatography
We use hydrophobic chromatography over the Source ISO resin (GE Healthcare) to remove the 43 kDa EF-Tu polypeptide from the Piccolo NuA4 complex. After adding ammonium sulfate to a final concentration of 1500 mM to the SourceS pool, the sample is centrifuged and the supernatant loaded onto the Source ISO column equilibrated in 20 mM Tris-Cl pH 8.0, 1500 mM ammonium sulfate, 0.1 mM EDTA, 10 mM 2-mercaptoethanol. The Piccolo NuA4 is then eluted with a gradient from 1500 mM to 0 mM ammonium sulfate. Unfortunately, we were unable to use this procedure to purify the EF-Tu contaminant from the Ada2/Ada3/Gcn5 complex because that complex dissociated during our hydrophobic chromatography experiments.
Since the interaction of EF-Tu to Ada2/Ada3/Gcn5 and Piccolo NuA4 is stable to the 300–500 mM NaCl conditions experienced during the metal affinity and ion-exchange chromatography, hydrophobic and not ionic interactions are likely to dominate the binding of EF-Tu to the two HAT subcomplexes. The utility of Source ISO hydrophobic interaction chromatography to purify EF-Tu from Piccolo NuA4 is consistent with this explanation. It is also possible that EF-Tu's reported chaperone-like properties [19] may also play a role in the binding of EF-Tu to either or both of Ada2/Ada3/Gcn5 and Piccolo NuA4. Given the wide abundance of EF-Tu in E. coli and its ability to bind tightly to different HAT subcomplexes, we suspect that EF-Tu could contaminate other recombinant proteins or complexes, particularly subcomplexes which might expose hydrophobic regions which would otherwise interact with other components of the native parent complex.
We usually obtain approximately 1 to 3 mg of the Piccolo NuA4 complex per liter of culture.
Troubleshooting
1. No or very low expression of complex subunit(s) detected
Excluding trivial reasons, the most likely explanation for low expression of the recombinant complex is that the culture has lost or rearranged the overexpression plasmid due to uninduced expression of the recombinant proteins. This is, of course, a problem common to almost all expression of heterologous proteins in E. coli and not one unique to expression of HAT complexes. The fundamental problem is that even stringently regulated bacterial expression systems are not completely turned off in the absence of the inducing agent and even small amounts of expressed heterologous protein could be toxic or at least slow down the growth of cells bearing the expression plasmid. Thus, there will be a selective pressure to lose or rearrange the plasmid to reduce or eliminate expression of the heterologous protein. In general, when using traditional liquid media such as 2xTY or LB and their solid media equivalents, one should use freshly transformed cells for protein expression. In addition, cultures should not be allowed to grow to saturation.
An exciting recent development from F. William Studier of the Brookhaven National Laboratory is the formulation of growth media that greatly reduces non-induced expression of IPTG-inducible promoters and which can also allow for auto-induction when the cells reach saturation [20]. This media, available commercially from Novagen under the trademark Overnight Express, can alleviate problems due to noninduced expression as well as increase yields per liter of media because the cells are grown to higher density before induction.
2. Incomplete lysis of cells
Cell lysates can be prepared from host strains that contain the pLysS plasmid simply by freeze-thawing the cells. Freeze-thawing damages the inner cell membrane, which exposes and therefore makes susceptible the outer cell membrane to the T7 lysozyme enzyme expressed from the pLysS plasmid. Freeze-thaw damage to the cell membrane is most effective if the cells are frozen in an aqueous solution. Thus, we resuspend the harvested cells in buffer before storing at −20°C or −80°C. Where possible, flash freeze the resuspended cells in liquid nitrogen before storage to avoid settling of the cells when placed in the appropriate freezer.
3. A significant fraction of the expressed proteins is not soluble and is found in the lysis pellet
Expression of the yeast Ada2/Ada3/Gcn5 complex is performed at 18°C to increase the solubility of the expressed complex. While we typically observe less than 20% of the yeast Ada2/Ada3/Gcn5 complex in the lysis pellet, we have also seen up to 50% insoluble complex in some cases. One potential problem to avoid is inducing cells for Ada2/Ada3/Gcn5 expression too soon after dropping the growth temperature from 37°C to 18°C. We allow the cells to grow at 18°C for at least one doubling time (1.5 to 2 h) to make sure the cells have adapted to the lower temperature before adding IPTG. A second potential problem is excessive sonication used to disrupt the cells. Attempts to maximize cell lysis by longer sonication times could increase the amount of insoluble protein by denaturating the protein..
Conclusion
The ability to express and purify recombinant HAT subcomplexes of the megadalton yeast SAGA and NuA4 complexes in milligram quantities greatly facilitates biochemical and biophysical studies into the molecular mechanism of nucleosome acetylation. We have detailed here expression and purification procedures to produce the yeast Ada2/Ada3/Gcn5 and the Piccolo NuA4 (Epl1/Yng2/Esa1) complexes which are highly active in histone H3/H2B and H4/H2A acetylation respectively. In contrast to Gcn5 and Esa1 proteins which acetylate histones and poorly at that, the Ada2/Ada3/Gcn5 and the Piccolo NuA4 complex are robust enzymes which acetylate histones, and importantly, nucleosomes in both mononucleosomes and in nucleosomal arrays.
The purification procedures for both Ada2/Ada3/Gcn5 and Piccolo NuA4 utilize affinity chromatography for rapid and efficient capture of the recombinant complexes directly from a crude E. coli extract. Subsequent conventional chromatography steps allow inexpensive, scalable purification of both complexes to near homogeneity. We do not remove the hexahistidine tag for these HAT enzymes because the presence of HIS tag does not affect the integrity or the activity of the HAT complexes (our unpublished data). In addition to detailing the expression and purification procedures for the two HAT complexes, we have also described an unexpected purification benefit of using the BL21-CodonPlus(DE3)-RIL strain for expressing the yeast Ada2/Ada3/Gcn5 complex. We observed that Talon metal affinity purified Ada2/Ada3/Gcn5 complex was much purer when isolated from BL21-CodonPlus(DE3)-RIL cells compared to the equivalent BL21 host strain. Although we do not have a molecular explanation for this result, other investigators may find it fruitful to test host strain such as BL21-CodonPlus(DE3)-RIL even if rare codon usage does not appear to be a problem and even if they observe strong expression of their recombinant protein.
We also document our finding that the E. coli translation factor EF-Tu copurifies with Ada2/Ada3/Gcn5 and very likely the Piccolo NuA4 complex over multiple columns including metal affinity, anion- and cation-exchange resins. The 43 kDa EF-Tu protein also coelutes with the ~150 kDa Ada2/Ada3/Gcn5 and Piccolo NuA4 complexes by size exclusion chromatography, suggesting that it actually binds to the complexes and does not simply coelute from each of the columns (our published data). EF-Tu can be separated from the Piccolo NuA4 complex by hydrophobic interaction chromatography, consistent with our hypothesis that EF-Tu binds to the two HAT complexes via hydrophobic interactions. Given the wide abundance of EF-Tu in E. coli and its suggested role as a molecular chaperone, it is possible that others may observe similar contamination of EF-Tu for other proteins expressed recombinantly in E. coli.
Figure 2.
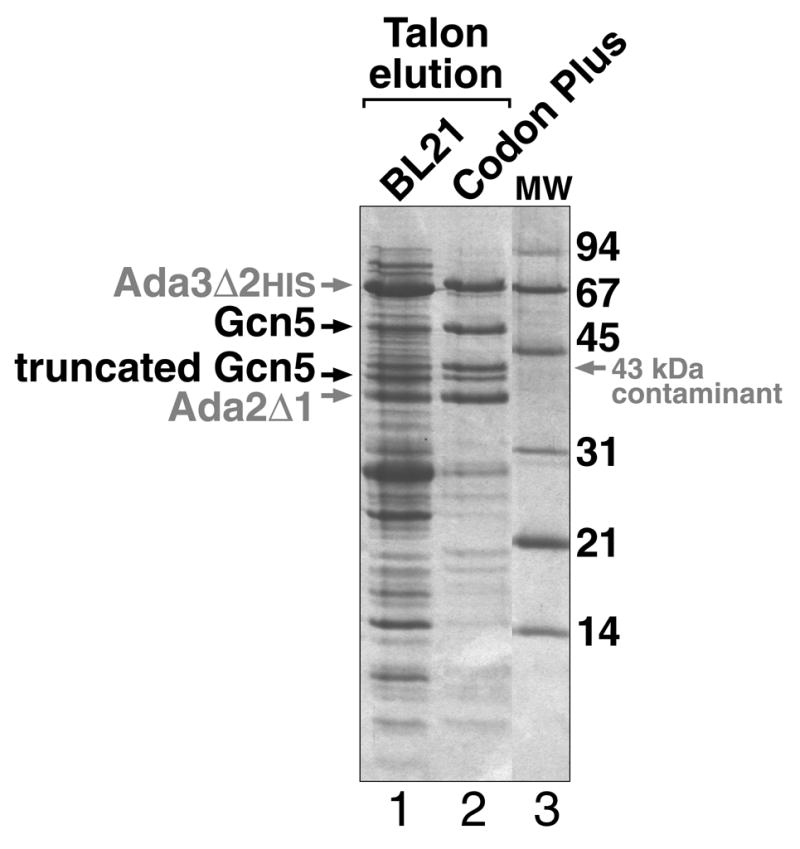
Use of the BL21-CodonPlu host strain improves Talon metal affinity purification of the recombinant yeast Ada2/Ada3/Gcn5 complex compared to BL21 cells. BL21(DE3)pLysS and BL21-CodonPlus(DE3)-RIL cells containing the pST44-yAda3Δ2HIS-yAda2Δ1-Gcn5 expression plasmid were induced at 18°C overnight, harvested by centrifugation, resuspended in buffer, Lysol by sonication and purified using Talon metal affinity resin with imidazole elution. Lanes 1 and 2 represent equivalent volume samples eluted from the Talon resin for BL21(DE3)pLysS and BL21-CodonPlus(DE3)pLysS cells respectively. The contaminating bands in lane 1 do not appear to result from prematurely truncated Ada3, Ada2 or Gcn5 polypeptides since anti-Ada3, anti-Ada2 or anti-Gcn5 antibodies do not recognize these bands in Western blots (data not shown). The 43 kDa contaminating band just below the truncated Gcn5 band was identified by mass spectrometry as the E. coli EF-Tu polypeptide.
Figure 3.

Purification of recombinant yeast Ada2/Ada3/Gcn5 complex expressed in E. coli. The whole cell extract of BL21-CodonPlus(DE3)-RIL cells with the pST44-yAda3Δ2HIS-yAda2-Gcn5 plasmid before induction with IPTG is shown in lane 1. Lane 2 shows the crude soluble extract of the same cells after IPTG induction while the pooled fractions after Talon metal affinity, Source Q anion-exchange and Source S cation-exchange chromatography are shown in lanes 3, 4 and 5 respectively. The major contaminant after Source Q and S chromatography is a 43 kDa contaminant identified by mass spectrometry as the E. coli translation elongation factor EF-Tu. Molecular weight markers are shown in lane 6.
Figure 4.

Purification of the recombinant yeast Piccolo NuA4 complex of Epl1/Yng2/Esa1 expressed in E. coli. The whole cell extract of BL21(DE3)pLysS cells containing the pST44-HISyEsa1-yEpl1Δ3-Yng2Δ1 plasmid before IPTG induction is shown in lane 1, while the soluble extract after IPTG induction is shown in lane 2. The pooled fractions after Talon metal affinity, Source Q anion-exchange, Source S cation-exchange and Source ISO hydrophobic interaction chromatography are shown in lanes 3, 4, 5 and 6 respectively. The 43 kDa contaminant removed by hydrophobic interaction chromatography is presumed to be EF-Tu.
Acknowledgments
We are grateful to other members of the Tan laboratory and the Penn State gene regulation community for helpful discussion and advice. This work was supported in part by NIH grant GM060489.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Khorasanizadeh S. Cell. 2004;116:259–272. doi: 10.1016/s0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- 2.Kuo MH, Zhou J, Jambeck P, Churchill ME, Allis CD. Genes Dev. 1998;12:627–639. doi: 10.1101/gad.12.5.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Liu L, Berger SL. Genes Dev. 1998;12:640–653. doi: 10.1101/gad.12.5.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang W, Bone JR, Edmondson DG, Turner BM, Roth SY. Embo J. 1998;17:3155–3167. doi: 10.1093/emboj/17.11.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grant PA, Duggan L, Cote J, et al. Genes Dev. 1997;11:1640–1650. doi: 10.1101/gad.11.13.1640. [DOI] [PubMed] [Google Scholar]
- 6.Allard S, Utley RT, Savard J, et al. Embo J. 1999;18:5108–5119. doi: 10.1093/emboj/18.18.5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Candau R, Berger SL. J Biol Chem. 1996;271:5237–5245. doi: 10.1074/jbc.271.9.5237. [DOI] [PubMed] [Google Scholar]
- 8.Horiuchi J, Silverman N, Marcus GA, Guarente L. Mol Cell Biol. 1995;15:1203–1209. doi: 10.1128/mcb.15.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balasubramanian R, Pray-Grant MG, Selleck W, Grant PA, Tan S. J Biol Chem. 2002;277:7989–7995. doi: 10.1074/jbc.M110849200. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Garcia AB, Sendra R, Pamblanco M, Tordera V. FEBS Lett. 1997;403:186–190. doi: 10.1016/s0014-5793(97)00049-5. [DOI] [PubMed] [Google Scholar]
- 11.Sendra R, Tse C, Hansen JC. J Biol Chem. 2000;275:24928–24934. doi: 10.1074/jbc.M003783200. [DOI] [PubMed] [Google Scholar]
- 12.Brown CE, Howe L, Sousa K, et al. Science. 2001;292:2333–2337. doi: 10.1126/science.1060214. [DOI] [PubMed] [Google Scholar]
- 13.Selleck W, Fortin I, Sermwittayawong D, Cote J, Tan S. Mol Cell Biol. 2005;25:5535–5542. doi: 10.1128/MCB.25.13.5535-5542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boudreault AA, Cronier D, Selleck W, et al. Genes Dev. 2003;17:1415–1428. doi: 10.1101/gad.1056603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan S, Kern RC, Selleck W. Protein Expr Purif. 2005;40:385–395. doi: 10.1016/j.pep.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Tan S. Protein Expr Purif. 2001;21:224–234. doi: 10.1006/prep.2000.1363. [DOI] [PubMed] [Google Scholar]
- 17.Studier FW. J Mol Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- 18.Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. Protein Expr Purif. 2005;41:98–105. doi: 10.1016/j.pep.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 19.Caldas TD, El Yaagoubi A, Richarme G. J Biol Chem. 1998;273:11478–11482. doi: 10.1074/jbc.273.19.11478. [DOI] [PubMed] [Google Scholar]
- 20.Studier FW. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]