

Abstract
This study addresses the role of venule-derived mediators in the arteriolar constriction that accompanies hypercholesterolemia. Constriction was assessed by measuring the tone of small arterioles closely paired with venules in the mesentery of normal cholesterol rats (NC), high cholesterol rats (HC), HC rats injected with antibodies against CD18 and P-selectin (HC/mAbs), HC rats treated with the thromboxane synthase inhibitor, ozagrel (HC/ozagrel), and HC rats pretreated with anti-platelet serum (HC/APS). Venule-paired arterioles in the untreated HC group demonstrated enhanced tone compared with arterioles in the NC group, while no difference was found between unpaired arterioles of the two groups. Perivascular nitric oxide (NO) concentrations were found to be significantly decreased in venule-paired arterioles of HC rats (238±14 nM) compared with those of NC rats (426±42 nM). The injection of anti-adhesion antibodies successfully attenuated the enhanced arteriolar tone and venular leukocyte adherence in the HC group, and tended to increase levels of NO in venule-paired arterioles by 33% (to 326±19 nM; still lower than that of the NC group). Ozagrel and platelet depletion attenuated the enhanced arteriolar tone by 53% and 33%, respectively, without affecting NO concentrations. These findings indicate that the mechanism of blood cell-dependent arteriolar constriction during hypercholesterolemia may be dependent on thromboxane, a decrease in NO, and the proximity of the arterioles to postcapillary venules.
Keywords: Nitric oxide, platelet, arteriolar tone, thromboxane, cholesterol
INTRODUCTION
Several recent studies reveal that vasoactive molecules released from venules play an important role in regulating the diameter of closely paired arterioles under both normal physiological and pathophysiological conditions. In healthy tissue, diffusional transport of vasoactive molecules from venules to paired arterioles contributes to arteriolar dilation during both stimulated [10;16;24;29] and resting conditions [27]. Nitric oxide (NO), prostaglandins, and adenosine all have been implicated as mediators of venule-induced arteriolar dilation. In contrast, during inflammatory conditions, venules have been shown to have the opposite effect on arterioles, which is, to induce vasoconstriction. The mechanism of venule-dependent constriction of arterioles appears to involve inflammatory cell adhesion (leukocyte and/or platelet) in the venules.
Zamboni et al. [42;44] found that ischemia-reperfusion induced the constriction of arterioles, but only when the arterioles were closely paired to venules. The venule-dependent constriction was significantly attenuated by a monoclonal antibody against CD18, a leukocyte adhesion molecule that allows firm adhesion to the endothelium. The authors hypothesized that the vasoconstricting mediator could be thromboxane, a potent vasoconstrictor released by platelets and macrophages, and tested this hypothesis with a thromboxane receptor antagonist. The antagonist partially reduced the vasoconstriction, suggesting that one or more other constricting mediators could be involved in their model of ischemia-reperfusion [22]. Thromboxane also appears to play a role in the venule-dependent arteriolar constriction observed in a model of intestinal inflammation induced by dextran sodium sulfate [14].
Hypercholesterolemia is one of the most critical cardiovascular risk factors associated with the progression of atherosclerosis in large arteries. In addition, the early stages of hypercholesterolemia induce endothelial dysfunction in microvessels well before the formation of atherosclerotic lesions in larger vessels [32;36]. Attenuated endothelium-dependent dilation is thought to be a result of a diminished production or availability of NO, possibly as a result of reaction with the increased levels of superoxide. This imbalance of superoxide and NO not only leads to deficient arteriolar dilation [6;17;21;33], but also enhances adhesion of leukocytes and platelets in postcapillary venules [11;31;35;37].
Nellore and Harris [26] recently collected data from venule-paired arterioles in hypercholesterolemic rats, and found that the resting diameters of second- and third-order arterioles in these rats were significantly smaller than those of normocholesterolemic rats. Decreases in capillary blood flow velocity were observed with hypercholesterolemia, but only when the feeding arteriolar pathway was closely paired to the inflamed venules. Moreover, depletion of circulating neutrophils with anti-neutrophil serum restored perfusion, specifically in the capillaries fed by closely venule-paired arterioles. However, use of the anti-neutrophil serum did not address the role of adhesion in the venule-dependent arteriolar constriction.
We hypothesize that hypercholesterolemia-induced adhesion of leukocytes and platelets may be responsible for arteriolar constriction via a mechanism that involves venulo-arteriolar communication. The main objectives of this study were to investigate in hypercholesterolemia (1) the role of adhesion molecules in decreasing arteriolar diameter and NO levels, and (2) the role of thromboxane in the venule-dependent mechanism of arteriolar constriction. To accomplish these goals, venular leukocyte adherence and arteriolar tone were observed via intravital microscopy in hypercholesterolemic rats.
MATERIALS AND METHODS
Animal preparation
Male Wistar rats were purchased in the 185~200 g weight range. At 7 to 8 weeks of age the rats were placed on either a normal diet or a high-fat diet (0.9% cholesterol, 0.3% cholic acid, and 2% olive oil) for a period of 2 weeks. Rats were housed 2–3 per cage in a controlled environment (12/12, light/dark cycle). Animal procedures were approved by the Institutional Animal Care and Use Committee of Louisiana State University Health Sciences Center. Rats were anesthetized by an intraperitoneal injection of 135 mg/kg thiobutabarbital (Inactin, Sigma T-133, St. Louis, MO). The right carotid artery was cannulated for systemic injection of saline during the experiment (~1 ml) and of euthanasia solution at the end of experiment. In some experiments, the right femoral vein was cannulated for injection of monoclonal antibodies against CD18 (WT-3, 0.5 mg/kg) and P-selectin (RMP-1, 2 mg/kg). A segment of the small intestine was exteriorized through a midline abdominal incision, and the spontaneously breathing rat was placed on its right side on a Plexiglas board so that a selected section of mesentery could be draped over a glass cover slip glued on a hole centered in the board. The exposed intestine, except for the selected mesenteric section under study, was covered with gauze soaked in bicarbonate-buffered saline (BBS) consisting of 131 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 20 mM NaHCO3, and 3.5 mM CaCl2. After the board was mounted onto the stage of an inverted microscope, the mesentery and intestine were kept moist with a 2 ml/min superfusion of BBS bubbled with a 95% N2 and 5% CO2 gas mixture and warmed to 37 °C. Rectal temperature was maintained near 37 °C by positioning an infrared heat lamp over the rat.
Intravital video microscopy
The mesenteric microvessels were observed through a x40 objective (Nikon Plan Apo, 0.95 N.A.) using a 100-W halogen light source, and brightfield images were captured with a color camera (CCD-IRIS, SONY). The images were directed into a videocassette recorder (SVO-9500 MD, SONY) and the taped images were used for playback analysis with an image grabber and image processor (Scion Image) for length and diameter measurements. Arteriolar and venular red blood cell velocities were measured with live images using an optical Doppler velocimeter (Microcirculation Research Institute, Texas A & M).
Arteriolar NO measurement using a NO-selective microelectrode
The peri-arteriolar NO concentration was monitored using a nitric oxide-selective microelectrode (ISO-NO-3005, World Precision Instruments, Sarasota, FL). In vitro calibration of the microelectrode was performed 30 min before each experiment. For calibration, the electrode was placed in a copper sulfate solution at 37 °C and a known concentration of S-Nitroso-N-acetyl-D, L-penicillamine (SNAP), a stable NO-containing compound, was added. SNAP decomposes to NO and a disulfide byproduct when dissolved in water in a reaction catalyzed by copper sulfate. The time course of current output was displayed on the instrument monitor. Fig. 1A demonstrates a typical example of an in vitro calibration, which shows linearity between the changes in output current (pA) and NO concentration (nM). The microelectrode attached to a micromanipulator was allowed to equilibrate for 30 min in BBS superfusate bathing the mesentery. The microelectrode then was placed over the arteriolar wall as close as possible and slightly pressed against the tissue. The slight touch of the microelectrode on the tissue resulted in a transient initial signal, which equilibrated to a stable value after 20–30 s.
Figure 1.
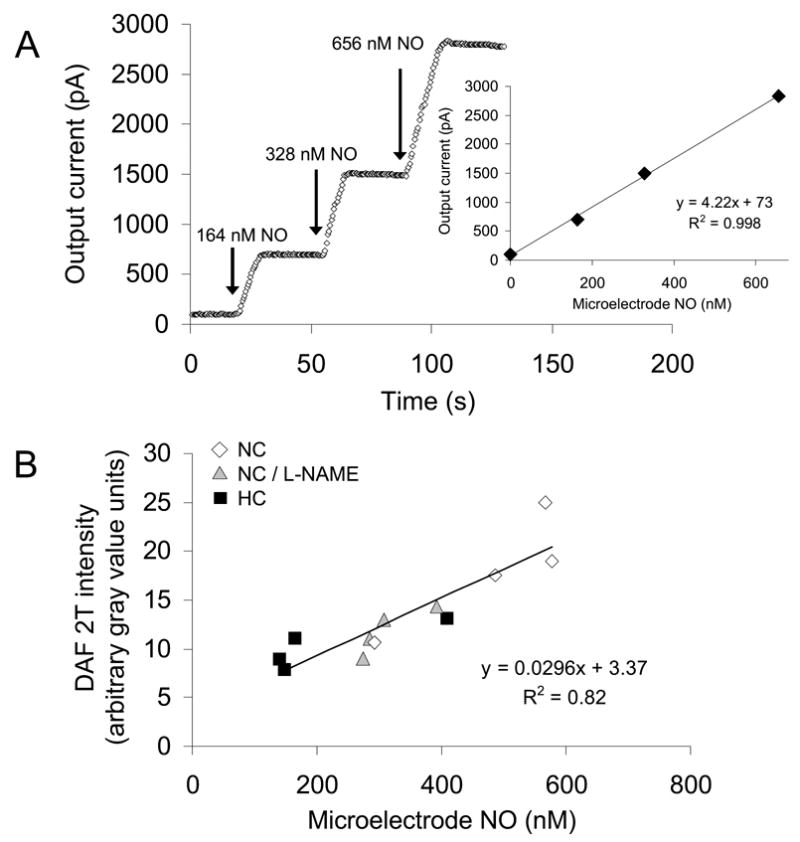
(A) Sample calibration between microelectrode output current (pA) and NO concentration (nM). (B) Similarity between DAF-2T fluorescence intensity (arbitrary gray value units) and microelectrode NO concentration (nM) in vessels in the mesentery of normal cholesterol rats (NC), high cholesterol rats (HC), and NC mesentery exposed to L-NAME. N=4 in each group.
To corroborate the measurements of the NO-selective microelectrode, simultaneous in vivo NO measurements were performed in a preliminary study of mesenteric venules using both the NO-selective microelectrode and DAF-2-DA (diaminofluorescein-2-diacetate). DAF-2-DA is membrane permeable and is converted to DAF-2 by esterase inside cells and then to fluorescent DAF-2T by intracellular NO. For fluorescence microscopy, the preparation was illuminated with a 100-W mercury lamp. The simultaneous NO measurements were performed for three experimental groups: (1) normal cholesterol rats (NC), (2) normal cholesterol rats treated with the NO synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME, 100 μM), and (3) high cholesterol rats (HC). In the NC and HC groups, after a 10 min baseline measurement using the NO-selective microelectrode, DAF-2-DA (5 μM) was superfused over the mesenteric tissue for 20 min and the fluorescent images were recorded. The exposure time was maintained less than 3 s to limit photobleaching of the dye. In the NC group with L-NAME treatment, after 10 min of BBS superfusion, L-NAME was superfused for 20 min and the NO concentrations were measured simultaneously using both the NO microelectrode and DAF-2-DA dye. The vessel wall DAF-2T intensities (subtracted from background tissue intensities) were analyzed using image processing software (Scion Image). Fig. 1B shows the close correspondence of gray value intensity of DAF-2T and NO microelectrode measurements.
Venular leukocyte adherence
Leukocyte adherence was defined as the number of leukocytes that remained stationary on the venule wall for a period of at least 30 s during a 2-min observation period and expressed as the number of cells per square millimeter of venular surface.
Platelet counting
Approximately 0.9 mL of blood was collected via the right carotid artery into acid-citrate-dextrous buffer (Sigma) at the end of experiment and the number of circulating platelets was counted using a hemocytometer.
Cholesterol measurements
At the end of each experiment, a blood sample (~400 μL) was withdrawn from a cannulated carotid artery and gently mixed with ~5 μL of 1000 U/mL heparin. Approximately 50 μL of the blood sample were dropped onto a test panel of a cholesterol measurement kit (Polymer Technology System, Indianapolis, IN).
Experimental protocols
Experimental groups were divided as follows: (1) untreated normal cholesterol-diet rats (NC), (2) untreated high cholesterol-diet rats (HC), (3) high cholesterol-diet rats with the injection of monoclonal antibodies against CD18 and P-selectin 30 min prior to surgery (HC/mAbs), (4) high cholesterol-diet rats with superfusion of the thromboxane synthase inhibitor, ozagrel, (HC/ozagrel), and (5) high cholesterol-diet rats with the injection of anti-platelet serum (0.5 ml/kg i.p; AI-AD51440, Accurate Chemicals, Westbury, NY) 3 hour prior to surgery, (HC/APS).
Both unpaired arterioles and closely paired, parallel arterioles and venules (arterio-venular distance <20 μm) in the mesentery were selected. In each experimental group, resting arteriolar diameter, leukocyte adherence, and NO microelectrode concentrations were measured simultaneously. To determine arteriolar tone, resting arteriolar diameter D was compared to the maximum diameter Dmax obtained after a 10 min superfusion of papaverine (1 mM, Sigma). Arterioles that did not respond to papaverine (by at least 0.4 μm, the limit of optical resolution) were not included in the data analysis. In the HC/ozagrel group, after the baseline measurement of arteriolar diameter and NO concentration for 10 min, ozagrel (1 mg/ml in BBS, Sigma) was superfused onto the mesenteric tissue for 20 min and changes in arteriolar diameter and NO concentration were measured simultaneously.
Statistics
GraphPad Instat (GraphPad Instat, San Diego, CA) software was used for statistical analysis. Data were compared with standard t-tests, or with ANOVA (using Bonferroni post-hoc corrections) with multiple groups or repeated measures. Minitab software (Minitab, State college, PA) was used for regression analysis. Error bars are presented as ± standard error (SE). Statistical significance was set at P<0.05.
RESULTS
To address the possible role of venular pairing on resting arteriolar tone and NO bioavailability, mesenteric arterioles were categorized as either venule-paired (close parallel pairing with a venule) or unpaired. In both NC and HC groups, paired and unpaired third-order arterioles with baseline diameters of 20–35 μm were selected and tone was defined as the percentage constriction compared with the maximum diameter measured following papaverine exposure (Fig 2). For unpaired arterioles, no significant difference in arteriolar tone was observed between NC (2.5 ± 0.2 %) and HC (3.3 ± 0.4 %, P>0.05 vs NC). However, for venule-paired arterioles, the resting arteriolar tone of HC (9.3 ± 1.0 %) was significantly higher than that of NC (2.2 ± 0.2 %, P<0.001 vs NC), indicating that venular pairing enhances basal arteriolar constriction in hypercholesterolemia.
Figure 2.
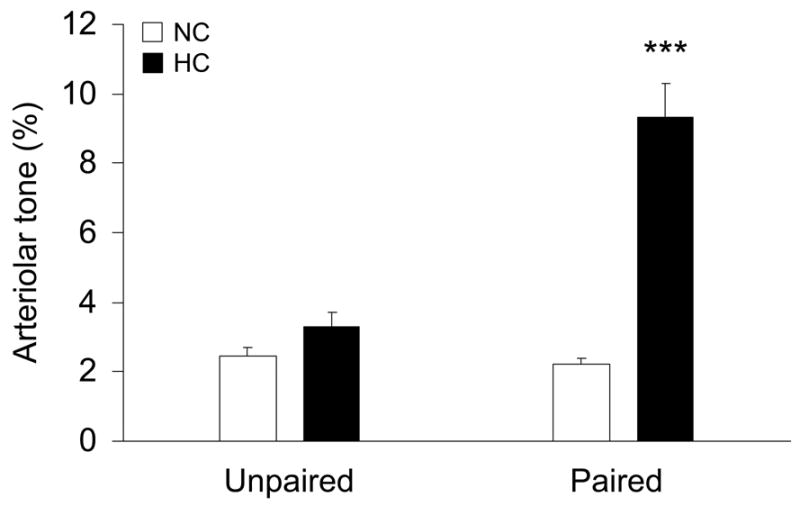
Influence of venular pairing on arteriolar tone for both normal cholesterol (NC) diet and high cholesterol (HC) diet rats. Arterioles were divided into two groups: unpaired and paired arterioles (number of data, N=5 for NC/unpaired, N=8 for NC/paired, N=8 for HC/unpaired, and N=9 for HC/paired). ***: P<0.001 vs paired NC.
To investigate the mechanism by which venular pairing affects vascular tone, 20–35 μm diameter third-order arterioles, closely paired with venules, were selected. Table 1 shows selected statistical data from all five groups. The total cholesterol level of HC (177 ± 18 mg/dL) was significantly higher than that of NC (77 ± 2 mg/dL). High cholesterol-diet rats treated with monoclonal antibodies, ozagrel, or APS had increases in total cholesterol similar to that of the untreated HC group. Arteriolar and venular red blood cell velocities were not significantly different in all groups, indicating that any changes in local hemodynamic factors were not implicated in the interpretation of our results.
Table 1.
Selected statistical data for the four experimental groups: NC (normal cholesterol), HC (high cholesterol), HC/mAb (HC injected with anti-adhesion molecules), HC/ozagrel (HC treated with ozagrel), and HC/APS (HC injected with anti-platelet serum).
| NC | HC | HC/mAbs | HC/ozagrel | HC/APS | |
|---|---|---|---|---|---|
| Number of rats | 8 | 9 | 6 | 6 | 6 |
| Age (weeks) | 9–10 | 9–10 | 9–10 | 9–10 | 9–10 |
| Weight (g) | 343 ± 6 | 339 ± 8 | 350 ± 10 | 336 ± 5 | 356 ± 8 |
| Cholesterol (mg/dl) | 77 ± 2 | 177 ± 18** | 175 ± 25** | 163 ± 15** | 148 ± 10** |
| Baseline arteriolar diameter (μm) | 23.0 ±0.9 | 23.7 ± 1.1 | 24.4 ±0.7 | 23.5 ± 1.8 | 24.5 ± 0.9 |
| Baseline venular diameter (μm) | 40.5 ± 4.2 | 40.1 ± 3.2 | 41.7 ±2.4 | 36.2 ± 4.0 | 39.9 ± 3.7 |
| Arterio-venular distance (μm) | 10.5 ± 1.3 | 12.4 ± 2.4 | 12.6±2.6 | 9.0 ± 1.8 | 7.2 ± 0.5 |
| Arteriolar red blood cell velocity (mm/s) | 4.1 ± 0.2 | 3.8 ± 0.4 | 4.3 ± 0.4 | 3.9 ± 0.2 | 4.2 ± 0.3 |
| Venular red blood cell velocity (mm/s) | 1.79 ± 0.1 | 1.66 ± 0.1 | 1.78 ± 0.2 | 1.68 ± 0.1 | 1.69 ± 0.2 |
P < 0.01 compared to NC group
Figure 3 shows the number of adherent leukocytes in venules. Hypercholesterolemia induced a significant increase in the number of adherent leukocytes in postcapillary venules compared with NC (724 ± 85/mm2 for NC vs 3033 ± 282/mm2 for HC, P<0.001). Pretreatment with monoclonal antibodies against CD18 and P-selectin successfully attenuated the number of adherent leukocytes in HC (262 ± 130/mm2, P<0.001 vs HC and P>0.05 vs NC). However, inclusion of ozagrel in the superfusate did not alter leukocyte adherence (2758 ± 351/mm2, P>0.05 vs HC and P<0.001 vs NC). Pretreatment with APS also did not significantly alter leukocyte adherence (2283 ± 347/mm2, P>0.05 vs HC and P<0.01 vs NC).
Figure 3.
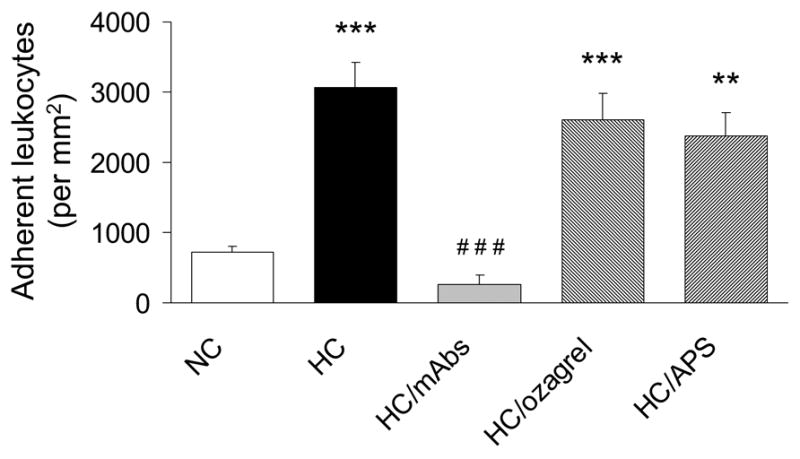
The number of adherent leukocytes in postcapillary venules for NC, HC, HC/mAbs, HC/ozagrel, and HC/APS groups. **: P<0.01 vs NC, ***: P<0.001 vs NC, and ###: P<0.001 vs HC.
The effect of the three treatments on arteriolar tone and NO concentration in HC are shown in Fig 4. Arteriolar tone and NO were measured simultaneously in each group, with perivascular NO measurements obtained using a NO-selective microelectrode placed near the arteriolar wall. The enhanced arteriolar tone induced by HC (9.3 ± 1.0% for HC vs 2.2 ± 0.2% for NC, P<0.001) was accompanied by a decrease in arteriolar NO (238 ± 14 nM for HC vs 426 ± 42 nM for NC, P<0.001). The injection of monoclonal antibodies against CD18 and P-selectin (given simultaneously) successfully attenuated the HC-induced increase in arteriolar constriction, with the resulting tone (2.5 ± 0.2%, P<0.001 vs HC) similar to that of NC. The antibodies also increased NO levels by 37% to 326 ± 19 nM, although this concentration of NO was not significantly different from either NC or untreated HC groups. The limited increase in NO could suggest that an additional pathway is involved in the HC-induced constriction. Addition of ozagrel attenuated the HC-induced enhancement of tone to 4.4 ± 0.5% (P<0.001 vs HC), to a value that still was not significantly different (P>0.05) than that of the NC group. The ozagrel-induced attenuation of arteriolar constriction occurred with no changes in NO levels (Fig 4B) or leukocyte adherence (Fig 3). To investigate whether platelets contributed to the increased arteriolar tone in HC, arteriolar tone and NO were measured during platelet depletion with APS in HC (HC/APS). Pretreatment with APS significantly decreased circulating platelet levels by 95 % (from 8.3 ± 0.8 × 105 platelets/μL to 0.4 ± 0.09 × 105 platelets/μL). Platelet depletion partially attenuated HC-induced arteriolar tone (6.2 ± 0.2% for HC/APS vs 9.3 ± 1.0% for HC, P<0.01) with no change in NO levels (252 ± 20 nM for HC/APS vs 238 ± 14 nM for HC, P>0.05).
Figure 4.
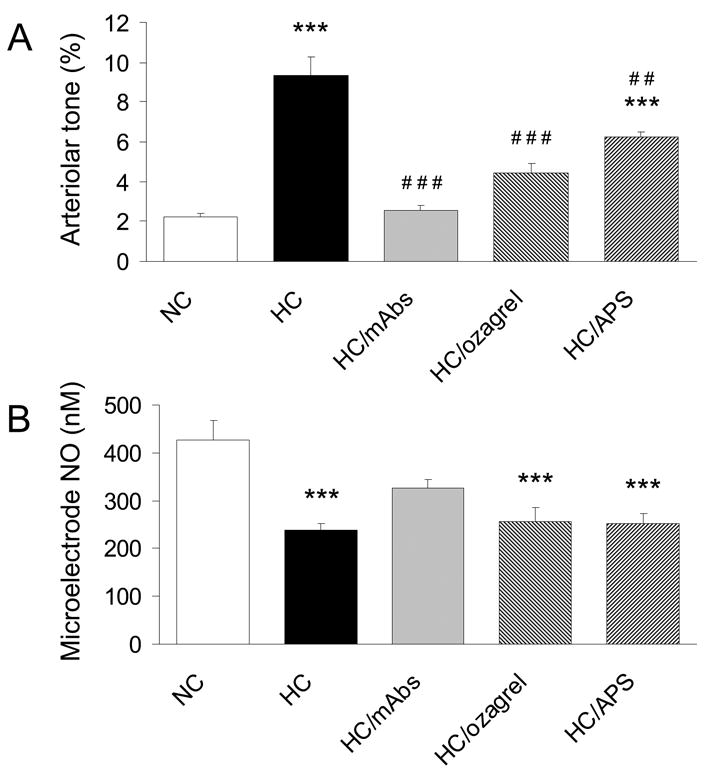
Arteriolar tone (A) and NO concentrations (B) in arterioles closely paired with postcapillary venules for NC, HC, HC/mAbs, HC/ozagrel, and HC/APS groups. ***: P<0.001 vs NC, ##: P<0.01 vs HC, and ###: P<0.001 vs HC.
DISCUSSION
Endothelial dysfunction often is defined as an attenuated dilation of arterioles, induced experimentally by endothelium-dependent mediators such as acetylcholine, bradykinin, or increased shear. Another endothelial alteration present in hypercholesterolemia is the increased adhesion of platelets and leukocytes to venular endothelial cells. This study addresses the role of venule-derived mediators in the arteriolar constriction that accompanies hypercholesterolemia. The results of our study indicate that the mechanism of blood cell-dependent arteriolar constriction may be dependent on several factors including thromboxane, a decrease in NO, and the proximity of the arterioles to postcapillary venules.
In most tissues, countercurrent venules and veins are found in an anatomical arrangement of close, parallel proximity to the feeding arteries and arterioles. Besides heat transfer, the functionality of this pairing is thought to include the delivery of venule-derived molecules involved in vasodilation or vasoconstriction [9;13;15;23;43] due to the short diffusion distance [40]. In resting conditions and in the presence of acetylcholine, venule-mediated arteriolar dilation depends on NO [8;25]. However, during inflammation, the presence of adherent leukocytes in venules may limit the concentration of NO in paired arterioles [20;28], and a monoclonal antibody against CD18 has been shown to prevent the venule-dependent arteriolar constriction induced by ischemia-reperfusion [45]. Leukocyte-endothelial cell interactions mediate the release of superoxide anion by activating leukocyte NADPH oxidase, which counteracts NO-mediated vasodilation [2;5].
With this background, we hypothesized that venular leukocyte adhesion could play a major role in arteriolar constriction induced by hypercholesterolemia via a mechanism that involves a decrease in NO. As described by others [30;34], we found venular leukocyte adherence to be enhanced in hypercholesterolemia, and in addition, we found a concomitant decrease in NO and constriction of venule-paired arterioles. However, injection of adhesion-blocking antibodies completely eliminated HC-induced constriction despite only partial restoration of arteriolar NO.
The finding that acute adhesion blockade does not restore NO completely to the values found in normocholesterolemic rats could indicate that emigrated platelets and/or leukocytes (that would not be eliminated by acute administration of the antibodies) could contribute to the HC-induced decrease of NO in venule-paired arterioles. In addition, other mechanisms of attenuated NO bioavailability can be considered. For example, during hypercholesterolemia, superoxide also is produced from NADPH oxidase localized in the arteriolar endothelium, which leads to increased degradation of NO [41]. Hypercholesterolemia also is associated with a reduced enzymatic production of NO due to either increased circulating levels of the endogenous NOS inhibitor asymmetric dimethyl-L-arginine (ADMA) [4] or reduced bioavailability of the cofactor tetrahydrobiopterin (BH4) [19].
The finding that adhesion blockade eliminates hypercholesterolemia-induced constriction without fully restoring NO also points to the potential importance of a NO-independent pathway of dysfunction. [7]. Our finding that thromboxane inhibition substantially restores arteriolar dilation with no change in NO level might elucidate such a mechanism. Thromboxane is generated from the metabolism of arachidonic acid through the cyclooxygenase pathway, and formed by the action of thromboxane synthase on the prostaglandin endoperoxide H2 (PGH2) in activated platelets and macrophages [7]. Thromboxane is known to play a crucial role in renal vasoconstriction induced by short-term hypercholesterolemia [18]. The importance of thromboxane activity in vasoconstriction has been implicated in other inflammatory models. In spontaneously hypertensive rats, treatment with cyclooxygenase inhibition or thromboxane receptor antagonists abolished the constriction observed in arterial rings [1]. Additionally, intestinal inflammation induced by dextran sodium sulfate (a model of colitis) was found to induce thromboxane-dependent arteriolar constriction, but only in arterioles closely paired with venules having adherent leukocytes and platelets [12].
The dependence of the thromboxane pathway on CD18- and/or P-selectin-mediatedadhesion may point to a role for platelets considering their potent ability to release this vasoconstrictor. Our finding that platelet depletion partially attenuated HC-induced arteriolar constriction through a NO-independent mechanism supports this hypothesis. Recent studies report elevated P-selectin expression on platelets and endothelial cells in animals fed a high cholesterol diet, and a significantly increased platelet adhesion that accompanies leukocyte adhesion to venular endothelium [38;39]. Additionally, it has been shown that platelet adhesion is dependent on the presence of adherent leukocytes through heterotypic binding of the two cell types. Therefore, it is plausible that our simultaneous injection of monoclonal antibodies against CD18 and P-selectin would attenuate both platelet and leukocyte adhesion, thus inhibit platelet and leukocyte-derived mediators that might contribute to arteriolar constriction.
The mechanism of venule- and blood cell-dependent constriction may extend to locations proximal to the microcirculation. Banda et al. [3] have demonstrated that mesenteric ischemia-reperfusion, which induces venular platelet and leukocyte adhesion, attenuates endothelium-dependent dilation of the superior mesenteric artery. Attenuated dilation was absent in mutant mice lacking the adhesion molecules CD11/CD18, intercellular adhesion molecule-1 (ICAM-1), and P-selectin. Whether venulo-arteriolar communication exists at the site of larger vessels is not clear: another possibility is that inflammatory mediators produced in the microcirculation become systemically perfused. However, the role of a potential systemic mechanism was likely to be small in the microvessels of our model, inasmuch as unpaired arterioles did not demonstrate the same level of enhanced tone as venule-paired arterioles (Fig 2).
In summary, we found evidence that blood cell-dependent adhesion in postcapillary venules contributes to arteriolar constriction in closely paired arterioles. The pathological importance of arteriolar constriction induced in the early stages of hypercholesterolemia is not completely clear. However, hypercholesterolemia-induced endothelial dysfunction in the microvasculature precedes atherosclerotic lesion development in large arteries. With arteriolar dilation limited, adequate capillary perfusion is at risk: even a relatively small change in arteriolar diameter can induce major changes in flow, since resistance is inversely proportional to the fourth power of diameter. The resulting reduction in flow and shear is known to promote the adhesion of leukocytes and platelets in postcapillary venules, which could aggravate the microvascular response in a positive feedback mechanism, and potentially initiate subsequent systemic inflammation via the blood cell-derived mediators. Such a systemic inflammation might contribute to the eventual pathogenesis of atherosclerosis.
Acknowledgments
The authors thank Georgia Morgan for proofreading and editing. This work was funded by the Juvenile Diabetes Research Foundation (NRH; 1-2003-159), by the Biomedical Research Foundation of Northwest Louisiana (NRH), and by the National Institutes of Health (DNG; HL26441).
Reference List
- Abeywardena MY, Jablonskis LT, Head RJ. Age- and hypertension-induced changes in abnormal contractions in rat aorta. J Cardiovasc Pharmacol. 2002;40:930–937. doi: 10.1097/00005344-200212000-00015. [DOI] [PubMed] [Google Scholar]
- Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- Banda MA, Lefer DJ, Granger DN. Postischemic endothelium-dependent vascular reactivity is preserved in adhesion molecule-deficient mice. Am J Physiol. 1997;273:H2721–H2725. doi: 10.1152/ajpheart.1997.273.6.H2721. [DOI] [PubMed] [Google Scholar]
- Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98:1842–1847. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Casino PR, Kilcoyne CM, Quyyumi AA, Hoeg JM, Panza JA. The role of nitric oxide in endothelium-dependent vasodilation of hypercholesterolemic patients. Circulation. 1993;88:2541–2547. doi: 10.1161/01.cir.88.6.2541. [DOI] [PubMed] [Google Scholar]
- Donge JM, de Leval X, Benoit P, Rolin S, Pirotte B, Masereel B. Therapeutic potential of thromboxane inhibitors in asthma. Expert Opin Investig Drugs. 2002;11:1–8. doi: 10.1517/13543784.11.2.275. [DOI] [PubMed] [Google Scholar]
- Falcone JC, Bohlen HG. EDRF from rat intestine and skeletal muscle venules causes dilation of arterioles. Am J Physiol. 1990;258:H1515–H1523. doi: 10.1152/ajpheart.1990.258.5.H1515. [DOI] [PubMed] [Google Scholar]
- Falcone JC, Bohlen HG. EDRF from rat intestine and skeletal muscle venules causes dilation of arterioles. Am J Physiol. 1990;258:H1515–H1523. doi: 10.1152/ajpheart.1990.258.5.H1515. [DOI] [PubMed] [Google Scholar]
- Falcone JC, Bohlen HG. EDRF from rat intestine and skeletal muscle venules causes dilation of arterioles. Am J Physiol. 1990;258:H1515–H1523. doi: 10.1152/ajpheart.1990.258.5.H1515. [DOI] [PubMed] [Google Scholar]
- Gauthier TW, Scalia R, Murohara T, Guo JP, Lefer AM. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1995;15:1652–1659. doi: 10.1161/01.atv.15.10.1652. [DOI] [PubMed] [Google Scholar]
- Harris NR, Whatley JR, Carter PR, Specian RD. Venular constriction of submucosal arterioles induced by dextran sodium sulfate. Inflamm Bowel Dis. 2005;11:806–813. doi: 10.1097/01.mib.0000178262.95980.65. [DOI] [PubMed] [Google Scholar]
- Harris NR, Whatley JR, Carter PR, Specian RD. Venular constriction of submucosal arterioles induced by dextran sodium sulfate. Inflamm Bowel Dis. 2005;11:806–813. doi: 10.1097/01.mib.0000178262.95980.65. [DOI] [PubMed] [Google Scholar]
- Harris NR, Whatley JR, Carter PR, Specian RD. Venular constriction of submucosal arterioles induced by dextran sodium sulfate. Inflamm Bowel Dis. 2005;11:806–813. doi: 10.1097/01.mib.0000178262.95980.65. [DOI] [PubMed] [Google Scholar]
- Hester RL. Venular-arteriolar diffusion of adenosine in hamster cremaster microcirculation. Am J Physiol. 1990;258:H1918–H1924. doi: 10.1152/ajpheart.1990.258.6.H1918. [DOI] [PubMed] [Google Scholar]
- Hester RL. Venular-arteriolar diffusion of adenosine in hamster cremaster microcirculation. Am J Physiol. 1990;258:H1918–H1924. doi: 10.1152/ajpheart.1990.258.6.H1918. [DOI] [PubMed] [Google Scholar]
- Jeremy RW, McCarron H. Effect of hypercholesterolemia on Ca(2+)-dependent K(+) channel-mediated vasodilatation in vivo. Am J Physiol Heart Circ Physiol. 2000;279:H1600–H1608. doi: 10.1152/ajpheart.2000.279.4.H1600. [DOI] [PubMed] [Google Scholar]
- Kaplan R, Aynedjian HS, Schlondorff D, Bank N. Renal vasoconstriction caused by short-term cholesterol feeding is corrected by thromboxane antagonist or probucol. J Clin Invest. 1990;86:1707–1714. doi: 10.1172/JCI114895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katusic ZS. Vascular endothelial dysfunction: does tetrahydrobiopterin play a role? Am J Physiol Heart Circ Physiol. 2001;281:H981–H986. doi: 10.1152/ajpheart.2001.281.3.H981. [DOI] [PubMed] [Google Scholar]
- Kim MH, Harris NR. Leukocyte adherence inhibits adenosine-dependent venular control of arteriolar diameter and nitric oxide. Am J Physiol Heart Circ Physiol. 2006;291:H724–H731. doi: 10.1152/ajpheart.01215.2005. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Merten R, Spiekermann S, Buttner K, Drexler H, Hornig B. Vascular extracellular superoxide dismutase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation. 2000;101:2264–2270. doi: 10.1161/01.cir.101.19.2264. [DOI] [PubMed] [Google Scholar]
- Mazolewski PJ, Roth AC, Suchy H, Stephenson LL, Zamboni WA. Role of the thromboxane A2 receptor in the vasoactive response to ischemia-reperfusion injury. Plast Reconstr Surg. 1999;104:1393–1396. doi: 10.1097/00006534-199910000-00023. [DOI] [PubMed] [Google Scholar]
- Mckay MK, Gardner AL, Boyd D, Hester RL. Influence of venular prostaglandin release on arteriolar diameter during functional hyperemia. Hypertension. 1998;31:213–217. doi: 10.1161/01.hyp.31.1.213. [DOI] [PubMed] [Google Scholar]
- Mckay MK, Gardner AL, Boyd D, Hester RL. Influence of venular prostaglandin release on arteriolar diameter during functional hyperemia. Hypertension. 1998;31:213–217. doi: 10.1161/01.hyp.31.1.213. [DOI] [PubMed] [Google Scholar]
- Nellore K, Harris NR. L-arginine and antineutrophil serum enable venular control of capillary perfusion in hypercholesterolemic rats. Microcirculation. 2002;9:477–485. doi: 10.1038/sj.mn.7800162. [DOI] [PubMed] [Google Scholar]
- Nellore K, Harris NR. L-arginine and antineutrophil serum enable venular control of capillary perfusion in hypercholesterolemic rats. Microcirculation. 2002;9:477–485. doi: 10.1038/sj.mn.7800162. [DOI] [PubMed] [Google Scholar]
- Nellore K, Harris NR. L-arginine and antineutrophil serum enable venular control of capillary perfusion in hypercholesterolemic rats. Microcirculation. 2002;9:477–485. doi: 10.1038/sj.mn.7800162. [DOI] [PubMed] [Google Scholar]
- Nellore K, Harris NR. Nitric oxide measurements in rat mesentery reveal disrupted venulo-arteriolar communication in diabetes. Microcirculation. 2004;11:415–423. doi: 10.1080/10739680490457809. [DOI] [PubMed] [Google Scholar]
- Saito Y, Eraslan A, Lockard V, Hester RL. Role of venular endothelium in control of arteriolar diameter during functional hyperemia. Am J Physiol Heart Circ Physiol. 1994;267:H1227–H1231. doi: 10.1152/ajpheart.1994.267.3.H1227. [DOI] [PubMed] [Google Scholar]
- Scalia R, Appel JZ, III, Lefer AM. Leukocyte-endothelium interaction during the early stages of hypercholesterolemia in the rabbit: role of P-selectin, ICAM-1, and VCAM-1. Arterioscler Thromb Vasc Biol. 1998;18:1093–1100. doi: 10.1161/01.atv.18.7.1093. [DOI] [PubMed] [Google Scholar]
- Scalia R, Appel JZ, III, Lefer AM. Leukocyte-endothelium interaction during the early stages of hypercholesterolemia in the rabbit: role of P-selectin, ICAM-1, and VCAM-1. Arterioscler Thromb Vasc Biol. 1998;18:1093–1100. doi: 10.1161/01.atv.18.7.1093. [DOI] [PubMed] [Google Scholar]
- Scalia R, Appel JZ, III, Lefer AM. Leukocyte-endothelium interaction during the early stages of hypercholesterolemia in the rabbit: role of P-selectin, ICAM-1, and VCAM-1. Arterioscler Thromb Vasc Biol. 1998;18:1093–1100. doi: 10.1161/01.atv.18.7.1093. [DOI] [PubMed] [Google Scholar]
- Shiode N, Kato M, Hiraoka A, Yamagata T, Matsuura H, Kajiyama G. Impaired endothelium-dependent vasodilation of coronary resistance vessels in hypercholesterolemic patients. Intern Med. 1996;35:89–93. doi: 10.2169/internalmedicine.35.89. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Clanton EC, Russell JM, Ross CR, Granger DN. NAD(P)H oxidase-derived superoxide mediates hypercholesterolemia-induced leukocyte-endothelial cell adhesion. Circ Res. 2001;88:499–505. doi: 10.1161/01.res.88.5.499. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Clanton EC, Russell JM, Ross CR, Granger DN. NAD(P)H oxidase-derived superoxide mediates hypercholesterolemia-induced leukocyte-endothelial cell adhesion. Circ Res. 2001;88:499–505. doi: 10.1161/01.res.88.5.499. [DOI] [PubMed] [Google Scholar]
- Stokes KY, Cooper D, Tailor A, Granger DN. Hypercholesterolemia promotes inflammation and microvascular dysfunction: role of nitric oxide and superoxide. Free Radic Biol Med. 2002;33:1026–1036. doi: 10.1016/s0891-5849(02)01015-8. [DOI] [PubMed] [Google Scholar]
- Tailor A, Granger DN. Hypercholesterolemia promotes P-selectin-dependent platelet-endothelial cell adhesion in postcapillary venules. Arterioscler Thromb Vasc Biol. 2003;23:675–680. doi: 10.1161/01.ATV.0000056742.97580.79. [DOI] [PubMed] [Google Scholar]
- Tailor A, Granger DN. Hypercholesterolemia promotes P-selectin-dependent platelet-endothelial cell adhesion in postcapillary venules. Arterioscler Thromb Vasc Biol. 2003;23:675–680. doi: 10.1161/01.ATV.0000056742.97580.79. [DOI] [PubMed] [Google Scholar]
- Tailor A, Granger DN. Hypercholesterolemia promotes leukocyte-dependent platelet adhesion in murine postcapillary venules. Microcirculation. 2004;11:597–603. doi: 10.1080/10739680490503393. [DOI] [PubMed] [Google Scholar]
- Tigno XT, Ley K, Pries AR, Gaehtgens P. Venulo-arteriolar communication and propagated response. A possible mechanism for local control of blood flow. Pflugers Arch. 1989;414:450–456. doi: 10.1007/BF00585056. [DOI] [PubMed] [Google Scholar]
- Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, Heitzer T, Stasch JP, Griendling KK, Harrison DG, Bohm M, Meinertz T, Munzel T. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–2033. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Roth AC, Russell RC, Graham B, Suchy H, Kucan JO. Morphologic analysis of the microcirculation during reperfusion of ischemic skeletal muscle and the effect of hyperbaric oxygen. Plast Reconstr Surg. 1993;91:1110–1123. doi: 10.1097/00006534-199305000-00022. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Roth AC, Russell RC, Graham B, Suchy H, Kucan JO. Morphologic analysis of the microcirculation during reperfusion of ischemic skeletal muscle and the effect of hyperbaric oxygen. Plast Reconstr Surg. 1993;91:1110–1123. doi: 10.1097/00006534-199305000-00022. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Stephenson LL, Roth AC, Suchy H, Russell RC. Ischemia-reperfusion injury in skeletal muscle: CD 18-dependent neutrophil-endothelial adhesion and arteriolar vasoconstriction. Plast Reconstr Surg. 1997;99:2002–2007. doi: 10.1097/00006534-199706000-00028. [DOI] [PubMed] [Google Scholar]
- Zamboni WA, Stephenson LL, Roth AC, Suchy H, Russell RC. Ischemia-reperfusion injury in skeletal muscle: CD 18-dependent neutrophil-endothelial adhesion and arteriolar vasoconstriction. Plast Reconstr Surg. 1997;99:2002–2007. doi: 10.1097/00006534-199706000-00028. [DOI] [PubMed] [Google Scholar]