

Abstract
Human choriogonadotropin (hCG) and follitropin (hFSH) have been shown to contact different regions of the extracellular domains of G-protein coupled lutropin (LHR) and follitropin (FSHR) receptors. We report here that hCG and hFSH analogs interact with an FSHR/LHR chimera having only two unique LHR residues similar to the manners in which they dock with LHR and FSHR, respectively. This shows that although the FSHR does not normally bind hCG, it contains a cryptic lutropin binding site that has the potential to recognize hCG in a manner similar to the LHR. The presence of this cryptic site may explain why equine lutropins bind many mammalian FSHR and why mutations in the transmembrane domain distant from the extracellular domain enable the FSHR to bind hCG. The leucine-rich repeat domain (LRD) of the FSHR also appears to contain a cryptic FSH binding site that is obscured by other parts of the extracellular domain. This will explain why contacts seen in crystals of hFSH complexed with an LRD fragment of the human FSHR are hard to reconcile with the abilities of FSH analogs to interact with membrane G-protein coupled FSHR. We speculate that cryptic lutropin binding sites in the FSHR, which are also likely to be present in thyrotropin receptors (TSHR), permit the physiological regulation of ligand binding specificity. Cryptic FSH binding sites in the LRD may enable alternate spliced forms of the FSHR to interact with FSH.
Introduction
hFSH2 and the other heterodimeric glycoprotein hormones are composed of a common α-subunit and a hormone-specific β-subunit that determines receptor binding specificity (Pierce & Parsons 1981). Both subunits are needed for hormone activity. Each subunit in hFSH (Fox, Dias, & Van Roey 2001), hCG (Lapthorn et al. 1994;Wu et al. 1994), and all related family members has a cystine knot motif. The hFSH and hCG heterodimers are stabilized by 20 β-subunit residues that restrict movements of α-subunit loop 2 (α2) like a “seatbelt” (Lapthorn, et al. 1994). The seatbelt is responsible for much of the influence of the β-subunit on receptor binding specificity (Campbell, Dean Emig, & Moyle 1991;Dias, Zhang, & Liu 1994;Grossmann et al. 1997;Moyle et al. 1994).
Glycoprotein hormone receptors have large extracellular domains, a rhodopsin-like transmembrane domain (TMD) typical of G-protein coupled receptors, and a short cytoplasmic tail. The extracellular domain has two subdomains that we termed the leucine-rich repeat domain (LRD) and the signaling-specificity domain (SSD) to reflect their structure and function, respectively (Moyle et al. 2004). The LRD is capable of binding some ligands per se and was shown to control some aspects of ligand binding specificity shortly after the LHR and FSHR sequences were determined (Braun, Schofield, & Sprengel 1991). Ligand binding specificity also depends on the SSD (Bernard, Myers, & Moyle 1998;Moyle, et al., 2004) and, in some cases, is influenced by the TMD (Smits et al. 2003b). Indeed, the latter studies showed that mutations in the TMD can allow the FSHR to bind hCG well enough to enable the high levels of hCG seen during pregnancy to cause hyperovarian stimulation, a potentially life threatening condition.
The most common view of glycoprotein hormone receptor interactions is based on a crystal structure of hFSH bound to a fragment of the human FSHR extracellular domain (Dias 2005;Fan & Hendrickson 2005) that confirmed an earlier notion that has dominated this field (Jiang et al. 1995). In the crystals, both hFSH subunits contact the concave surface of the LRD in an orientation that causes the long axis of hFSH to be roughly “perpendicular” to the long axis of the LRD. These contacts between hFSH and the LRD are thought to stabilize the hormone in a position that enables α-subunit loops 1 and/or 3 to contact the outer loops of the transmembrane domain (TMD), thereby initiating signal transduction. The finding that the hormone-receptor complex crystallized as a dimer in which two LRD contact one another (Fan & Hendrickson 2005) was considered as support for the notion that signal transduction occurs by ligand-induced receptor dimerization. Unlike other G-protein coupled receptors that are thought to function as homodimers or heterodimers (Bulenger, Marullo, & Bouvier 2005;Javitch 2004), dimerization of the glycoprotein hormone receptors as proposed by Fan & Hendrickson (2005) would prevent contacts between their TMD.
As we have reviewed (Moyle et al. 2005), this model does not explain many aspects of FSHR function. The discrepancies between the crystalline and membrane FSHR forms would be explained if the FSHR contains “cryptic” ligand docking sites – i.e., those that have the potential to bind ligands, but are prevented from doing so normally. We had observed that the SSD portion of the FSHR extracellular domain is needed for the abilities of LHR/FSHR chimeras to bind FSH and initiate signal transduction (Moyle, et al., 1994). In our models of the receptors (Moyle, et al., 2004), the SSD would block contacts seen between FSH and the LRD observed in the crystals (Fan & Hendrickson 2005).
Efforts to resolve differences in the manner in which ligands bind membrane FSHR and crystalline FSHR fragments led us to test the possibility that the FSHR contains cryptic ligand docking sites. We tested the possibility that the FSHR contains a cryptic hCG binding site by monitoring the manner in which hCG and hFSH bind to an FSHR/LHR chimera that contains only two unique LHR residues (Smits et al. 2003a). We found that this chimera, which we term FSHR2 to reflect its content of LHR residues, binds hCG and hFSH analogs in orientations that are characteristic of LHR and FSHR, respectively. This finding suggests the FSHR contains at least one cryptic hCG binding site and supports the notion that the glycoprotein hormone receptors may contain hidden binding sites, most likely a consequence of their organization. By comparing hFSH/FSHR contacts seen in the crystal structure with the properties of membrane receptors, we conclude that the extracellular domain of the FSHR also contains a cryptic FSH docking site. As we discuss, cryptic docking sites may permit the physiological regulation of receptor binding specificity.
Experimental Procedures
Pure recombinant hFSH was a gift of Dr. Robert Campell, Serono Research Institute, Rockland, MA. Clones encoding the hFSH β-subunit and the full-length hFSHR were gifts of Dr. Christine Kelton, also of the Serono Research Institute. By co-transfecting the latter with pSV2Neo (Southern & Berg 1982) into Chinese hamster cells and selecting cells that grew in the presence of 1 mg G418/ml, we obtained a stable line that expressed a functional full-length hFSHR. We used PCR mutagenesis to prepare an hFSH β-subunit construct lacking codons for residues Asp41-Pro42-Ala43-Arg44 (hFSHδ4) in an expression vector (pCI, Promega, Madison, WI) that had been modified as described (Lin et al. 1999). After the sequences of the coding and anti-coding strands of the plasmid preparation that was to be used for expressing the hFSHRδ4 subunit were confirmed, we prepared the hFSHRδ4 heterodimer by co-expressing this plasmid with a plasmid encoding the human α-subunit transiently in COS-7 cells (Campbell, Dean Emig, & Moyle 1991). The hFSHδ4 heterodimer in the medium was quantified using a sandwich immunoassay (Moyle, Ehrlich, & Canfield 1982) employing α-subunit monoclonal antibody A113 for capture, radioiodinated β-subunit hFSH antibody B603 for detection, and pure recombinant hFSH as a standard. Both monoclonal antibodies were obtained from Dr. William Munroe (Hybritech Incorporated, San Diego, CA, a subsidiary of Beckman Coulter, Inc.). The culture media were concentrated by ultrafiltration and requantified by A113/B603 sandwich assay with pure recombinant hFSH as the standard before being tested in ligand binding and signaling assays. Radioiodinated hFSH and B603 were prepared as described (Bernard et al. 2004). To monitor receptor binding, we compared the abilities of pure recombinant hFSH and medium containing hFSHδ4 to inhibit the binding of 125I-hFSH to cells expressing the hFSHR. To monitor signal transduction, we compared the abilities of pure recombinant hFSH and medium containing hFSHδ4 to stimulate cyclic AMP accumulation. Cyclic AMP was determined by radioimmunoassay as described (Brooker et al. 1979). Statistical analyses were performed with the graphics program Prism 4 (GraphPad Software Inc., San Diego, CA). All assays were performed three times and gave essentially identical results. Models of the hormone receptor complexes based on the coordinates determined by Fan and Hendrickson (Fan & Hendrickson 2005) were prepared using LOOK (Lee & Irizarry 2001). The structures were energy minimized using Sybyl force field in and visualized using Sybyl (Tripos, St. Louis, MO).
We prepared a construct encoding the FSHR/LHR chimera that had been described by (Smits, et al., 2003a) and that contains only 2 residues unique to the LHR. Stable CHO cell lines that expressed this receptor chimera were prepared in the same fashion as those that expressed the FSHR as described in the preceding paragraph. Knobbed analogs of hCG, hFSH, and an hCG/hFSH chimera (CFC101-114) were prepared as described earlier (Moyle, et al., 2004;Xing et al. 2004a). We monitored the abilities of these analogs to block the binding of 125I-hFSH and 125I-hCG to the FSHR2 expressing cells as described in the preceding paragraph to determine the abilities of hFSH and hFSHδ4 to inhibit the binding of 125I-hFSH to CHO cells that express the hFSHR.
Results
The FSHR contains a cryptic hCG binding site
Several years ago we reported that some hCG/hFSH chimeras bound LHR and FSHR and that some LHR/FSHR chimeras bound hCG and hFSH (Moyle, et al., 1994). hCG bound best to LHR/FSHR chimeras in which the central region of the LRD was derived from the LHR. hFSH bound well only to those chimeras in which the N- and C-terminal ends of the LRD and most of the SSD was derived from the FSHR. Indeed, the FSHR SSD was required to observe FSH binding to all these chimeras. LHR/FSHR chimeras in which the central region of the LRD was derived from the FSHR and the remainder of the receptor was derived from the LHR did not bind either hormone (Moyle, et al., 1994). These observations suggested that hCG and hFSH bound to different sites in the extracellular domain of LHR and FSHR. Other findings suggested that different residues in hCG and hFSH contacted the receptors. For example, the small seatbelt loop of hCG has a much greater influence on LHR binding than FSHR binding. Thus, changing the small seatbelt loop of hFSH enabled it to bind LHR without disrupting its ability to bind FSHR (Dias, Zhang, & Liu 1994); changing the small seatbelt loop of hCG/hFSH chimeras reduced their abilities to bind LHR, but not FSHR (Han, Bernard, & Moyle 1996;Moyle, et al., 1994). The C-terminal end of the seatbelt is required for FSHR binding but not for LHR binding (Moyle, et al., 1994). The surface of α2 near Arg42 has a key role in binding to FSHR but not to LHR. In the orientation favored by hFSH, α2 residue 42 is buried; in that favored by hCG, α2 residue 42 is not buried. Thus, the addition of a protein knob to α2 residue 42 nearly eliminated binding of hFSH and hCG/hFSH chimeras to the FSHR whereas addition of a protein knob to this site of hCG and hCG/hFSH chimeras had relatively little influence on the binding of these ligands to the LHR (Moyle, et al., 2004).
The finding that an FSHR/LHR chimera having only 2 unique LHR residues (i.e., FSHR2) binds both ligands with high affinity (Smits, et al., 2003a) challenged the notion that hCG and hFSH docked with different sites of their receptors. Due to its minimal LHR content, FSHR2 would be expected to be almost identical in conformation to the FSHR. We expected that measurements of the abilities of knobbed hCG, hFSH, and hCG/hFSH chimera analogs to FSHR2 would enable us to determine if glycoprotein hormone receptors contained multiple potential ligand recognition sites and if the FSHR has a cryptic site that has properties seen in the LHR. There were three possible outcomes of this experiment as outlined next.
Outcome 1
Since FSH analogs containing a knob at α2 residue 42 do not bind the FSHR (Moyle, et al., 2004), if hCG and hFSH recognized the same FSH contact region in FSHR2, then the presence of an α2 knob at residue 42 would block binding of hCG and hFSH to FSHR2. Therefore, knobbed analogs of hFSH or hCG would be unable to block 125I-hCG or 125I-hFSH binding to FSHR2 nearly as well as either the hCG and hFSH controls.
Outcome 2
If the FSHR contains a cryptic lutropin-like binding site that had the potential to distinguish hCG and hFSH, then analogs of hCG that have a knob at α2 residue 42 would recognize the exposed lutropin binding site in FSHR2 and, similar to hCG and hFSH, prevent it from binding of 125I-hCG or 125I-hFSH. In contrast, similar knobbed analogs of hFSH would fail to recognize either the LHR-like site or the endogenous FSHR site in FSHR2. Therefore, they would not bind FSHR2 and be unable to keep it from binding of 125I-hCG or 125I-hFSH.
Outcome 3
If the FSHR has a cryptic binding site that does not distinguish hCG or hFSH, then we expected that knobbed analogs of hCG and hFSH would bind to this site in FSHR2. Consequently, each would be capable of blocking the binding of 125I-hCG or 125I-hFSH to FSHR2.
To test these possibilities we introduced protein knobs into sites of α2 that enabled us to identify differences in the hCG binding domain of the LHR and the hFSH binding domain in the FSHR (Moyle, et al., 2004;Xing, et al., 2004a) and monitored the abilities of these analogs to inhibit binding of 125I-hCG or 125I-hFSH to FSHR2. We observed that the data were consistent with Outcome 2, but not Outcomes 1 or 3. Thus, the presence of a protein knob on α-subunit residue 42 or 48 of hFSH greatly reduced its ability to inhibit the binding of 125I-hCG or 125I-hFSH to FSHR2 (Figs. 1A, D). Similar knobs on hCG were much less effective in inhibiting its ability to block binding of 125I-hCG or 125I-hFSH to FSHR2 (Figs. 1B, E). Remarkably the presence of knobs on the CFC101-114 chimeras did not block the abilities of these hCG/hFSH chimeras to inhibit binding of 125I-hCG or 125I-hFSH to FSHR2 (Figs. 1C, F).
Figure 1. Interactions of knobbed analogs with FSHR2 revealed the presence of cryptic receptor that recognizes hCG in a fashion similar to that of the LHR.
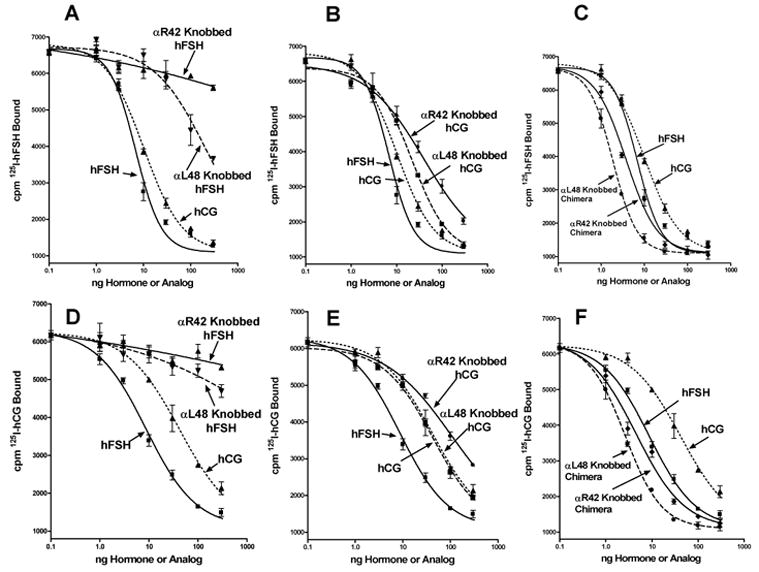
CHO cells that express FSHR2 were incubated with 125I-hFSH (panels A, B, C) or 125I-hCG (panels D, E, F) and increasing amounts of hCG, hFSH, hFSH analog containing knobs at α2 residues 42 or 48 (panels A, D), hCG containing knobs at α2 residues 42 or 48 (panels B, E), or chimera CFC101-109 containing knobs at α2 residues 42 or 48 (panels C, F). The results described in the left panels were obtained in a single experiment. Those described in the right panels were obtained in a single experiment done two days later using the same hormone and analog preparations to make the results comparable. Note also, that the dose response curves illustrated for hCG and hFSH standards in panels A, B, C are the same data; those for hCG and hFSH standards in panels D, E, F are the same data. We copied the hCG and hFSH dose response curves into each panel to make it easier for readers to determine the potencies of the knobbed analogs relative to those of the hormone standards. All values are triplicates and the vertical lines extend to the limits of the SEM.
These observations showed clearly that FSHR2 contained at least two ligand binding sites, one that behaved as expected for an FSHR and one that behaved as expected for an LHR. The fact that hCG and hFSH blocked the binding of both 125I-hCG or 125I-hFSH showed that hCG and hFSH cannot bind to the receptor at the same time, which is consistent with our model of hormone receptor interaction (Moyle, et al., 2004). An interesting aspect of these studies was the observation that the knobbed chimera analogs inhibited the binding of both 125I-hCG and 125I-hFSH to the receptor in nearly identical fashions. The CFC101-114 chimera has similar affinity for LHR as hCG and roughly one third the affinity for FSHR as hFSH (Moyle, et al., 2004). Attaching knobs to this chimera did not prevent it from binding LHR similar to hCG, but it did prevent it from binding to FSHR (Moyle, et al., 2004). This showed that the chimera did not recognize the cryptic binding site in the FSHR. In contrast, the fact that knobbed analogs of the chimera bound to FSHR2 and blocked the binding of 125I-hCG and 125I-hFSH showed that it recognized the hCG binding site in FSHR2. These data strongly support the notion that the FSHR has a cryptic lutropin-specific binding site.
As had been observed earlier (Moyle, et al., 2004;Xing, et al., 2004a), the presence of a knob on FSH α-subunit residue 42 was more inhibitory than a knob on α-subunit residue 48 (Fig. 1, all panels). This was also seen in studies of signal transduction (Fig. 2) and showed that FSH interacted with FSHR2 similar to the fashion that we had found it to interact with the FSHR (Moyle, et al., 2004;Xing, et al., 2004a).
Figure 2. Knobbed analogs of hFSH have low signaling activity in FSHR2 assays.
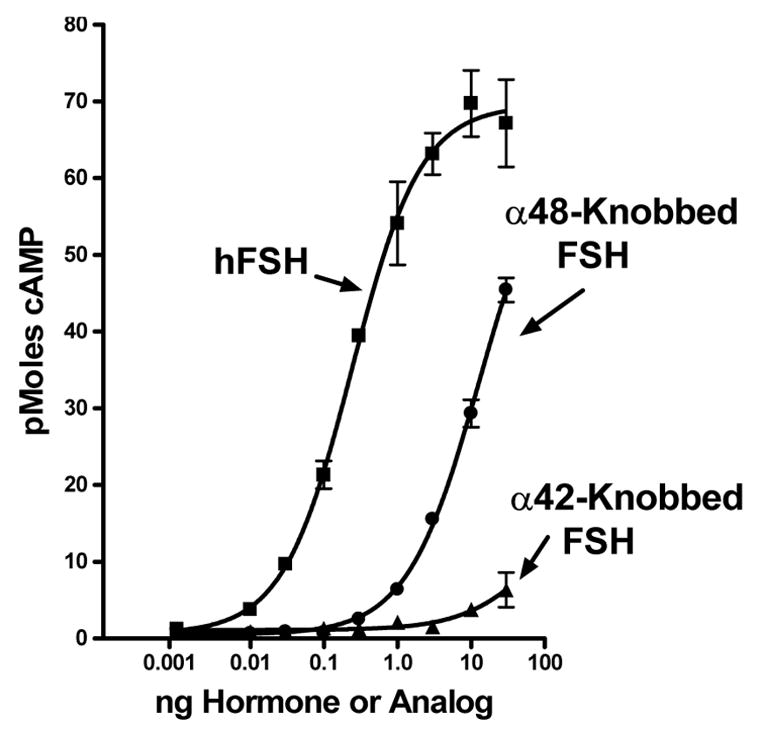
CHO cells that express the FSHR2 were incubated with increasing concentrations of hFSH and hFSH analogs containing knobs at α2 residues 42 or 48 as indicated on the figure. Cyclic AMP in the cells and medium was measured following a 20 minute incubation at 37°C. Values are means of triplicate incubations and the vertical lines extend to the limits of the SEM.
A cryptic FSH binding site appears to be present in the LRD of the FSHR
hFSH and hCG have at least 1,000 – 10,000 fold higher affinities for FSHR and LHR than for LHR and FSHR, respectively, a phenomenon controlled by differences in their β-subunits (Pierce & Parsons 1981). The portions of the β-subunit that contact the receptor fragment in the crystals are loop 2 (β2) and the seatbelt (Fan & Hendrickson 2005). Previous studies showed that β-subunit loop 2 could be swapped between the hormones without disrupting receptor binding specificity or ligand binding affinity (Campbell, Dean Emig, & Moyle 1991). Although this suggested that β-subunit loop 2 does not contact the receptor, it is conceivable that some residues in this loop of both ligands are capable of interacting with each receptor. Since this type of contact is much less likely to be present in an analog that lacked residues in the receptor contact site, we characterized the receptor binding and signaling activity of an hFSH analog (hFSHδ4) that lacked four residues in the region of loop β2 that contacts the receptor. We designed hFSHδ4 by considering the structure of hFSH (Fox, Dias, & Van Roey 2001), the locations of hFSH β-subunit loop 2 residues that contact the hFSHR in the crystals (Fan & Hendrickson 2005), and residues in β-subunit loop 2 that we had found to be important for heterodimer assembly (Xing et al. 2004b). To maximize the likelihood that hFSHδ4 would combine with the α-subunit, we did not alter β-subunit loop 2 residues that contact the α-subunit (Fox, Dias, & Van Roey 2001) and that do not contact the hFSHR (Fan & Hendrickson 2005). Models based on the crystal structure (Fan & Hendrickson 2005) suggested that deletion of hFSH residues Asp41-Pro42-Ala43-Arg44 would disrupt contacts between loop 2 and the receptor without altering the overall conformation of the heterodimer or interfering with other hormone receptor contacts (Fig. 3A–C).
Figure 3. Contacts of the hFSH β-subunit with its FSHR fragment.
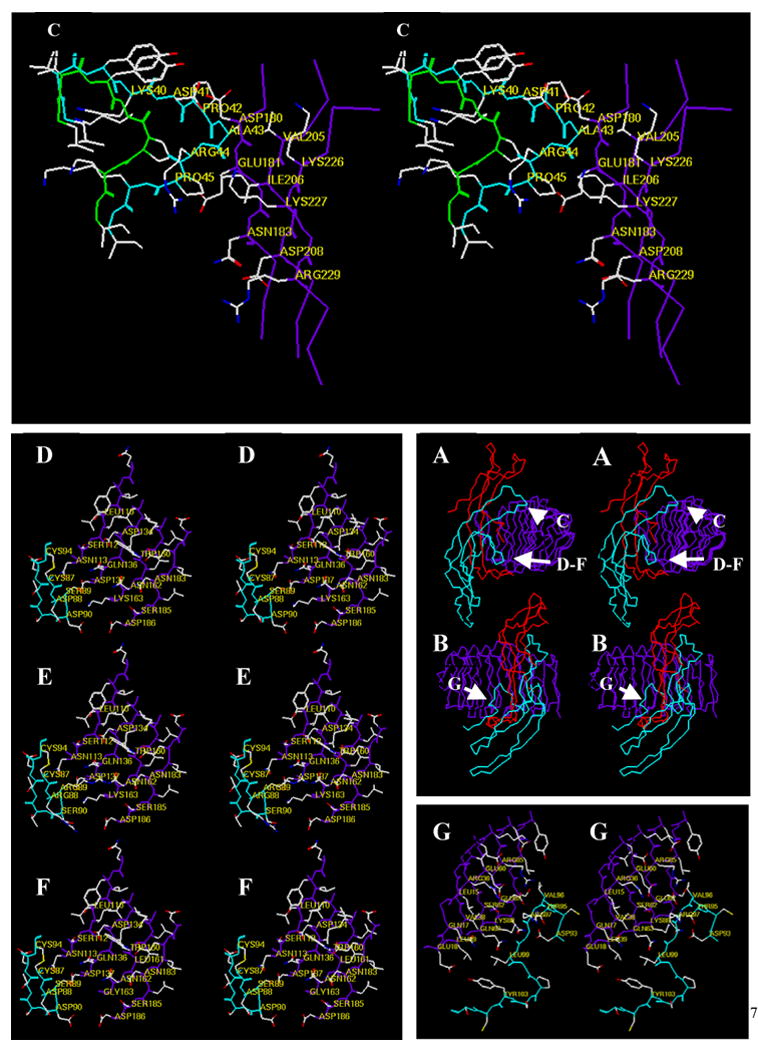
(Panels A and B, center right) Cα coordinates of the hFSH-FSHR fragment shown in relaxed stereo view. Panel A depicts the complex turned in an orientation that shows the relative positions of the enlarge regions highlighted in Panel C and in Panels D, E, and F. Panel B depicts the complex in an orientation that shows the position of the enlarged region highlighted in Panel G. Color scheme: red, α-subunit; cyan, β-subunit; purple, FSHR fragment. (Panel C) Relaxed stereo view of the tip of β-subunit loop 2 as it is found in the FSH-FSHR fragment complex (cyan backbone) and as it is modeled in the deletion mutant hFSHδ4 (green backbone). The Cα carbons of several residues in the native β-subunit and in the FSHR fragment are labeled. Those in the analog are not labeled to make the figure easier to visualize. The residues that have been deleted are: Asp41-Pro42-Ala43-Arg44. Thus, in the mutant Lys40 is joined to Pro45. Deletion of residues 41–44 was expected to eliminate contacts between the sidechains of Pro42 and Ala43 and between the backbone of Ala43 with the receptor. Note that this region of the β-subunit appears not to contribute to receptor binding specificity. (Panel D). Relaxed stereo view of the “determinant loop” (cyan backbone) and the region of the receptor that it contacts in the FSHR fragment (purple backbone). Note that this region of FSH has little or no influence on binding to cell surface FSHR. (Panel E) Replacing FSH residues Asp88, Ser89, Asp90, and unlabeled Ser91 (Panel D) with Arg-Arg-Ser-Thr, its hCG counterpart, enhances binding of FSH to LHR; the converse swap in hCG does not enhance binding of hCG to FSHR, but reduces binding of hCG to LHR. (Panel F) Replacing FSHR residues Lys163 with glycine and Lys88 (c.f. Panel G) with asparagine enables hCG to bind to FSHR, but has little influence on the binding of FSH to FSHR. Together, the findings that FSH binding to the cell surface FSHR is not reduced or improved substantially by changes in “determinant loop” residues that face this region of the receptor suggest that these contacts are either not present in the cell surface receptor or that they contribute little to FSH-FSHR interactions. (Panel G) Relaxed stereo view of residues in the region of the FSH seatbelt that controls FSH receptor binding specificity (cyan backbone) and adjacent residues of the FSHR fragment (purple backbone). It is difficult to see how contacts between these hormone and receptor residues confer ligand binding specificity. Receptor residues Lys58 (not labeled)-Glu60-Ser62-Gln63, the central receptor leucine-rich repeat that contacts this portion of the seatbelt, are identical in most mammalian FSHR and LHR. Receptor residue Lys88, which faces Asp93, a residue found at the end of the seatbelt loop in nearly all β-subunit residues can be changed to asparagine without altering FSH or LHR binding. Glu87 is a glutamine in mammalian LHR and would be expected to make similar contacts. Arg36 is serine in mammalian LHR, which would be expected to facilitate contacts with FSH residue Arg97.
COS-7 cells transfected with vectors encoding the human α-subunit and the hFSHδ4 β-subunit analog secreted hFSHδ4 heterodimer into the medium. The amount of hFSHδ4 in unconcentrated medium three days after transfection was 103 ± 13 ng/ml (n = 3), which is similar to those observed typically following transient co-transfection of COS-7 cells with constructs encoding the human α-subunit and either the hFSH or the hCG β-subunit. This showed that hFSH residues Asp41-Pro42-Ala43-Arg44 are not required for hFSH β-subunit folding, for combination with the α-subunit, or for secretion.
hFSHδ4 and hFSH had equal abilities to inhibit the binding of 125I-hFSH to CHO cells that express the hFSHR (Fig. 4). Both stimulated cyclic AMP accumulation to the same maximal extent with equal potencies (Fig. 5). The high potency of hFSHδ4 in these assays relative to hFSH showed that residues missing from hFSHδ4 are not required for ligand binding or signaling in assays employing hFSHR expressed at the cell surface.
Figure 4. Ability of hFSH and hFSHδ4 to inhibit binding of 125I-hFSH to human FSHR.
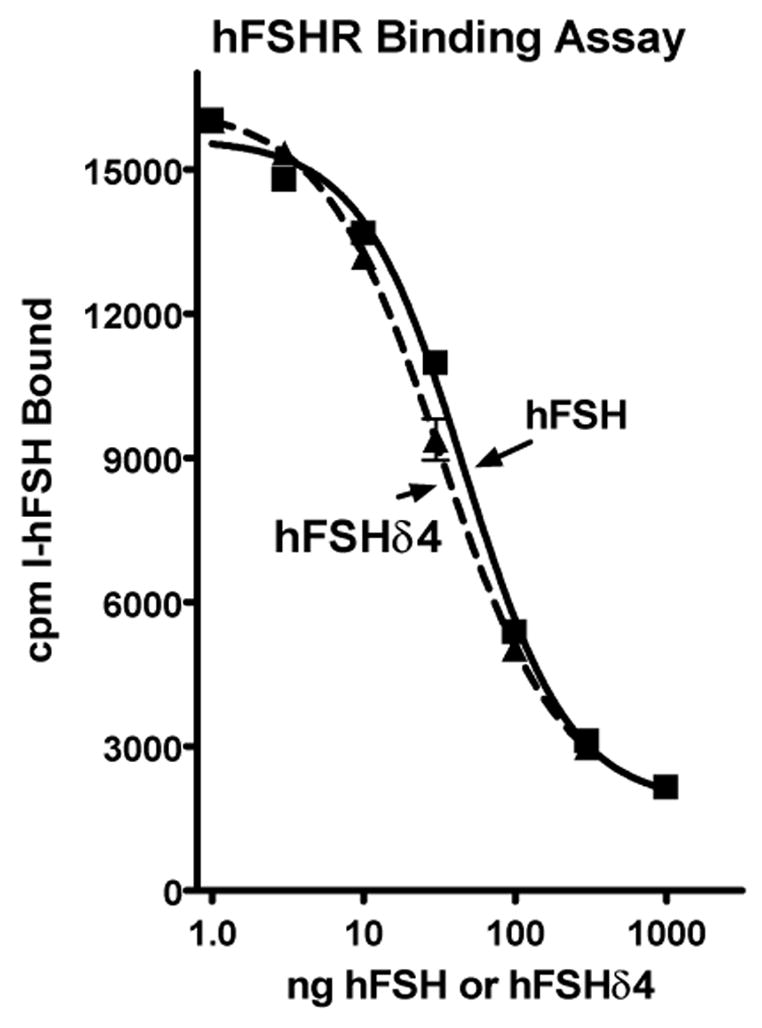
CHO cells (200,000) that express the hFSHR were incubated with 125I-hFSH (100,000 cpm; approximately 1.5 ng) in 100 μl of Krebs-Ringer solution buffered with 20 mM HEPES (pH 7.4) and the indicated amount of hFSH or hFSHδ4 for 1 hour at 37°C. The reaction mixture was diluted with 2 ml of an ice cold aqueous solution of 1 mg/ml bovine serum albumin in 0.9% NaCl and the cells were sedimented into a pellet at 1000 x g. After the supernate was aspirated, the cell pellet was counted in a γ-counter. The values are means of triplicates and the vertical bars extend to the limits of the SEM. There was no significant difference between either treatment in any of the three independent assays performed.
Figure 5. Ability of hFSH and hFSHδ4 to stimulate cyclic AMP accumulation.
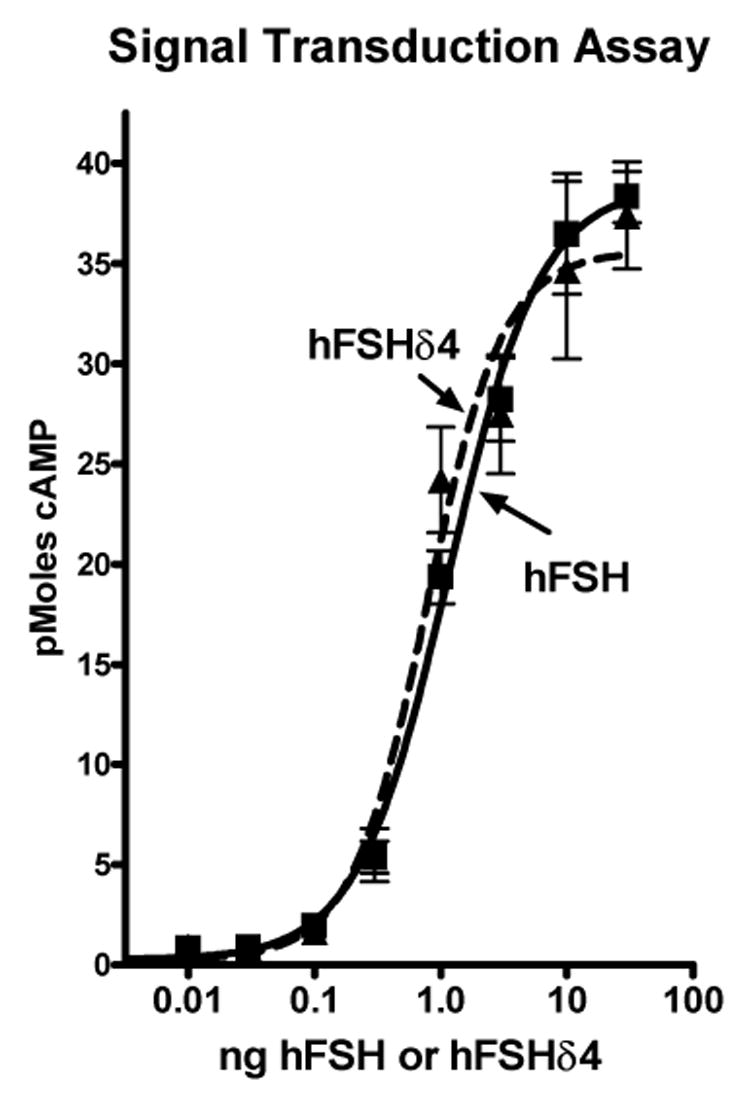
CHO cells (50,000) that express the hFSHR were incubated with hFSH or hFSHδ4 in 60 μl of Krebs-Ringer solution buffered with 20 mM HEPES (pH 7.4) for 20 min at 37°C. The incubation tubes were heated at 75°C for one minute to stop the reaction and cause the release of all cyclic AMP. The cyclic AMP was measured using an RIA employing a rabbit antiserum and a radioiodinated cyclic AMP derivative prepared as described (Brooker, Harper, Terasaki, & Moylan 1979). The values are means of triplicates and the vertical bars extend to the limits of the SEM. There was no significant difference between either treatment in any of the three independent assays performed.
The finding that hFSHδ4 and hFSH had equal activity in assays employing cell surface receptors is consistent with the notion that β-subunit loop 2 does not contact the common physiologically active form of the receptor. Nonetheless, since residues in β-subunit loop 2 that contact the crystallized receptor are at the periphery of the hormone-receptor interface (Fig. 3A–C), the finding that hFSHδ4 is as active as hFSH does not preclude the possibility that loop 2 contacts the FSHR. Therefore, we turned our attention to interactions that involve the seatbelt, the only other portion of the hFSH β-subunit that contacts the receptor fragment in the crystal structure. Since this region of hFSH has been studied extensively, it was possible to analyze the ability of the crystal structure to explain these observations by mapping the existing data onto the crystal structure. As described next, this analysis revealed that contacts between hFSH residues Ser89-Asp90-Asp93-Cys94-Thr95-Val96-Arg97-Gly98-Leu99-Tyr103-Ser105 and the receptor (Fan & Hendrickson 2005) are hard to rationalize with the known activities of FSH and FSHR analogs.
Based on contacts between hFSH and its receptor fragment in the crystals, Fan and Hendrickson suggested that Ser89, Arg97, and Leu99 have the greatest role in ligand binding specificity (Fan & Hendrickson 2005). Ser89 and an adjacent residue (Asp90) that is also near the receptor are found in a part of the seatbelt that was termed the “determinant loop” due only to the fact that its sequence differs in lutropins and follitropins (Ward & Moore 1979). In hFSH and hCG these residues are Asp88-Ser89-Asp90-Ser91 and Arg94-Arg95-Ser96-Thr97, respectively. Ser89 and Asp90 are adjacent to FSHR residue Lys163 in the crystals (Fig. 3A,B,D). The notion that hFSHR residue Lys163 functions as a key contact is consistent with the fact that it is conserved in all five classes of vertebrate follitropin receptors. Furthermore, its replacement by glycine is one of two substitutions that enable the FSHR to bind hCG (Smits, et al., 2003a).
Remarkably, contacts between hFSH residues Ser89-Asp90 and hFSHR residue Lys163 observed in the crystals are not required for docking or signaling of hFSH analogs in assays employing cell surface FSHR. Replacing Ser89 and Asp90 with Arg and Ser (Dias, Zhang, & Liu 1994) – Figure 3E -or replacing FSHR residue Lys163 with Gly (Smits, et al., 2003a) – Figure 3F – did not interfere with binding or signaling observed in cell surface FSHR assays. Residues in the “determinant loop” influence binding to LHR substantially, but have little influence on the abilities of ligands to interact with the FSHR (Han, Bernard, & Moyle 1996;Moyle, et al., 1994). Indeed, bifunctional hCG/hFSH chimeras that have Arg-Arg-Arg, Asp-Asp-Asp, or several other residues including some that have hydrophobic sidechains in place of the hFSH specific sequence Asp88-Ser89-Asp90 had similar abilities to interact with FSHR (Han, Bernard, & Moyle 1996). Since these residues differ substantially in charge, size, and hydrophobicity, it is difficult to see how the interactions between this region of the hormone and FSHR residue Lys163 make major contributions to hFSH-FSHR interactions as proposed (Fan & Hendrickson 2005). Furthermore, substitution of hFSH-specific residues for their hCG counterparts of the “determinant loop” does not increase the ability of hCG to bind to FSHR (Campbell, Dean Emig, & Moyle 1991). Thus, interactions between these ligand and receptor residues do not appear to specify hormone interactions with G-protein coupled FSHR. Indeed, the minimal influence of these residues on interactions of the chimeras with FSHR led us to conclude that they do not make important contacts with FSHR that are expressed at the cell surface (Han, Bernard, & Moyle 1996).
In contrast to the small seatbelt loop, which forms most of the N-terminal half of the seatbelt and is much more important for the activities of lutropins than follitropins, the C-terminal half of the seatbelt has a key role in the activities of follitropins and a lesser role in the activities of lutropins (Moyle, et al., 1994). Replacing the C-terminal half of the hCG seatbelt with its hFSH counterpart leads to a bifunctional chimera – i.e., one that interacts with LHR and FSHR. This hCG/hFSH chimera has roughly 30–50% the affinity of hFSH for FSHR and 100% the affinity of hCG for the full-length LHR (Moyle, et al., 1994). A cysteine at the C-terminal end of the seatbelt forms a disulfide with a cysteine in the β-subunit core that latches the seatbelt to loop 1 in most glycoprotein hormones, although it is latched to a cysteine in the N-terminal end of the FSH β-subunit in many teleost fish. The hCG seatbelt can also be latched to a cysteine in place of some residues in α-subunit loop 2 – e.g., residue 41 – without disrupting LHR binding (Xing et al. 2001), a finding that suggests the C-terminal half of the seatbelt does not make key contacts with mammalian LHR. The seatbelts of the bifunctional chimera described above can also be latched to α-subunit loop 2 residue 41, but this eliminates its FSH activity (Bernard, Lin, Myers, and Moyle, unpublished observations). Together, these observations support the notion that the C-terminal half of the seatbelt has a more important role in FSHR than in LHR interactions, at least in mammals. They suggest also that this region of the seatbelt might participate in important FSHR contacts.
As would be expected from its role in follitropin activity, some contacts between the C-terminal half of the seatbelt and the receptor were observed in the crystals (Fan & Hendrickson 2005). Nonetheless, it is difficult to see how these would be sufficient to enable bifunctional hCG/hFSH chimeras to interact with FSHR. In fact, hFSH β-subunit residue Arg97, a residue postulated to have a key role in ligand-receptor binding specificity (Fan & Hendrickson 2005), is near FSHR residue Arg36 (Fig. 3A,B,G). Why would this contact not reduce the affinity of hFSH for the FSHR? Although it could be argued that these residues could move away from one another during ligand binding, it is hard to visualize how this would contribute to the high affinity observed between hFSH and its cell surface receptor. Thus, the influence of the C-terminal end of the FSH seatbelt on FSHR interactions is not evident from the crystal structure.
We turned our attention to interactions between the FSH α-subunit and the receptor. The crystal structure shows that several residues in α2 – e.g., αArg42 – do not contact the FSHR or, at most, make minimal contacts with the receptor (Fan & Hendrickson 2005). This observation is difficult to reconcile with the finding that αArg42 appears to have a significant role in the binding of hFSH to cell surface FSHR since its replacement by glutamate reduced FSHR binding significantly (Moyle, et al., 2004) and since the addition of a protein “knob” to this site nearly eliminated hFSHR binding (Xing, et al., 2004a). One could argue that this observation was due to an influence of the mutation on the β-subunit that led to a change in the conformation of the hFSH heterodimer, particularly in view of the fact that similar changes in hCG did not alter its ability to bind to LHR (Moyle, et al., 2004;Xing, et al., 2004a). Nonetheless, this could not have explained the influence of these mutations on hCG/hFSH chimeras since these contained the same β-subunit. Substitutions for αArg42 in the chimeras interfered with their abilities to recognize FSHR much more than their abilities to recognize LHR. This led us to devise models of hCG-LHR and hFSH-FSHR complexes in which the ligands docked with their receptors in different ways (Moyle, et al., 2004), a phenomenon consistent with the finding that different residues of the seatbelt influence hCG and hFSH activities.
Discussion
Implications for the structure of the glycoprotein hormone receptors
Crystal structures usually provide a good platform for interpreting diverse biochemical phenomena. As shown here, contacts between the hFSH β-subunit and the FSHR fragment seen in the crystal structure (Fan & Hendrickson 2005) do not account for the ability of the FSH β-subunit and the FSHR to control receptor and ligand binding specificity. Although the experimental data do not exclude a role for contacts between hFSH residues in β-subunit loop 2 and the receptor, they do not support them. Contacts seen between the seatbelt and the receptor do not explain the influence of the seatbelt on receptor binding specificity. As we have reviewed recently (Moyle et al. 2005), the manner in which hFSH binds to the FSHR fragment makes it difficult to envision how receptor-bound ligand can initiate signal transduction. Taken together, these considerations lead us to conclude that the manner in which FSH and related ligands contact their cell surface receptors is likely to differ from that in which hFSH contacts the fragment of the hFSHR in crystals. We propose this is because the crystals contain hFSH bound to a cryptic receptor site.
Our view of ligand binding to cell surface glycoprotein hormone receptors (Moyle, et al., 2004) differs considerably from that seen in crystals that contain a receptor fragment (Fan & Hendrickson 2005). As discussed next, several investigators, including us, have found that the SSD portion of the extracellular domain has a substantial role in ligand binding. The SSD is often presumed to be a “linker” or “hinge” that tethers the LRD to the TMD (Ji et al. 2002;Nakabayashi et al. 2000), a role for the SSD that was also proposed by Fan and Hendrickson in their model of signaling (Fan & Hendrickson 2005). Although the SSD cannot bind ligands in the absence of the LRD, it makes important contributions to ligand binding specificity (Bernard, Myers, & Moyle 1998;Gromoll et al. 2000) and efficacy (Moyle, et al., 2004). It contributes to the binding of some lutropins to LHR (Moyle, et al., 2004) and is capable of blocking the binding of bovine LH to the human LHR (Bernard, Myers, & Moyle 1998). The SSD also influences signal transduction and some SSD mutations cause constitutive receptor activity (Nakabayashi, et al., 2000). Disulfide-rich parts of the SSD are highly conserved in all vertebrate receptors and in receptor-like proteins found in invertebrates, an observation suggesting that these regions have important structural and/or physiological functions. Exon 10 of the LHR, which encodes residues in the variable part of its SSD is known to influence ligand binding (Moyle, et al., 2004;Muller et al. 2004a;Simula et al. 1995). Indeed, the co-evolution of lutropins and their receptors in marmoset monkeys led to the development of LHR that lack exon 10 and the replacement of the LH β-subunit with the CG β-subunit (Muller et al. 2004b). Therefore, even though the LRD can bind some ligands per se, it is clearly not the only region of the receptor that has a role in hormone-receptor interactions. The role of the SSD in ligand binding, binding specificity, and efficacy suggests that it is likely to be located adjacent to the LRD (Moyle, et al., 2004). In this location it would be expected to prevent binding of ligands to the concave surface of the LRD, the manner in which it was found to bind to the FSHR fragment that lacks the SSD (Fan & Hendrickson 2005).
Potential roles of the cryptic binding sites
Why does the FSHR contain a cryptic hCG docking site? While this question cannot be answered, there are at least three possible explanations. One is that the cryptic docking site is the result of an evolutionary phenomenon involving receptor duplication followed by the divergence of binding specificity, a phenomenon that we proposed based on the abilities of hormone and receptor chimeras to recognize ligands and receptors (Moyle, et al., 1994;Moyle, et al., 2004). Once the lutropin binding site of the FSHR had been suppressed sufficiently, there may have been little evolutionary pressure to eliminate it. We do not favor this notion, however, because the two lysines that were replaced in the FSHR by their LHR counterparts to create FSHR2 (Moyle, et al., 2004) are highly conserved in all vertebrates except fish. The FSHR of most fish differ from those of other vertebrates and usually have a much smaller SSD. Therefore, the FSHR residues that are present in most vertebrate FSHR species may have been conserved for a reason. Another possibility is that residues in the cryptic hCG binding site of the FSHR are essential for receptor folding and expression. We also consider this explanation highly unlikely since it is possible to create functional LHR/FSHR chimeras in which many residues in this region have been altered. The third explanation, which we favor, is the notion that the cryptic lutropin binding site in the FSHR has been highly conserved in amphibians, reptiles, birds, and mammals because it has an important function. The ability of TMD mutations to alter the ligand binding specificity of the human FSHR (Smits, et al., 2003b) raises the possibility that post-translational modifications of its TMD or the interaction of the FSHR with other proteins may enable it to bind lutropins in certain physiological conditions. For example, it might enable the FSHR in the Sertoli cells of the testis or the granulosa cells of the ovary to respond to lutropins and thereby augment the response to FSH. An example of this type of regulation may involve the role of equine CG. The horse ovary depends on the ability of equine CG to stimulate follicle development and steroidogenesis during pregnancy (Murphy & Martinuk 1991). Equine CG has been found not to bind to the horse FSHR (Richard et al., 1997), but it has remained unclear as to how this potent lutropin can stimulate both the LHR and FSHR in the ovaries of pregnant mares in ways needed to initiate significant folliculogenesis. The notion that the TMD can alter ligand binding to the FSHR raises the possibility that factors that alter the TMD of the horse FSHR during pregnancy may expose a cryptic lutropin binding site that can be activated by equine CG.
Finally, the finding that chimeras of TSHR/LHR having only 8 LHR specific residues bind both hCG and TSH (Smits, et al., 2003a;Vassart, Pardo, & Costagliola 2004) suggests that the TSHR may have a cryptic hCG binding site similar to the FSHR. The central region of the LRD in the TSHR can be replaced without disrupting its ability to interact functionally with TSH (Nagayama et al. 1991), which suggests that the organization of the TSHR may be similar to that of the FSHR. Furthermore, we have found that modification of similar regions of hTSH and hFSH α-subunits prevents binding of both hormones to their receptors and proposed that TSH docks with the TSHR in a fashion that is similar to that by which FSH docks with the FSHR (Moyle, et al., 2004). These considerations suggest that it may be worth searching for physiological factors that regulate the receptor binding specificities of both the FSHR and TSHR. Exon 10 of the LHR has been shown to have a potential role in altering ligand binding and signaling and its absence in some conditions can lead to infertility (Gromoll, et al., 2000). Thus, it is conceivable that all glycoprotein hormone receptors contain intrinsic mechanisms that would permit their ligand binding specificity to be regulated.
Acknowledgments
We are indebted to Robert Campbell, Serono Research Institute, Rockland MA for the hFSHR for helpful discussions early in this work. We thank Dr. William Munroe, Hybritech Incorporated, San Diego, CA, a subsidiary of Beckman Coulter, Inc. for antibodies A113 and B603 used in these studies. We thank Christine Kelton, Serono Research Institute, Rockland MA for the hFSHR cDNA used in these studies.
Abbreviations
- hFSH
human follitropin
- hFSHδ4
hFSH analog lacking 4 residues in β-subunit loop 2
- FSHR
follitropin receptor
- LHR
lutropin receptor
- hCG
human choriogonadotropin
- hLH
human lutropin
- LRD
leucine-rich repeat domain of receptor extracellular domain
- SSD
signaling-specificity domain of the receptor extracellular domain, a portion of the receptor often termed the “hinge”
- TMD
transmembrane domain of the receptor
- FSHR2
chimera of FSHR and LHR that contains only two LHR specific residues (rFSHR-K88N,K163G)
- CFC101-114
hCG/hFSH chimera heterodimer containing the human α-subunit, hCG β-subunit residues 1-100 plus 115-145, and hFSH β-subunit residues 95-108 in place of their hCG counterparts (i.e., residues 101-114)
- α2
α-subunit loop 2
- β2
β-subunit loop 2
Footnotes
Supported by grant HD14907 from the NIH Institute of Child Health and Human Development and personal funds from one of the authors (WRM).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Win Lin, Departments of OBGYN, UMDNJ Robert Wood Johnson (Rutgers) Medical School, 675 Hoes Lane, Piscataway, NJ 08854.
Michael P. Bernard, Departments of OBGYN, UMDNJ Robert Wood Johnson (Rutgers) Medical School, 675 Hoes Lane, Piscataway, NJ 08854
Donghui Cao, Departments of OBGYN, UMDNJ Robert Wood Johnson (Rutgers) Medical School, 675 Hoes Lane, Piscataway, NJ 08854.
Rebecca V. Myers, Departments of OBGYN, UMDNJ Robert Wood Johnson (Rutgers) Medical School, 675 Hoes Lane, Piscataway, NJ 08854
John E. Kerrigan, Academic Systems and Technologies, UMDNJ, 65 Bergen Street, Newark, NJ 07101
William R. Moyle, Departments of OBGYN, UMDNJ Robert Wood Johnson (Rutgers) Medical School, 675 Hoes Lane, Piscataway, NJ 08854
References
- Bernard MP, Cao D, Myers RV, Moyle WR. Tight Attachment Of Chitin Binding Domain Tagged Proteins To Surfaces Coated With Acetylated Chitosan. AnalBiochem. 2004;327:278–283. doi: 10.1016/j.ab.2003.12.029. [DOI] [PubMed] [Google Scholar]
- Bernard MP, Myers RV, Moyle WR. Lutropins Appear To Contact Two Independent Sites In The Extracellular Domain Of Their Receptors. Biochemical Journal. 1998;335:611–617. doi: 10.1042/bj3350611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T, Schofield PR, Sprengel R. Amino-terminal leucine-rich repeats in gonadotropin receptors determine hormone selectivity. EMBOJ. 1991;10:1885–1890. doi: 10.1002/j.1460-2075.1991.tb07714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooker J, Harper JF, Terasaki WL, Moylan RD. Radioimmunoassay of cyclic AMP and cyclic GMP. Advances in Cyclic Nucleotide Research. 1979;10:1–33. [PubMed] [Google Scholar]
- Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends PharmacolSci. 2005;26(3):131–137. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Campbell RK, Dean Emig DM, Moyle WR. Conversion of human choriogonadotropin into a follitropin by protein engineering. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:760–764. doi: 10.1073/pnas.88.3.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias JA. Endocrinology: fertility hormone in repose. Nature. 2005;433(7023):203–204. doi: 10.1038/433203a. [DOI] [PubMed] [Google Scholar]
- Dias JA, Zhang Y, Liu X. Receptor binding and functional properties of chimeric human follitropin prepared by an exchange between a small hydrophilic intercysteine loop of human follitropin and human lutropin. Journal of Biological Chemistry. 1994;269:25289–25294. [PubMed] [Google Scholar]
- Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature. 2005;433(7023):269–277. doi: 10.1038/nature03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox KM, Dias JA, Van Roey P. Three-dimensional structure of human follicle-stimulating hormone. Molecular Endocrinology. 2001;15:378–389. doi: 10.1210/mend.15.3.0603. [DOI] [PubMed] [Google Scholar]
- Gromoll J, Eiholzer U, Nieschlag E, Simoni M. Male hypogonadism caused by homozygous deletion of exon 10 of the luteinizing hormone (LH) receptor: differential action of human chorionic gonadotropin and LH. JClinEndocrinolMetab. 2000;85(6):2281–2286. doi: 10.1210/jcem.85.6.6636. [DOI] [PubMed] [Google Scholar]
- Grossmann M, Szkudlinski MW, Wong R, Dias JA, Ji TH, Weintraub BD. Substitution of the seat-belt region of the thyroid-stimulating hormone (TSH) beta-subunit with the corresponding regions of choriogonadotropin or follitropin confers luteotropic but not follitropic activity to chimeric TSH. Journal of Biological Chemistry. 1997;272(24):15532–15540. doi: 10.1074/jbc.272.24.15532. [DOI] [PubMed] [Google Scholar]
- Han Y, Bernard MP, Moyle WR. hCGβ Residues 94–96 alter LH activity without appearing to make key receptor contacts. Molecular & Cellular Endocrinology. 1996;124:151–161. doi: 10.1016/s0303-7207(96)03936-6. [DOI] [PubMed] [Google Scholar]
- Javitch JA. The ants go marching two by two: oligomeric structure of G-protein-coupled receptors. MolPharmacol. 2004;66(5):1077–1082. doi: 10.1124/mol.104.006320. [DOI] [PubMed] [Google Scholar]
- Ji I, Lee C, Song Y, Conn PM, Ji TH. Cis- and trans-activation of hormone receptors: the LH receptor. Molecular Endocrinology. 2002;16(6):1299–1308. doi: 10.1210/mend.16.6.0852. [DOI] [PubMed] [Google Scholar]
- Jiang X, Dreano M, Buckler DR, Cheng S, Ythier A, Wu H, Hendrickson WA, Tayar NE, el Tayar N. Structural predictions for the ligand-binding region of glycoprotein hormone receptors and the nature of hormone-receptor interactions. Structure. 1995;3(12):1341–1353. doi: 10.1016/s0969-2126(01)00272-6. [DOI] [PubMed] [Google Scholar]
- Lapthorn AJ, Harris DC, Littlejohn A, Lustbader JW, Canfield RE, Machin KJ, Morgan FJ, Isaacs NW. Crystal structure of human chorionic gonadotropin. Nature. 1994;369:455–461. doi: 10.1038/369455a0. [DOI] [PubMed] [Google Scholar]
- Lee C, Irizarry K. The GeneMine System for genome/proteome annotation and collaborative data mining. IBM Systems Journal. 2001;40:592–603. [Google Scholar]
- Lin W, Ransom MX, Myers RV, Bernard MP, Moyle WR. Addition of an N-terminal dimerization domain promotes assembly of hCG analogs: implications for subunit combination and structure-function analysis. Molecular & Cellular Endocrinology. 1999;152:91–98. doi: 10.1016/s0303-7207(99)00056-8. [DOI] [PubMed] [Google Scholar]
- Moyle WR, Campbell RK, Myers RV, Bernard MP, Han Y, Wang X. Co-evolution of ligand-receptor pairs. Nature. 1994;368:251–255. doi: 10.1038/368251a0. [DOI] [PubMed] [Google Scholar]
- Moyle WR, Ehrlich PH, Canfield RE. Use of monoclonal antibodies to hCG subunits to examine the orientation of hCG in the hormone-receptor complex. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:2245–2249. doi: 10.1073/pnas.79.7.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle WR, Lin W, Myers RV, Cao D, Kerrigan JE, Bernard MP. Models of glycoprotein hormone receptor interaction. Endocrine. 2005;26(3):189–205. doi: 10.1385/ENDO:26:3:189. [DOI] [PubMed] [Google Scholar]
- Moyle WR, Xing Y, Lin W, Cao D, Myers RV, Kerrigan JE, Bernard MP. Model of glycoprotein hormone receptor ligand binding and signaling. Journal of Biological Chemistry. 2004;279(43):44442–44459. doi: 10.1074/jbc.M406948200. [DOI] [PubMed] [Google Scholar]
- Muller T, Gromoll J, Simula AP, Norman R, Sandhowe-Klaverkamp R, Simoni M. The carboxyterminal peptide of chorionic gonadotropin facilitates activation of the marmoset LH receptor. ExpClinEndocrinolDiabetes. 2004a;112(10):574–579. doi: 10.1055/s-2004-830409. [DOI] [PubMed] [Google Scholar]
- Muller T, Simoni M, Pekel E, Luetjens CM, Chandolia R, Amato F, Norman RJ, Gromoll J. Chorionic gonadotrophin beta subunit mRNA but not luteinising hormone beta subunit mRNA is expressed in the pituitary of the common marmoset (Callithrix jacchus) JMolEndocrinol. 2004b;32(1):115–128. doi: 10.1677/jme.0.0320115. [DOI] [PubMed] [Google Scholar]
- Murphy BD, Martinuk SD. Equine chorionic gonadotropin. Endocrine Reviews. 1991;12:27–44. doi: 10.1210/edrv-12-1-27. [DOI] [PubMed] [Google Scholar]
- Nagayama Y, Wadsworth HL, Chazenbalk GD, Russo D, Seto P, Rapoport B. Thyrotropin-luteinizing hormone/chorionic gonadotropin receptor extracellular domain chimeras as probes for thyrotropin receptor function. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:902–905. doi: 10.1073/pnas.88.3.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi K, Kudo M, Kobilka B, Hsueh AJ. Activation of the luteinizing hormone receptor following substitution of Ser-277 with selective hydrophobic residues in the ectodomain hinge region. Journal of Biological Chemistry. 2000;275:30264–30271. doi: 10.1074/jbc.M005568200. [DOI] [PubMed] [Google Scholar]
- Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annual Review of Biochemistry. 1981;50:465–495. doi: 10.1146/annurev.bi.50.070181.002341. [DOI] [PubMed] [Google Scholar]
- Richard F, Martinat N, Remy JJ, Salesse R, Combarnous Y. Cloning, sequencing and in vitro functional expression of recombinant donkey follicle-stimulating hormone receptor: a new insight into the binding specificity of gonadotrophin receptors. JMolEndocrinol. 1997;18(3):193–202. doi: 10.1677/jme.0.0180193. [DOI] [PubMed] [Google Scholar]
- Simula AP, Amato F, Faast R, Lopata A, Berka J, Norman Luteinizing hormone/chorionic gonadotropin bioactivity in the common marmoset (Callithrix jacchus) is due to a chorionic gonadotropin molecule with a structure intermediate between human chorionic gonadotropin and human luteinizing hormone. Biology of Reproduction. 1995;53(2):380–389. doi: 10.1095/biolreprod53.2.380. [DOI] [PubMed] [Google Scholar]
- Smits G, Campillo M, Govaerts C, Janssens V, Richter C, Vassart G, Pardo L, Costagliola S. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. The EMBO Journal. 2003a;22:2692–2703. doi: 10.1093/emboj/cdg260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits G, Olatunbosun O, Delbaere A, Pierson R, Vassart G, Costagliola S. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. NEnglJMed. 2003b;349(8):760–766. doi: 10.1056/NEJMoa030064. [DOI] [PubMed] [Google Scholar]
- Southern PJ, Berg P. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. JMolApplGenet. 1982;1(4):327–341. [PubMed] [Google Scholar]
- Vassart G, Pardo L, Costagliola S. A molecular dissection of the glycoprotein hormone receptors. TIBS. 2004;29(3):119–126. doi: 10.1016/j.tibs.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Ward DN, Moore WT. Comparative studies of mammalian glycoprotein hormones. In: Alexander NJ, editor. Animal models for research on contraception and fertility. Harper and Row; New York: 1979. pp. 151–164. [Google Scholar]
- Wu H, Lustbader JW, Liu Y, Canfield RE, Hendrickson WA. Structure of human chorionic gonadotropin at 2.6Å resolution from MAD analysis of the selenomethionyl protein. Structure. 1994;2:545–558. doi: 10.1016/s0969-2126(00)00054-x. [DOI] [PubMed] [Google Scholar]
- Xing Y, Lin W, Jiang M, Cao D, Myers RV, Bernard MP, Moyle WR. Use of Protein Knobs to Characterize the Position of Conserved α-Subunit Regions in Lutropin Receptor Complexes. Journal of Biological Chemistry. 2004a;279(43):44427–44437. doi: 10.1074/jbc.M406931200. [DOI] [PubMed] [Google Scholar]
- Xing Y, Lin W, Jiang M, Myers RV, Cao D, Bernard MP, Moyle WR. Alternatively folded choriogonadotropin analogs. Implications for hormone folding and biological activity. Journal of Biological Chemistry. 2001;276(50):46953–46960. doi: 10.1074/jbc.M108374200. [DOI] [PubMed] [Google Scholar]
- Xing Y, Myers RV, Cao D, Lin W, Jiang M, Bernard MP, Moyle WR. Glycoprotein hormone assembly in the endoplasmic reticulum: IV. Probable mechanism of subunit docking and completion of assembly. Journal of Biological Chemistry. 2004b;279(34):35458–35468. doi: 10.1074/jbc.M403055200. [DOI] [PubMed] [Google Scholar]