

Abstract
Increased transcriptional activity of β -catenin resulting from Wnt/Wingless dependent or independent signaling has been detected in many types of human cancer, but the underlying mechanism of Wnt-independent regulation is poorly understood. We have demonstrated that AKT, which is activated downstream from EGFR signaling, phosphorylates β-catenin at Ser552 in vitro and in vivo. AKT-mediated phosphorylation of β -catenin causes its disassociation from cell-cell contacts and accumulation in both the cytosol and nucleus, and enhances its interaction with 14-3-3 ζ via a binding motif containing Ser552. Phosphorylation of β-catenin by AKT increases its transcriptional activity and promotes tumor cell invasion, indicating that AKT-dependent regulation of β-catenin plays a critical role in tumor invasion and development.
β-catenin, originally identified as a component of cell-cell adhesion structures, interacts with the cytoplasmic domain of E-cadherin and links E-cadherin to α-catenin, which in turn mediates anchorage of the E-cadherin complex to the cortical actin cytoskeleton (1–3). Genetic and embryologic studies have revealed that β-catenin is also a component of the Wnt signaling pathway and that it exhibits signaling functions (4). In the absence of a Wnt/Wingless signal, cytoplasmic β-catenin interacts with axin/conductin, glycogen synthase kinase-3β (GSK-3β), and the adenomatous polyposis coli protein (APC), which competes with E-cadherin for binding to the armadillo (ARM)-like repeats of β-catenin (5). β-catenin is phosphorylated in its N-terminal domain by GSK-3β, which leads to its degradation via the SCF/ubiquitin/proteasome pathway (6–11). Activation of the Wnt/Wingless pathway inhibits GSK-3β-dependent phosphorylation of β-catenin. Stabilized, hypophosphorylated β-catenin translocates to the nucleus, where it interacts with transcription factors of the TCF/LEF-1 family, leading to the increased expression of genes, such as c-myc and cyclin D1 (12–14). Hepatocyte growth factor (HGF) and epidermal growth factor (EGF) induce β-catenin signaling under conditions where they stimulate cell motility (15,16). Gain of function mutations in β-catenin, including N-terminal mutations that lead to stabilization of the protein or loss of function mutations in proteins that participate in the regulated turnover of β-catenin, such as the tumor suppressor APC, result in upregulation of β-catenin protein levels, thus increasing its transcriptional activity. Increased β-catenin-TCF/LEF-1 transactivation by enhanced β-catenin stability, resulting from mutations in APC, AXIN1, or CTNNB1 (which encodes β-catenin), is found in a wide variety of human cancers, including colon cancer, desmoid tumor, gastric cancer, hepatocarcinoma, medulloblastoma, melanoma, ovarian cancer, pancreatic cancer, and prostate cancer (17–19).
Mutations of Wnt pathway proteins that alter the stability of β-catenin is not the only factor that contributes to β-catenin activation. For instance, in 12 of 20 (60.0%) endometrial cancers, β-catenin was found to accumulate in the nucleus, which is a hallmark of β-catenin activation, whereas there were only 2 instances of mutations in the CTNNB1 gene (20). Similarly, only 1 of 65 primary melanomas had detectable CTNNB1 mutations, with a third of the cases displaying nuclear accumulation of β-catenin (21). Moreover, nearly 50% of hepatocellular carcinomas, in which the APC gene is rarely mutated, reveal nuclear accumulation of β-catenin protein, and genetic alterations in CTNNB1 are detected only in 16%–26% of the tumors (22–25). Clearly, more than one mechanism regulates the activity of β-catenin (26). In response to EGF stimulation, β-catenin translocates into the nucleus and increases its transactivation without altering its stability and phosphorylation level by GSK-3β (16). Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis chronic myelogenous leukemia (CML) have high nuclear β-catenin accumulation presumably driven by Bcr-Abl (27).
In this report, we demonstrate that AKT phosphorylates β-catenin at serine (Ser, S) 552, which leads to its disassociation from cell-cell contacts, increases its binding to 14-3-3ζ and its transcriptional activity, and enhances invasion by tumor cells.
EXPERIMENTAL PROCEDURES
Cells and cell culture conditions
A431 human epidermoid carcinoma cells, CHO AA8 cells, and 293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% bovine calf serum (HyClone, Logan, UT). Cell cultures were made quiescent by growing them to confluence and then replacing the medium with fresh medium containing 0.5% serum for 1 day.
Materials
Rabbit monoclonal antibody recognizing phosphorylated RRXS/T peptide was obtained from Cell Signaling Technology (Danvers, MA). Polyclonal antibody for 14-3-3ζ, and monoclonal antibodies for β-catenin (E-5) and HA were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal phospho-β-catenin (Ser33/Ser37)-specific antibody, mouse monoclonal antibodies for FLAG and tubulin, and EGF were purchased from Sigma (St. Louis, MO), along with Hygromycin, AKT inhibitor IV, active His-tagged AKT1 (purified from insect cells), and protein kinase A (purified from bovine heart) from EMD Biosciences San Diego, CA). Active GST-AKT2 (purified from insect cells) was obtained from Biomol International (Plymouth Meeting, PA), Hoechst 33342 and FITC-conjugated anti-mouse antibody and Texas red-conjugated anti-rabbit antibody from Molecular Probes (Eugene, OR), and HyFect transfection reagents from Denville Scientific (Metuchen, NJ). GelCode Blue Stain Reagent was obtained from Pierce Biotechnology (Rockford, IL).
Transfection
Cells were plated at a density of 4x105 per 60-mm-diameter dish 18 h prior to transfection. Transfection was performed using either calcium phosphate or HyFect reagents (Deveville Scientific), according to the vendor’s instructions. Transfected cultures were selected with hygromycin (200 μg/ml) for 10 to 14 days at 37°C. At that time, antibiotic-resistant colonies were picked, pooled, and expanded for further analysis under selective conditions.
Immunoprecipitation and immunoblotting analysis
Extraction of proteins with a modified buffer from cultured cells was followed by immunoprecipitation and immunoblotting with corresponding antibodies, as described previously (28).
DNA constructs and mutagenesis
A PCR-amplified human β-catenin cDNA was cloned either into pColdI vector (TaKaRa, Shiga, Japan) between BamHI and HindIII or into pcDNA3.1/hygro(+)-FLAG vector between BamHI and NotI. pColdI-β-catenin S552A or S675A, and pcDNA3.1-FLAG-β-catenin S552A, S552D, or S675A were made using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). pcDNA3.1 HA-AKT1 DD, HA-AKT2 DD, and HA-AKT1 AAA have been described previously (29).
Purification of recombinant proteins
The WT and mutants of His-β-catenin protein were expressed in bacteria and purified, as described previously (30).
In vitro invasion assay
Cell invasion was assessed by the invasion of the cells through Matrigel-coated Transwell inserts. Briefly, Transwell inserts with a 12 μm pore size were coated with 100 μl of a final concentration of 0.78 mg/mL Matrigel in cold serum-free medium. Cells were trypsinized, and cell suspension (500 μL; 1 x 106 cells/mL) was added in triplicate wells. After a 24-hour incubation, cells that invaded the Matrigel and passed through the filter were stained with crystal violet and photographed using a digital camera mounted onto a microscope with 50X magnification. The membranes were dissolved in 4% deoxycholic acid and read colorimetrically at 590 nm.
In vitro kinase assays
The kinase reactions were done by mixing purified His-β-catenin with or without purified active AKT or PKA in kinase assay buffer containing 10 μCi of [gamma-32P] ATP, 10 mM Tris-HCl (pH 7.4), 5 mM MnCl2, 1 mM dithiothreitol, and 20 μM ATP for 20 min at 30°C. Reactions were stopped by adding an equal volume of 2X SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer and boiling for 5 min. Samples were then separated by 6% SDS-PAGE and transferred onto nitrocellulose membranes for exposing to X-ray film.
Luciferase reporter gene assay
In order to measure the transcriptional activity of TCF/LEF-1, CHO cells were seeded in 24-well plates at 1.5x104 cells/well. 24 h after seeding, TCF/LEF-1 reporter (pTOP-FLASH) or control vector (pFOP-FLASH) were transiently transfected with 0.2 μg WT or mutants of β-catenin together with or without 0.2 μg AKT1 DD, 0.2 μg 14-3-3ζ, or 0.2 μg of pEGFR. Twelve h after transfection, the medium was replaced with 0.1% serum for another 12–24 h, and EGF (100 ng/ml) was added 6 h before harvesting. Ten μl out of the 100 μl cell extract were used for measuring luciferase activity.
Immunofluorescence analysis
Cells were fixed and incubated with primary antibodies, Alexa Fluor dye-conjugated secondary antibodies, and Hoechst 33342, according to standard protocols. Cells were examined using a deconvolutional microscope (Zeiss, Thornwood, NY) with a 63-Å oil immersion objective. Axio Vision software from Zeiss was used to deconvolute Z-series images.
Mass spectrometry analysis
The GelCode Blue-stained gel band was digested in-gel with 200 ng modified trypsin (sequencing grade, Promega) at 37°C for 24 h. Resulting peptides were analyzed by Nano-LC-MS/MS with on-line desalting on a system consisting of a Famos® autosampler, Ultimate® Nano-LC module and a Switchos® pre-column switching device (Dionex Corp., Sunnyvale CA) on a 75umX150 mm C-18 column (Dionex Corp.). Electrospray ion trap mass spectrometry was performed on a linear ion-trap mass spectrometer (LTQ, Thermo, San Jose, CA). A portion of the digest was desalted (C18-ZipTip, Millipore, MA) and analyzed by Maldi-TOF (Voyager DE-STR, Applied Biosystems, Framingham, MA). Proteins were identified by database search of the fragment spectra against the NCBI non-redundant protein database using Mascot (Matrix Science, London, UK) and Sequest (Thermo, San Jose, CA).
Isolation of nuclei, cytosol and membrane
Nuclei, cytosol, and membrane of cells were isolated using the Nuclear Extract Kit from Active Motif North America (Carlsbad, CA) and the ProteoExtract® Subcellular Proteome Extraction Kit from Calbiochem (San Diego, CA).
Pulse-chase analysis
293T cells transfected with FLAG-tagged WT or mutants of β-catenin for 2 days were incubated in the absence of methionine for 20 min, pulse-labeled for 1 h with 250 μCi/ml [35S] methionine, washed twice, and incubated in medium containing excess unlabeled methionine for 0, 24, 36, or 48 h. Proteins were immunoprecipitated with anti-FLAG antibodies. Samples were then separated on 6% SDS-PAGE and transferred to nitrocellulose membranes for exposure to X-ray film.
RESULTS
AKT phosphorylates β-catenin at Ser552 in vitro and in vivo
We previously showed that EGF stimulation results in translocation of β-catenin into the nucleus and increased transcriptional activity without altering its stability and phosphorylation level by GSK-3β (16). To examine whether the increased transcriptional activity of β-catenin is due to a possible unknown posttranslational modification, we analyzed a tryptic digest of β-catenin immunoprecipated from EGF-stimulated A431 cells with mass spectrometry. One candidate phosphopeptide spanning amino acids 550–565 was detected by MALDI-TOF mass spectrometry. S552 was then identified as a phosphorylation site in this β-catenin peptide by liquid chromatography-coupled ion trap mass spectrometry (LC-MS/MS) (Fig. 1A) (The presence of Arg at the N terminus of the peptide is due to the known inefficient tryptic digestion of the RX bond in RXpS). The 487-TQRRTS-552 sequence of β-catenin is closely related to the AKT phosphorylation motif RXRXXS/T (31) and the PKA phosphorylation consensus sequence (R/K)2XS/T (32). Furthermore, this sequence was also recognized as an AKT or PKA-putative phosphorylation site by using the motif-based profile-scanning ScanSite program (http://scansite.mit.edu/). To identify the kinase phosphorylating β-catenin at S552, we performed in vitro kinase assays with purified active AKT1, AKT2, or PKA mixed with purified wild-type (WT) His-β-catenin, His-β-catenin S552 alanine (Ala, A), or His-β-catenin S675A, in which the known PKA phosphorylation residue was mutated to Ala (33,34). As expected, and consistent with previous publications (33,34), PKA phosphorylated β-catenin, and this phosphorylation was largely abrogated by mutation at S675 but not at S552 (Fig. 1B). In contrast, both active AKT1 and AKT2 were able to phosphorylate β-catenin, and incubation with AKT inhibitor IV (35) or mutation at S552 but not at S675 abolished these effects. Furthermore, phosphorylated WT β-catenin and β-catenin S675A, but not β-catenin S552A, could be detected by a commercial antibody recognizing phosphorylated RRXS/T peptide, indicating that this antibody can specifically detect phosphorylated β-catenin at S552. (This antibody did not recognize PKA-phosphorylated S675. Data not shown). These results indicate that AKT, but not PKA, phosphorylates β-catenin at S552 in vitro.
Fig. 1. AKT phosphorylates β-catenin at S552 in vitro and in vivo.
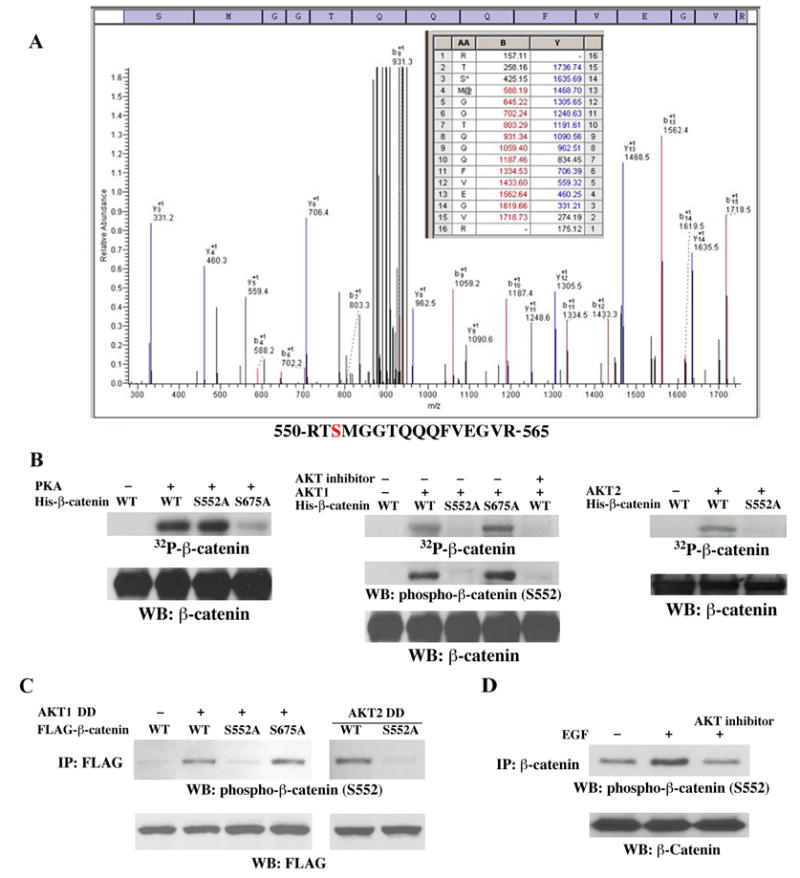
A, β-catenin immunoprecipated from EGF-stimulated A431 cells was analyzed with mass spectrometry. Mass spectrometric analysis of a tryptic fragment R550–R565 indicates Ser-552 is phosphorylated. The detected b- and y- ions are indicated in red and blue, respectively. The m/z difference between y-14 and y-13 matched with phospho-Ser. B–D, Immunoblotting analyses with the indicated antibodies. B, In vitro kinase assays were performed with purified active PKA (left panel), AKT1 (middle panel), or AKT2 (right panel), with purified bacterially expressed His-β-catenin, His-β-catenin S552A, or His-β-catenin S675A with or without AKT inhibitor. C, AKT1 DD or AKT2 DD was co-transfected with FLAG-tagged WT β-catenin, β-catenin S552A, or β-catenin S675A into 293T cells. Anti-FLAG antibody was used for immunoprecipitation. D, Serum-starved A431 cells were pretreated with the AKT inhibitor IV (10 μm) for 30 min before EGF treatment (100ng/ml). Anti-β-catenin antibody was used for immunoprecipitation.
To test whether AKT phosphorylates β-catenin in vivo, constitutively active AKT1 DD (T308D/S473D) was co-transfected with FLAG-tagged WT β-catenin, β-catenin S552A, or β-catenin S675A into 293T cells. Immunoblotting of immunoprecipitated FLAG-tagged protein with the phospho-β-catenin S552 antibody showed that active AKT1 induces phosphorylateion of WT β-catenin and β-catenin S675A, but not the β-catenin S552A mutant (Fig. 1C). Similarly, constitutively active AKT2 DD (T309D/S474D) was capable of inducing phosphorylation of WT β-catenin but not the β-catenin S552A mutant. These results indicate that β-catenin was phosphorylated by AKT at S552 but not S675 in vivo. EGF treatment activates AKT (36). To test the effect of EGF stimulation on β-catenin phosphorylation, A431 cells were treated with EGF for 30 min. As shown in Fig. 1D, EGF induced phosphorylation of endogenous β-catenin at S552, which was blocked by pretreatment with AKT inhibitor IV. This is consistent with β-catenin phosphorylation at S552 in response to EGF treatment, as detected by mass spectrometry.
β-catenin phosphorylated by AKT disassociates from cell contacts and translocates into the cytosol and nucleus
β-catenin localized in different cellular compartments forms distinct complexes and executes differential cellular functions. To examine whether β-catenin phosphorylation by AKT affects its subcellular distribution, a constitutively active AKT1 DD or an AKT1 AAA (K179A/T308A/S473A) kinase-dead mutant was transfected into A431 cells. Expression of AKT1 DD, but not AKT1 AAA, resulted in translocation of a portion of β-catenin from cell-cell contacts into the cytosol and nucleus (Fig. 2A). Similar data were also observed following expression of AKT2 DD and AKT2 AAA (data not shown). Furthermore, a phosphorylation-mimic β-catenin S552D mutant, in which Ser was mutated into aspartic acid (Asp, D), showed significantly increased cytosolic and nuclear localization in contrast to WT or S552A mutant of β-catenin that was primarily localized in cell-cell contacts (Fig. 2B). Consistently, immunoblotting experiments detected an increase in cytosolic and nuclear β-catenin S552D mutant in contrast to its WT, whereas the nuclear accumulation of β-catenin S552A mutant was reduced (Fig. 2C). Moreover, the amount of the nuclear protein lamin B remained unchanged (Fig. 2C) and we did not detect any E-cadherin in the nuclear fractions, indicating that the nuclear preparations were free of cytosolic contaminants (data not shown). Correlating with the differential redistribution of β-catenin S552D and S552A mutants in the cytosol and nuclei, reduced membrane-bound β-catenin S552D and increased membrane-bound β-catenin S552A were detected in contrast to WT β-catenin (Fig. 2C). To test whether the differential distribution of the WT and mutants of β-catenin affected their phosphorylation by GSK3β, which leads to proteasomal degradation of β-catenin, immunoprecipitated FLAG-tagged WT β-catenin, β-catenin S552A, and β-catenin S552D were immnuoblotted with phospho-β-catenin (S33/S37) antibody. As shown in Fig. 2D, β-catenin S552A and β-catenin S552D have comparable phosphorylation by GSK3β to WT β-catenin. Furthermore, pulse-chase analyses revealed that transiently expressed FLAG-tagged β-catenin S552A and β-catenin S552D had a comparable half-life to WT β-catenin (quantified by scanning densitometry) (Fig. 2E). These data indicate that β-catenin phosphorylation by AKT leads to its disassociation from cell-cell contacts without altering its phosphorylation by GSK-3β.
Fig. 2. β -catenin phosphorylated by AKT disassociates from cell contacts and translocates into the cytosol and nucleus.
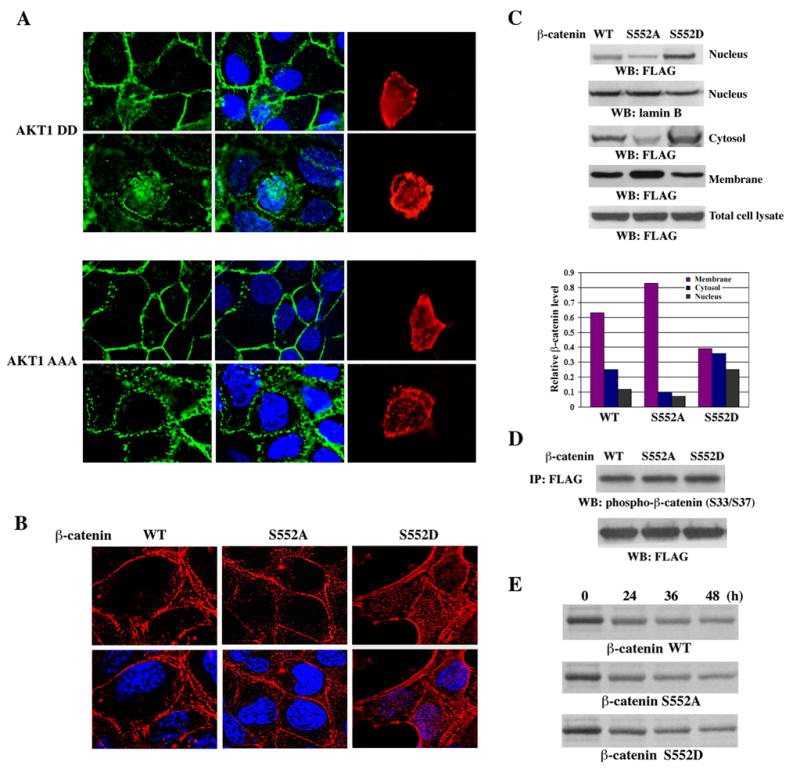
A, A431 cells transiently expressing HA-AKT1 DD or HA-AKT1 AAA were stained with an anti-β-catenin antibody (green), an anti-HA antibody (red), and Hoechst 33342 (blue). White color indicates overlap of green and blue colors. B, A431 cells stably expressing FLAG-tagged β-catenin, β-catenin S552A, or β-catenin S552D were stained with an anti-FLAG antibody (red) and Hoechst 33342 (blue). Pink color indicates overlap of red and blue colors. C, Nuclear, cytosolic, and membrane fractions of A431 cells stably expressing FLAG-tagged β-catenin, β-catenin S552A, or β-catenin S552D were processed for immunoblotting with anti-FLAG or anti-lamin B antibody (upper group panels). The data were quantified by scanning densitometry (lower panel). D, Immunoprecipitated FLAG-tagged WT β-catenin, β-catenin S552A, and β-catenin S552D, which were transiently expressed in 293T cells, were immnuoblotted with phospho-β-catenin (S33/S37) antibody. E, Pulse-chase analyses of transiently expressed FLAG-tagged β-catenin S552A and β-catenin S552D in 293T cells.
β-catenin phosphorylated by AKT increases its association with 14-3-3 ζ
14-3-3 proteins function as key regulators of signal transduction by binding to specific phospho-Ser/Thr-containing sequence motifs within a wide range of target proteins (37). It has been shown that 14-3-3ζ is a β-catenin-associated protein and can facilitate β-catenin transactivation by AKT (38). 14-3-3 binding motif RX1–2pS/pTX2–3, in which a Ser or a Thr at position −1 and/or −2 with respect to phosphoserine is found in many 14-3-3-binding proteins (39), and R/K(X)XXpS/TXP (37) is closely related to 489-RRTSMG-554 of β-catenin. To examine the effect of β-catenin phosphorylated by AKT on its association with 14-3-3ζ, FLAG-tagged WT β-catenin was co-transfected with and without a kinase-dead AKT1 AAA mutant or AKT1 DD into 293T cells. Immunoblotting of immunoprecipitated 14-3-3ζ with anti-FLAG antibody showed that expression of constitutively active AKT1, in contrast to its kinase-dead mutant, significantly increased the interaction between 14-3-3ζ and β-catenin (Fig. 3A). Consistent with the activity of AKT in facilitating this association, the β-catenin S552A mutant, in contrast to WT β-catenin, was reduced in its binding to 14-3-3ζ, whereas the phosphorylation-mimic β-catenin S552D mutant bound more 14-3-3ζ (Fig. 3B). These data indicate that phosphorylation of β-catenin at S552 by AKT increases the association between β-catenin and 14-3-3ζ.
Fig. 3. β -catenin phosphorylated by AKT increases its association with 14-3-3 ζ.
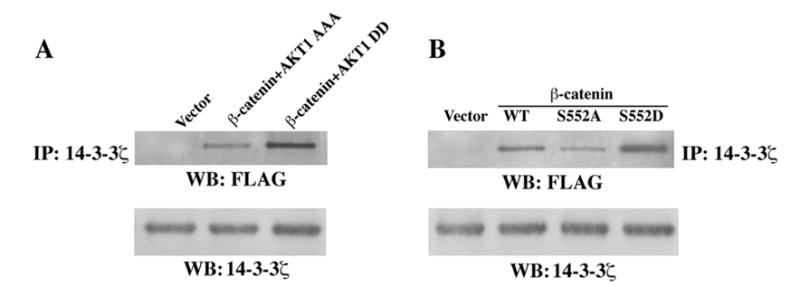
Immunoblotting analyses with the indicated antibodies. A, FLAG-tagged WT β-catenin or pFLAG was co-transfected with or without AKT1 DD or AKT1 AAA into 293T cells, followed by immunoprecipitation with anti-14-3-3ζ antibody. B, FLAG-tagged WT β-catenin, β-catenin S552A, or β-catenin S552D was transfected into 293T cells, followed by immunoprecipitation with anti-14-3-3ζ antibody.
Phosphorylation of β-catenin by AKT increases transcriptional activity of β-catenin and enhances tumor cell invasion
Nuclearly-localized β-catenin interacts with transcription factors of the TCF/LEF-1 family, leading to the increased expression of downstream genes. β-catenin phosphorylated by AKT translocated into the nucleus. To test the effect of β-catenin phosphorylation by AKT on TCF/LEF-1 transcriptional activity, the TCF/LEF-1 luciferase reporter TOP-FLASH or a control vector FOP-FLASH was co-transfected with and without constitutively active AKT1 DD with WT β-catenin or β-catenin S552A mutant. Expression of active AKT1 increased TCF/LEF-1 transcriptional activity (Fig. 4A). In contrast to WT β-catenin, β-catenin S552A had reduced transcriptional activity, either in the absence or presence of active AKT1. Furthermore, expression of β-catenin S552A reduced the 14-3-3ζ-ehancing effect of active AKT1 DD (Fig. 4B) or EGF-induced TCF/LEF-1 transcriptional activity (Fig. 4C). Consistent with the increased nuclear localization, expression of phosphorylation-mimic mutant β-catenin S552D significantly increased TCF/LEF-1 transcriptional activity, in contrast to WT β-catenin (Fig. 4D).
Fig. 4. Phosphorylation of β-catenin by AKT increases transcriptional activity of β-catenin and enhanced tumor cell invasion.
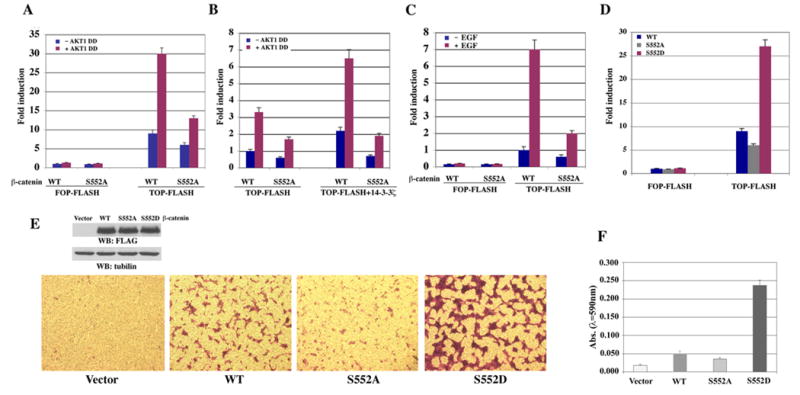
A–D, The relative levels of luciferase activity were normalized to the levels of untreated cells and to the levels of luciferase activity of the Renilla control plasmid. Data represent the means ± SD of three independent experiments. A, TOP-FLASH or FOP-FLASH with WT β-catenin or β-catenin S552A mutant was co-transfected with or without constitutively active AKT1 DD into CHO cells. The luciferase activity was determined. B, TOP-FLASH with WT β-catenin or β-catenin S552A mutant was co-transfected with or without constitutively active AKT1 DD together with or without 14-3-3ζ into CHO cells. The luciferase activity was determined. C, TOP-FLASH or FOP-FLASH was co-transfected with EGFR with WT β-catenin or β-catenin S552A mutant into CHO cells. The luciferase activity was determined. D, TOP-FLASH or FOP-FLASH was co-transfected with WT β-catenin, β-catenin S552A, or β-catenin S552D mutant into CHO cells. The luciferase activity was determined. E, A pool of A431 cells stably transfected with a vector, FLAG-tagged WT β-catenin, β-catenin S552A, or β-catenin S552D were processed for immunoblotting with indicated antibodies (upper panel). The cells were plated at the top surface of the Matrigel. One day after plating, cells that migrated to the opposite side of the insert were stained with crystal violet. Representative microphotographs are shown (lower panel). F, The membranes with invaded cells were dissolved in 4% deoxycholic acid and read colorimetrically at 590 nm for quantification of invasion. Data represent the mean ± standard deviation of three independent experiments.
β-catenin transcriptional activity has been closely related to tumor development. To examine the effect of phosphorylation of β-catenin by AKT on tumor cell invasion, A431 cells were stably transfected with WT β-catenin, β-catenin S552A, or β-catenin S552D, and a Matrigel invasion assay was carried out. While expression of β-catenin S552A moderately reduced cell invasion, in contrast to cell expression of WT β-catenin, expression of β-catenin S552D significantly enhanced tumor cell invasion (Fig. 4E, F). These results indicate that β-catenin phosphorylation by AKT promotes tumor cell invasion.
DISCUSSION
β-catenin, functioning as a major component of both Wnt signaling and cell-cell adhesion, plays a central role in cell proliferation, differention, polarity, morphogenesis, and development (40–42). β-catenin deficiency arrests mouse embryo development at gastrulation (43). β-catenin localized in different cellular compartments forms distant complexes that carry out differential cellular functions. We show here that β-catenin distribution can be dynamically regulated, and phosphorylation of β-catenin by AKT results in the relocalization of some β-catenin from cell-cell contracts and increase in its transcriptional activity.
Activating mutations of Wnt components lead to nuclear localization of β-catenin and are involved in tumor formation and development (44). Wnt-independent signaling, nevertheless, is also involved in regulation of β-catenin transactivation and tumorigenesis (26). β-catenin-TCF/LEF-1 signaling can be activated by growth factors, such as EGF, HGF/scatter factor (SF), insulin-like growth factor (IGF)-I, IGF-II, and insulin (15,16,45,46). In response to insulin stimulation, phosphatidylinositol (PI) 3-kinase-activated AKT phosphoryates GSK-3β at Ser9, which leads to inactivation of GSK-3β and augmentation of β-catenin-TCF/LEF-1 transcriptional activity (47,48). In addition to this indirect regulation, our results show that AKT phosphorylates β-catenin both in vitro and in vivo. The mutation of S552 of β-catenin into Ala or Asp did not change its phosphorylation level by GSK-3β or its half-life, in contrast to WT, indicating that phosphorylation of β-catenin at S552 by AKT does not alter β-catenin protein stability. Instead, phosphorylation at this site enables some β-catenin to disassociate from cell-cell contacts and accumulate in the nucleus. Mutation of the AKT phosphorylation site reduced total transcriptional activity of TCF/LEF-1 induced by AKT (Fig. 4A) about 57%. This partial effect of AKT on TCF/LEF-1 transcriptional activity is likely because AKT can still indirectly enhance upregulation of β-catenin transcriptional activity by phosphorylation and inhibition of GSK-3β. Thus, AKT can activate β-catenin-TCF/LEF-1 transcriptional activity both by indirect stabilization of β-catenin through inhibition of GSK-3β and direct phosphorylation of β-catenin, which enhances β-catenin nuclear accumulation.
In addition to sequential phosphorylation of β-catenin at its N-terminus by CK1 and GSK-3β (44,49–51), β-catenin can be phosphorylated by CK2 at Thr393 (52) and PKA at Ser675 (33,34), which enhance β-catenin transctivation by stabilizing β-catenin protein. Paradoxically, it was reported that CK2-dependent phosphorylation of the E2 ubiquitin conjugating enzyme UBC3B induces the interaction between UBC3B and β-TrCP and enhances β-catenin degradation (53). PKA-phosphorylated β-catenin at S675 promotes the association with its co-activator, CREB-binding protein, thus enhancing β-catenin-TCF/LEF-1 transcriptional activity, although different effects of this phosphorylation on β-catenin stability were observed (33,34). S552 was shown to be a weak phosphorylation site of PKA (33). However, we did not detect measurable reduction of phosphorylation levels of the β-catenin S552A mutant by PKA compared to WT. In contrast, chemical inhibition of AKT and mutation of S552 of β-catenin, but not PKA phosphorylation site S675, abolished phosphorylation by both AKT1 and AKT2 and detection by anti-phospho-RRXS/T antibody, although this antibody may also recognize other –3 position R-bearing phospho-S/T peptides, such as the substrate motif for PKA and PKC. Active AKT phosphorylates proteins containing the consensus sequence RXRXXS/T (31). However, several reports indicate that AKT is also able to phosphorylate Ser or Thr when present in the consensus sequence RXXS/T (54,55) or other non-consensus sequences (56). We demonstrated that AKT phosphorylates S552 in the 489-RRTS-552 sequence of β-catenin, providing additional evidence showing that RXXS/T can be a phosphorylation sequence by AKT.
As a family of conserved regulatory proteins that mainly bind to a plethora of functionally diverse signaling molecules that are phosphorylated at the Ser/Thr residues (39), 14-3-3 can regulate the subcellular distribution of its associated proteins. This is exemplified by the nuclear import of Chk2-phosphorylated the MDM2 homolog MDMX promoted by 14-3-3 (57). 14-3-3ζ, in association with β-catenin, can facilitate transactivation of β-catenin by AKT (38). 489-RRTSMG-554 of β-catenin is a binding motif for 14-3-3ζ. β-catenin S552A weakens its binding to 14-3-3ζ and has a reduced transcriptional activity, whereas β-catenin S552D enhances this binding and increases its transactivation, in contrast to WT β-catenin. Furthermore, S552D β-catenin has accumulated nuclear localization, which supports that 14-3-3ζ may mediate dynamic nucleus-cytosol transportation of its binding proteins (58). It has been shown that AKT1 and AKT2 have distinct roles in tumor cell growth and migration, while AKT1 and AKT2 can either have positive or negative roles, depending on the cell types and experimental setting (59–64). Our results show that activated forms of AKT1 and AKT2 are able to phosphorylate β-catenin in vitro and in vivo. Furthermore, expression of constitutively activated AKT1 or AKT2 results in the relocalization of β-catenin from cell–cell contacts and subsequent transactivation. Thus, it seems that AKT1 and AKT2, can affect the same substrate for regulation of its functions.
S552D β-catenin mutant has a higher transcriptional activity than WT or S552A β-catenin, which correlates with their differential levels of nuclear accumulation. Furthermore, A431 cells stably expressing S552D β-catenin mutant showed much enhanced invasion in contrast to the cells stably expressing either WT or S552 β-catenin. Transactivation of β-catenin increases the transcription of genes that promote tumor cell growth, such as MYC (65), CCND1 (which encodes cyclin D) (66,67), and JUN (68), and genes that promote tumor cell invasion, such as matrix metalloproteinase-7 (MMP7) (69,70) and TWIST1 (71). Further investigation is needed to determine whether phosphorylation of β-catenin at S552 induces a specific set of downstream genes that promote tumor development.
EGFR overexpression or mutation has been found in many types of cancers, with activation of AKT by EGFR or other growth factor receptors, Ras, and regulation of PI 3-K and PTEN contributing to tumor development (72–74). EGF-induced phosphorylation and transactivation of β-catenin are dependent on the activity of AKT. We have previously shown that EGFR activation results in Wnt-independent β-catenin transactivation by downregulation of caveolin-1, whereas overexpression of caveolin-1 suppresses the effect of EGFR on β-catenin (16). Downregulation and internalization of caveolin-1 by EGF will release inhibited signaling molecules normally sequestered in caveolae, such as growth factor receptors, Ha-Ras, and PI 3-K (16,75), which may in turn contribute to AKT activation. Activated AKT can stabilize β-catenin through inhibition of GSK-3β and/or directly phosphorylate β-catenin, resulting in its disassociation from cell-cell contacts and nuclear accumulation. The combined effects enhance β-catenin-TCF/LEF-1 transcriptional activity, which in turn contributes to tumor cell invasion and tumor development.
Footnotes
We thank Hans Clevers (Netherlands Institute for Developmental Biology, Hubrecht Laboratory) for pTOP-FLASH and pFOP-FLASH, Haian Fu (Emory University School of Medicine), and Anthony Muslin (Washington University School of Medicine) for 14-3-3ζ plasmidsζ and Lei Li (M.D. Anderson Cancer Center) for CHO AA8 cells. We thank Hehua Chen for technical help and Theresa Willis for critical reading of this manuscript.
This work was supported by the Pediatric Brain Tumor Foundation (Z.L.), the Charlotte Geyer Foundation (Z.L.), an institutional research grant from The University of Texas M. D. Anderson Cancer Center (Z.L.), Cancer Center Support Grant CA16672 from the National Cancer Institute, and National Cancer Institute grants 1R01CA109035-01A1 (Z.L.), CA55418 (T.H.), and CA82683 (T.H.). T.H. is a Frank and Else Schilling American Cancer Society Research Professor.
References
- 1.Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Proc Natl Acad Sci U S A. 1995;92(19):8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasioukhin V, Fuchs E. Curr Opin Cell Biol. 2001;13(1):76–84. doi: 10.1016/s0955-0674(00)00177-0. [DOI] [PubMed] [Google Scholar]
- 3.Nagafuchi A. Curr Opin Cell Biol. 2001;13(5):600–603. doi: 10.1016/s0955-0674(00)00257-x. [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi A. Cancer Sci. 2003;94(3):225–229. doi: 10.1111/j.1349-7006.2003.tb01424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hulsken J, Birchmeier W, Behrens J. J Cell Biol. 1994;127(6):2061–2069. doi: 10.1083/jcb.127.6.2061. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Embo J. 1997;16(13):3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orford K, Orford CC, Byers SW. J Cell Biol. 1999;146(4):855–868. doi: 10.1083/jcb.146.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Science. 1996;272(5264):1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 9.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. Genes Dev. 1996;10(12):1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 10.Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Science. 1998;280(5363):596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Embo J. 1998;17(5):1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Science. 1998;281(5382):1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 13.Tetsu O, McCormick F. Nature. 1999;398(6726):422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 14.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. Proc Natl Acad Sci U S A. 1999;96(10):5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller T, Bain G, Wang X, Papkoff J. Exp Cell Res. 2002;280(1):119–133. doi: 10.1006/excr.2002.5630. [DOI] [PubMed] [Google Scholar]
- 16.Lu Z, Ghosh S, Wang Z, Hunter T. Cancer Cell. 2003;4(6):499–515. doi: 10.1016/s1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- 17.Polakis P. Genes Dev. 2000;14(15):1837–1851. [PubMed] [Google Scholar]
- 18.Peifer M, Polakis P. Science. 2000;287(5458):1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 19.Morin PJ. Bioessays. 1999;21(12):1021–1030. doi: 10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 20.Ashihara K, Saito T, Mizumoto H, Nishimura M, Tanaka R, Kudo R. Med Electron Microsc. 2002;35(1):9–15. doi: 10.1007/s007950200001. [DOI] [PubMed] [Google Scholar]
- 21.Rimm DL, Caca K, Hu G, Harrison FB, Fearon ER. Am J Pathol. 1999;154(2):325–329. doi: 10.1016/s0002-9440(10)65278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Proc Natl Acad Sci U S A. 1998;95(15):8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T, Yamaoka Y, Nakamura Y. Nat Genet. 2000;24(3):245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 24.Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y. Cancer Res. 1998;58(12):2524–2527. [PubMed] [Google Scholar]
- 25.Ihara A, Koizumi H, Hashizume R, Uchikoshi T. Hepatology. 1996;23(6):1441–1447. doi: 10.1053/jhep.1996.v23.pm0008675162. [DOI] [PubMed] [Google Scholar]
- 26.Lu Z, Hunter T. Cell Cycle. 2004;3(5):571–573. [PubMed] [Google Scholar]
- 27.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL. N Engl J Med. 2004;351(7):657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 28.Lu Z, Liu D, Hornia A, Devonish W, Pagano M, Foster DA. Mol Cell Biol. 1998;18(2):839–845. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe S, Mills GB, Unate H. Oncogene. 2002;21(13):1945–1954. doi: 10.1038/sj.onc.1205117. [DOI] [PubMed] [Google Scholar]
- 30.Lu Z, Xu S, Joazeiro C, Cobb MH, Hunter T. Mol Cell. 2002;9(5):945–956. doi: 10.1016/s1097-2765(02)00519-1. [DOI] [PubMed] [Google Scholar]
- 31.Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. FEBS Lett. 1996;399(3):333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- 32.Pinna LA, Ruzzene M. Biochim Biophys Acta. 1996;1314(3):191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 33.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. J Biol Chem. 2006;281(15):9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 34.Hino S, Tanji C, Nakayama KI, Kikuchi A. Mol Cell Biol. 2005;25(20):9063–9072. doi: 10.1128/MCB.25.20.9063-9072.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. Cancer Cell. 2003;4(6):463–476. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
- 36.Henson ES, Gibson SB. Cell Signal. 2006;18(12):2089–2097. doi: 10.1016/j.cellsig.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 37.Yaffe MB. FEBS Lett. 2002;513(1):53–57. doi: 10.1016/s0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- 38.Tian Q, Feetham MC, Tao WA, He XC, Li L, Aebersold R, Hood L. Proc Natl Acad Sci U S A. 2004;101(43):15370–15375. doi: 10.1073/pnas.0406499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu H, Subramanian RR, Masters SC. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 40.Wodarz A, Nusse R. Annu Rev Cell Dev Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- 41.Cox RT, Kirkpatrick C, Peifer M. J Cell Biol. 1996;134(1):133–148. doi: 10.1083/jcb.134.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. J Cell Biol. 2000;148(3):567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Development. 1995;121(11):3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 44.Giles RH, van Es JH, Clevers H. Biochim Biophys Acta. 2003;1653(1):1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 45.Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C, Capeau J. Oncogene. 2001;20(2):252–259. doi: 10.1038/sj.onc.1204064. [DOI] [PubMed] [Google Scholar]
- 46.Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L. Oncogene. 2001;20(36):4942–4950. doi: 10.1038/sj.onc.1204660. [DOI] [PubMed] [Google Scholar]
- 47.Weston CR, Davis RJ. Science. 2001;292(5526):2439–2440. doi: 10.1126/science.1063279. [DOI] [PubMed] [Google Scholar]
- 48.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378(6559):785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 49.Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I. Genes Dev. 2002;16(9):1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Cell. 2002;108(6):837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 51.Yanagawa S, Matsuda Y, Lee JS, Matsubayashi H, Sese S, Kadowaki T, Ishimoto A. Embo J. 2002;21(7):1733–1742. doi: 10.1093/emboj/21.7.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song DH, Dominguez I, Mizuno J, Kaut M, Mohr SC, Seldin DC. J Biol Chem. 2003;278(26):24018–24025. doi: 10.1074/jbc.M212260200. [DOI] [PubMed] [Google Scholar]
- 53.Semplici F, Meggio F, Pinna LA, Oliviero S. Oncogene. 2002;21(25):3978–3987. doi: 10.1038/sj.onc.1205574. [DOI] [PubMed] [Google Scholar]
- 54.Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Mol Cell. 2003;11(1):11–23. doi: 10.1016/s1097-2765(02)00776-1. [DOI] [PubMed] [Google Scholar]
- 55.Hoyal CR, Gutierrez A, Young BM, Catz SD, Lin JH, Tsichlis PN, Babior BM. Proc Natl Acad Sci U S A. 2003;100(9):5130–5135. doi: 10.1073/pnas.1031526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson JL, Pacquelet S, Lane WS, Eam B, Catz SD. Traffic. 2005;6(8):667–681. doi: 10.1111/j.1600-0854.2005.00308.x. [DOI] [PubMed] [Google Scholar]
- 57.LeBron C, Chen L, Gilkes DM, Chen J. Embo J. 2006;25(6):1196–1206. doi: 10.1038/sj.emboj.7601032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB. J Cell Biol. 2002;156(5):817–828. doi: 10.1083/jcb.200112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. J Cell Biol. 2005;171(6):1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang BH. Cell Signal. 2006;18(12):2262–2271. doi: 10.1016/j.cellsig.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 61.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Mol Cell. 2005;20(4):539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 62.Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. J Biol Chem. 2006 doi: 10.1074/jbc.M600788200. [DOI] [PubMed] [Google Scholar]
- 63.Stambolic V, Woodgett JR. Trends Cell Biol. 2006;16(9):461–466. doi: 10.1016/j.tcb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 64.Heron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, Fernandez A, Lamb NJ. Mol Cell Biol. 2006;26(22):8267–8280. doi: 10.1128/MCB.00201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Science. 1998;281(5382):1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 66.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. Proc Natl Acad Sci U S A. 1999;96(10):5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tetsu O, McCormick F. Nature. 1999;398(6726):422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 68.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C. Proc Natl Acad Sci U S A. 1999;96(4):1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T. Am J Pathol. 1999;155(4):1033–1038. doi: 10.1016/s0002-9440(10)65204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJ, Rubinfeld B, Polakis P, Matrisian LM. Oncogene. 1999;18(18):2883–2891. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]
- 71.Howe LR, Watanabe O, Leonard J, Brown AM. Cancer Res. 2003;63(8):1906–1913. [PubMed] [Google Scholar]
- 72.Downward J. Semin Cell Dev Biol. 2004;15(2):177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 73.Giehl K. Biol Chem. 2005;386(3):193–205. doi: 10.1515/BC.2005.025. [DOI] [PubMed] [Google Scholar]
- 74.Gullick WJ. Br Med Bull. 1991;47(1):87–98. doi: 10.1093/oxfordjournals.bmb.a072464. [DOI] [PubMed] [Google Scholar]
- 75.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. J Biol Chem. 1998;273(10):5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]