

Abstract
Hepatitis B virus (HBV) replication requires the viral polymerase to reverse transcribe the 3.5-kb pregenomic viral RNA within the nucleocapsid. It has been proposed that a sequence element designated phi (φ), which is located 32 nucleotides upstream of the 3′ DR1 pregenomic RNA sequence and is complementary to ε, is required for efficient minus-strand synthesis because it may mediate the translocation of the viral polymerase plus the three nucleotide primer from ε to DR1. A mutation in φ has been identified which can be compensated for with a complementary mutation in ε. This observation supports the suggestion that ε and φ base pair during the process of polymerase translocation from ε to DR1. However, additional mutations in φ were not complemented by the corresponding mutations in ε indicating that the functional recognition of ε and ε/φ stem-loop structures by polymerase probably requires both sequence- and structure-specific information.
Keywords: hepatitis B virus, replication, epsilon, phi
INTRODUCTION
The replication strategy of hepadnaviruses is unique for a DNA virus (Ganem and Schneider, 2001). Although the viral genome is a 3.2-kb partially double-stranded DNA molecule, it is generated by the reverse transcription of a greater-than-genome length 3.5-kb pregenomic RNA (Will et al., 1987; McLachlan, 1991). The 3.5-kb pregenomic RNA is one of several viral transcripts synthesized from nuclear HBV covalently closed circular (CCC) DNA by the cellular RNA polymerase (Cattaneo et al., 1984; Yokosuka et al., 1986; Imazeki et al., 1987; Su et al., 1989; Russnak and Ganem, 1990). These nuclear genomes are derived from virions infecting the hepatocytes or newly synthesized viral capsids cycling viral genomic DNA back into the nucleus as part of the HBV CCC DNA intracellular amplification pathway (Tuttleman et al., 1986; Raney and McLachlan, 1991).
The 3.5-kb pregenomic RNA is translated into the viral core and polymerase polypeptides (Will et al., 1987; Ou et al., 1990). The HBV polymerase recognizes and binds to a stem-loop structure called epsilon (ε) located at the 5′-end of the 3.5-kb pregenomic RNA (Junker-Niepmann et al., 1990; Knaus and Nassal, 1993; Pollack and Ganem, 1994; Wang et al., 1994). This ribonucleoprotein complex is subsequently encapsidated by the core polypeptide generating an immature capsid (Hirsch et al., 1990; Bartenschlager et al., 1990; Bartenschlager and Schaller, 1992; Pollack and Ganem, 1993; Pollack and Ganem, 1994; Fallows and Goff, 1995). Initially, the HBV polymerase utilizes a tyrosine residue within its own amino-terminal domain as a primer to synthesize three nucleotides using the six nucleotide bulge region located in the stem of ε as a template (Gerlich and Robinson, 1980; Wang and Seeger, 1992; Wang and Seeger, 1992; Wang and Seeger, 1993; Zoulim and Seeger, 1994; Weber et al., 1994; Tavis et al., 1994; Lanford et al., 1995; Lanford et al., 1997; Nassal and Rieger, 1996; Jiang and Loeb, 1997). The polymerase with the covalently attached trinucleotide sequence subsequently translocates to the DR1 element located at the 3′-end of the 3.5-kb pregenomic RNA (Seeger and Maragos, 1991; Tavis et al., 1994; Tavis and Ganem, 1995; Loeb and Tian, 1995; Nassal and Rieger, 1996). The mechanism governing this step in the replication cycle is not completely defined but a sequence called phi (φ) which is complementary to the 5′-half of ε has been shown to influence an early step in the viral replication cycle (Tang and McLachlan, 2002a). Interestingly the covalent attachment of the initial three nucleotides to the polymerase appears to be associated with a conformational change in the pregenomic RNA plus polymerase ribonucleoprotein complex (Tavis and Ganem, 1996; Tavis et al., 1998; Beck and Nassal, 1998). This conformational change may facilitate the exchange of the 3′-half of ε, which is initially base paired with the 5′-half of ε, for the φ sequence located 32 nucleotides upstream of the DR1 element at the 3′-end of the pregenomic RNA (Tang and McLachlan, 2002a). This would bring the three nucleotides covalent attached to the HBV polymerase in proximity to their complementary sequence within the DR1 sequence. Association of these three nucleotides with the DR1 element would permit elongation of HBV minus-strand DNA by reverse transcription of the 3.5-kb pregenomic RNA (Wang and Seeger, 1993). Elongation of minus-strand DNA to the end of the pregenomic RNA generates the complete HBV minus-strand DNA. As a result of the RNaseH activity present in the carboxy-terminal domain of the HBV polymerase, the pregenomic RNA is degraded generating an approximately 17 nucleotide primer derived from the 5′-end of the pregenomic RNA (Lien et al., 1986; Will et al., 1987; Loeb et al., 1991). This sequence includes the 5′-end copy of the DR1 sequence which is translocated to the complementary DR2 sequence within the newly synthesized HBV minus-strand DNA (Will et al., 1987; Seeger et al., 1986; Staprans et al., 1991; Loeb et al., 1996; Seeger and Maragos, 1989). This RNA primer serves to initiate plus-strand DNA synthesis leading ultimately to the production of the relaxed-circular 3.2-kb DNA present in the mature virions (Will et al., 1987; Loeb et al., 1997; Lien et al., 1987; Havert and Loeb, 1997).
In this study, the early steps in the viral replication cycle are examined further and the importance of the complementarity between ε and φ are evaluated. Extensive mutational analysis within the φ sequence demonstrates the critical importance of this sequence element in the early steps of viral replication. Analysis of complementary mutations between ε and φ indicate that base-pairing between these two sequence elements does occur and is essential for efficient viral replication. Although base-pairing between ε and φ likely contribute to efficient viral replication, it is apparent that there are also sequence specific requirements within these regulatory elements that are essential for efficient viral replication. Similar conclusions have recently been reported by others [=(Abraham and Loeb, 2006). Finally it is apparent that the φ sequence element cannot substitute for the 3′-half of ε in the initiation of viral replication despite its complementarity to the 5′-half of ε. This observation supports the suggestion that polymerase recognizes the stem-loop structure of ε and ε/φ in a functionally distinct manner.
RESULTS
Effect of mutations in the φ sequence on the efficiency of HBV replication
The φ sequence is located 32 nucleotides upstream of DR1 in the 3′-end of the pregenomic RNA (Fig. 1) and has been shown to contribute to the efficiency of HBV replication in cell culture (Tang and McLachlan, 2002a). In addition, it was noted that the φ sequence is complementary to the 5′-half of the ε sequence suggesting a model for the translocation of the HBV polymerase from ε at the 5′-end of the pregenomic RNA to the DR1 element at the 3′-end of the pregenomic RNA (Tang and McLachlan, 2002a). In this model, the pregenomic RNA and possibly the HBV polymerase would undergo a conformational change after the first three nucleotides of the minus-strand are synthesized (Beck and Nassal, 1998; Tang and McLachlan, 2002a; Tavis and Ganem, 1996; Tavis et al., 1998). During this conformational change, the 3′-half of the ε sequence at the 5′-end of the pregenomic RNA would be exchanged for the φ sequence at the 3′-end of the pregenomic RNA (Tang and McLachlan, 2002a). This would presumably bring the three nucleotide primer into proximity with DR1 allowing it to initiate synthesis of the minus-strand DNA from this location (Wang and Seeger, 1993).
Figure 1.
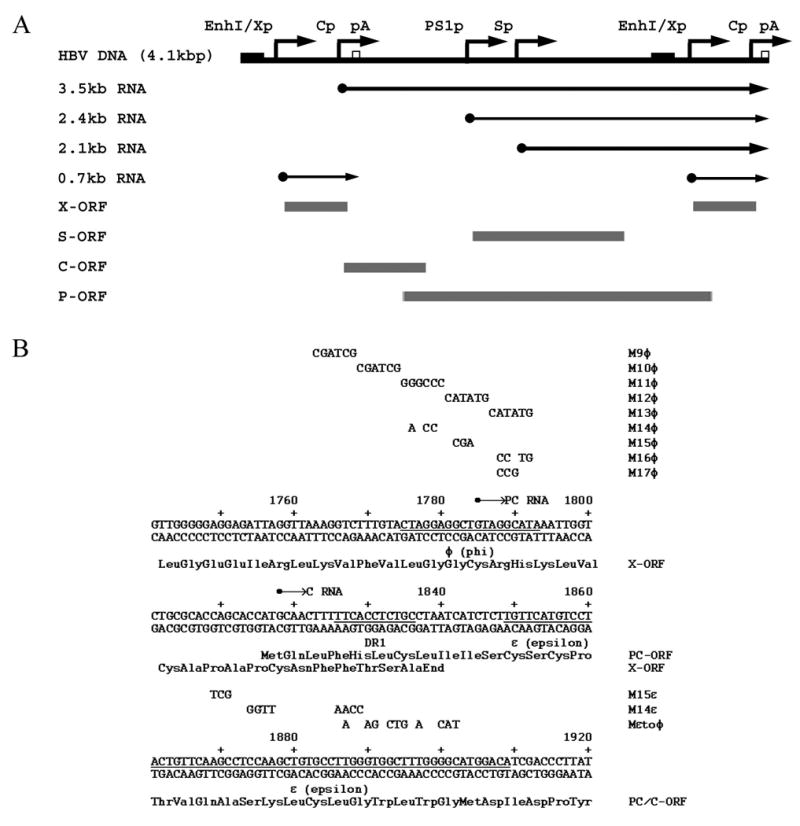
Structure and sequence of the HBV constructs supporting viral transcription and replication in the human HepG2 hepatoma cell line. (A) Structure of the HBV DNA (4.1kbp) construct used in transient transfection analysis. The 4.1-kbp greater-than-genome length HBV DNA sequence in this construct spans coordinates 1072–3182/1–1990 of the HBV genome (subtype ayw). The locations of the HBV 3.5-kb, 2.4-kb, 2.1-kb and 0.7-kb transcripts are indicated. EnhI/Xp, enhancer I/X-gene promoter region; Cp, nucleocapsid or core promoter; pA, polyadenylation site; PS1p, presurface antigen promoter; Sp, major surface antigen promoter; X, X-gene; S, surface antigen gene; C, core gene; P, polymerase gene; ORF, open reading frame. (B) Sequence of the HBV phi (φ), DR1 and epsilon (ε) region of the viral genome. The M9φ to M17φ mutations spanning the φ region are indicated above the HBV nucleotide sequence. These mutations were only introduced into the 3′-end of the HBV DNA (4.1kbp) construct. The M14ε, M15ε and Mεtoφ mutations spanning the ε sequence are also indicated above the HBV nucleotide sequence. The sequence of the precore and X-gene translation initiation codons were mutated from ATG to GTG in the HBV DNA (4.1kbp) PC-X- constructs to prevent the expression of the precore and X-gene polypeptides from the HBV genomes containing ε and φ mutations. The location of the precore (PC) and pregenomic or core (C) RNA initiation sites are indicated. Precore/core open reading frame, PC/C-ORF; X-gene open reading frame, X-ORF.
The original mutagenesis analysis of the φ sequence was performed using a wild-type HBV genome (Tang and McLachlan, 2002a). As the φ sequence is located in the X-gene open reading frame, the types of mutations that could be introduced in the original study were limited to those which did not alter the X-gene encoded polypeptide amino acid sequence (Tang and McLachlan, 2002a). In this study a comprehensive set of mutations was introduced into the φ sequence (Fig. 1). Consequently, all mutagenesis analysis performed in this study used an HBV genome defective in X-gene polypeptide expression due to the conversion of the translation initiation codon from AUG to GUG. In addition, the HBV DNA (4.1kbp) X-PC- construct used to characterize both the ε and φ mutations contained an additional mutation in the precore initiation codon which was also mutated to GUG to prevent the expression of the precore polypeptide (Tang and McLachlan, 2002b). As a result of these mutations, the HBV DNA (4.1kbp) X-PC-construct does not express either the X-gene or the precore polypeptide. Therefore, it is possible to mutate both ε and φ in this HBV genome without having to be concerned about altering the expression of these two viral gene products. Fortunately, it is apparent that the absence of the X-gene and precore polypeptides has a limited effect on HBV transcription and replication in HepG2 cells (Figs. 2–4).
Figure 2.
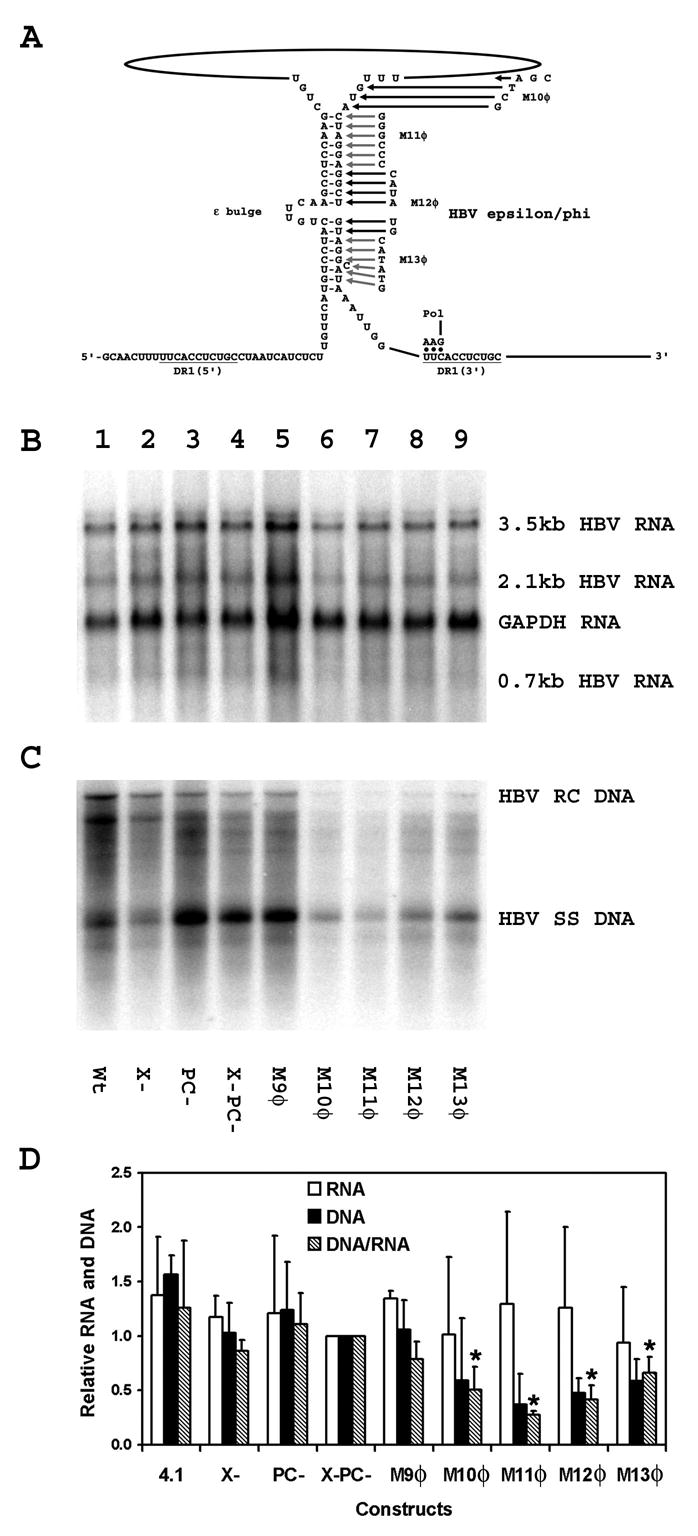
Effect of mutations in the φ sequence on HBV transcription and replication in HepG2 cells. (A) Structure of the putative HBV ε/φ stem-bulge sequence (Tang and McLachlan, 2002a). The location of the HBV polymerase with the three covalently attached nucleotides after translocation from ε to DR1 is indicated. The locations of the M10φ to M13φ mutations are also indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (B) RNA (Northern) filter hybridization analysis of HBV transcripts. Cells were transiently transfected with the wild type HBV DNA (4.1kbp) construct (lane 1) and the mutant HBV DNA (4.1kbp) constructs, X- (lane 2), PC-(lane 3), X-PC- (lane 4), M9φ (lane 5), M10φ (lane 6), M11φ (lane 7), M12φ (lane 8) and M13φ (lane 9). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (C) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA. (D) Quantitative analysis of the 3.5-kb HBV RNA and HBV DNA replication intermediates. The levels of the 3.5-kb HBV RNA and HBV DNA replication intermediates are reported relative to the HBV DNA (4.1kbp) X-PC- construct (lane 4) which are designated as having a relative activity of 1.0. The mean RNA and DNA levels plus standard deviations from two independent analyses are shown. The lower DNA/RNA ratios observed with the φ sequence mutant constructs that are statistically different by Student’s t test (p<0.05) from the DNA/RNA ratios observed with the constructs lacking φ sequence mutants are indicated with asterisks (*).
Figure 4.
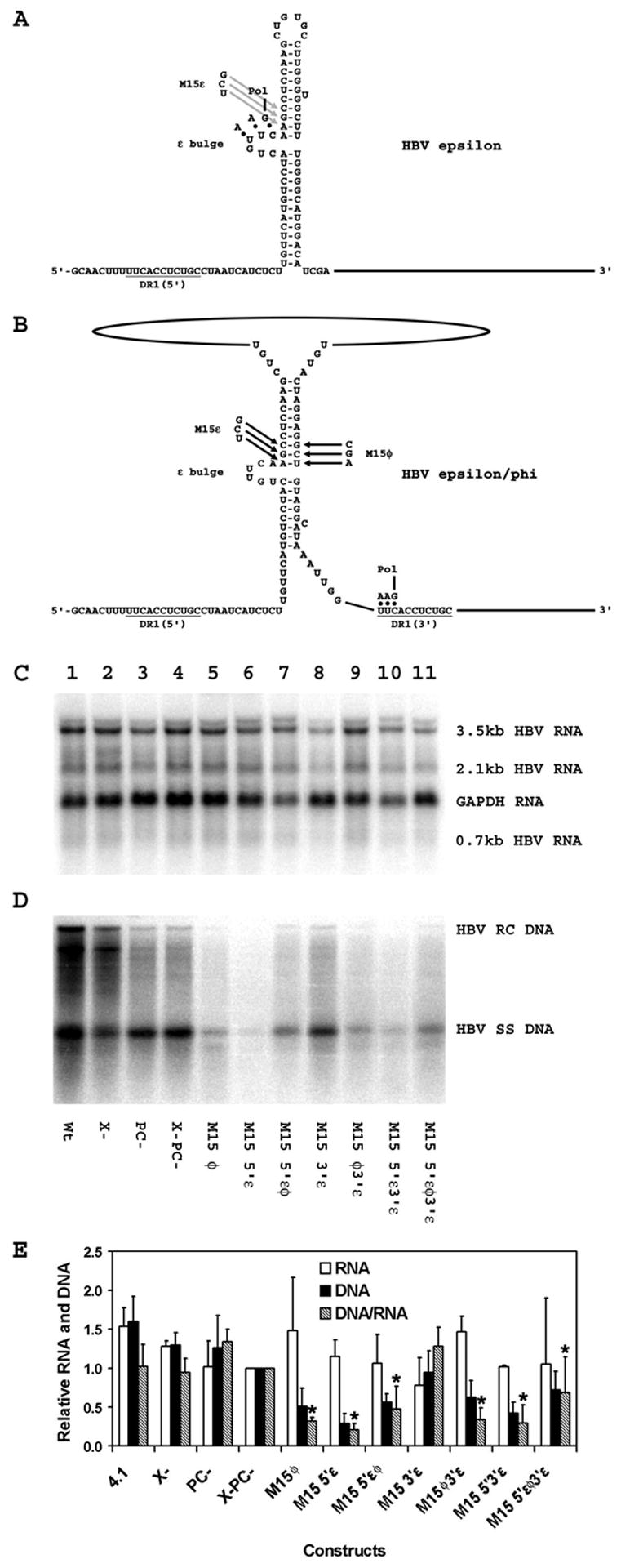
Effect of compensatory mutations in the ε and φ sequences on HBV transcription and replication in HepG2 cells. (A) Structure of the HBV ε stem-loop sequence (Junker-Niepmann et al., 1990). The location of the HBV polymerase with the three covalently attached nucleotides prior to translocation from ε to DR1 is indicated. The location of the M15ε mutation is also indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (B) Structure of the putative HBV ε/φ stem-bulge sequence (Tang and McLachlan, 2002a). The location of the HBV polymerase with the three covalently attached nucleotides after translocation from ε to DR1 is indicated. The location of the M15ε and M15φ mutations are also indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (C) RNA (Northern) filter hybridization analysis of HBV transcripts. Cells were transiently transfected with the wild type HBV DNA (4.1kbp) construct (lane 1) and the mutant HBV DNA (4.1kbp) constructs, X- (lane 2), PC- (lane 3), X-PC- (lane 4), M15 φ (lane 5), M15 5′ε (lane 6), M15 5′εφ (lane 7), M15 3′ε (lane 8), M15 φ3′ε (lane 9), M15 5′ε3′ε (lane 10) and M15 5′εφ3′ε (lane 11). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (D) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA. (E) Quantitative analysis of the 3.5-kb HBV RNA and HBV DNA replication intermediates. The levels of the 3.5-kb HBV RNA and HBV DNA replication intermediates are reported relative to the HBV DNA (4.1kbp) X-PC- construct (lane 4) which are designated as having a relative activity of 1.0. The mean RNA and DNA levels plus standard deviations from three independent analyses are shown. The lower DNA/RNA ratios observed with the ε and φ sequence mutant constructs that are statistically different by Student’s t test (p<0.05) from the DNA/RNA ratios observed with the constructs lacking ε and φ sequence mutants are indicated with asterisks (*).
In an initial attempt to further characterize the φ sequence elements contributing to efficient viral replication, a series of six nucleotide substitutions, M9φ, M10φ, M11φ, M12φ and M13φ, were introduced into the region of the φ sequence located at the 3′-end of the HBV DNA (4.1kbp) X-PC- construct and their effect on HBV RNA and DNA synthesis was determined in HepG2 cells (Figs. 1 and 2). The pregenomic RNA transcribed from these constructs was mutated only at the 3′-end of the transcript and the levels of the HBV transcripts were not influenced by the mutations in the φ sequence (Fig. 2). The level of replication per 3.5-kb viral transcript was not affected by the M9φ mutation which was not located within the φ sequence (Fig. 2). In contrast, the M10φ, M12φ and M13φ mutations, reduce the level of replication per 3.5-kb viral transcript approximately two-fold, whereas the M11φ mutation reduced the level of replication per 3.5-kb viral transcript approximately four-fold (Fig. 2). These results are consistent with our previous mutational analysis of the φ sequence performed using the wild-type HBV genome (Tang and McLachlan, 2002a). In combination, they indicate that the complete φ sequence plus as many as an additional six nucleotides located upstream of the φ sequence (M10φ) contribute to efficient HBV DNA synthesis and the central region of this sequence appears to be the most sensitive to nucleotide substitutions.
Additional mutational analysis of the φ sequence was performed in an attempt to find φ mutations that might permit the analysis of complementary mutations in the ε sequence (Fig. 3). For this analysis, a series of limited clustered point mutations, M14φ, M15φ, M16φ and M17φ, were introduced into the region of the φ sequence located at the 3′-end of the HBV DNA (4.1kbp) X-PC- construct and their effect of HBV RNA and DNA synthesis was determined in HepG2 cells (Figs. 1 and 3). The pregenomic RNA transcribed from these constructs was mutated only at the 3′-end of the transcript and the levels of the HBV transcripts were not influenced by these mutations in the φ sequence (Fig. 3B). The level of replication per 3.5-kb viral transcript observed with the M14φ, M16φ and M17φ mutations was reduced less than two-fold, whereas the M15φ mutation reduced replication per 3.5-kb viral transcript more than two-fold (Fig. 3). These results are consistent with all the previous mutational analysis of the φ sequence and support the suggestion that the central region of the φ sequence is the most sensitive to nucleotide substitutions. In addition, it appears that the greater the region of disruption to the base pairing between the 5′-half of ε and the φ sequence, the greater the effect on viral replication (Figs. 2 and 3) (Tang and McLachlan, 2002a).
Figure 3.
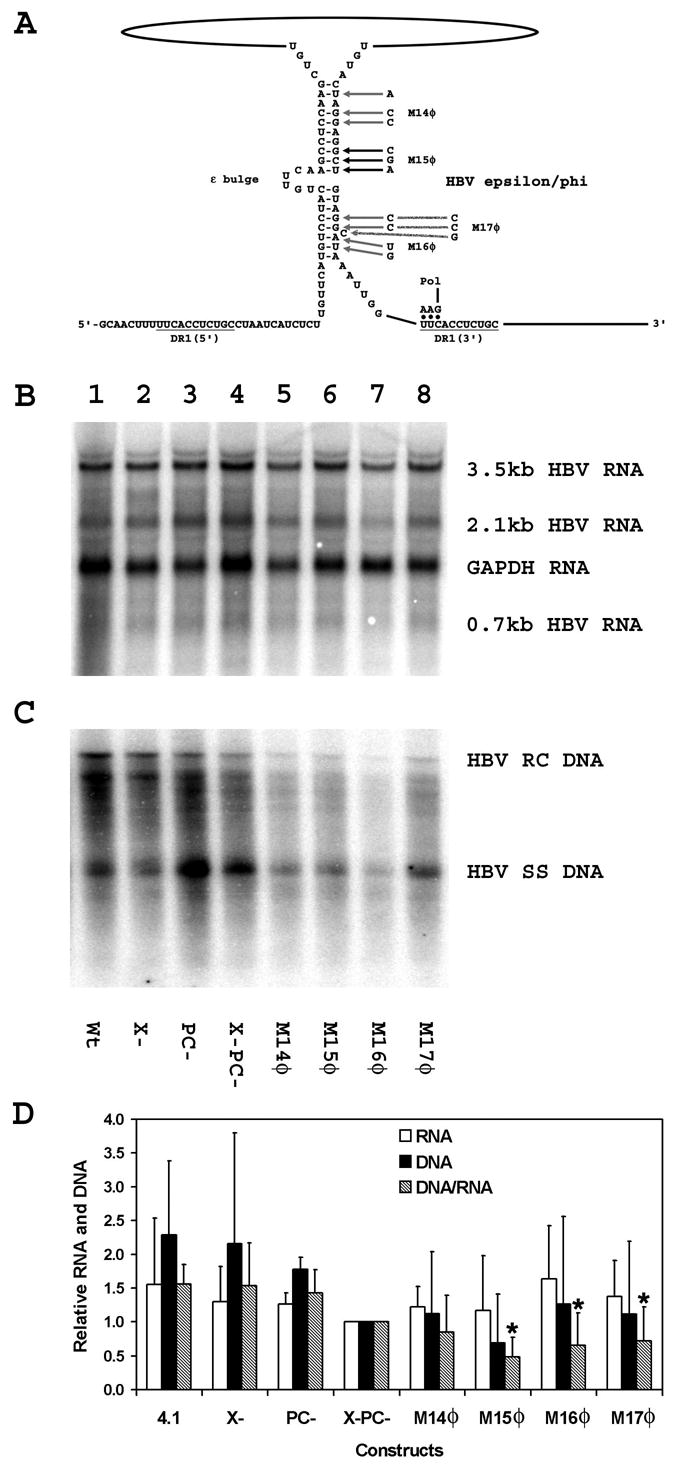
Effect of additional mutations in the φ sequence on HBV transcription and replication in HepG2 cells. (A) Structure of the putative HBV ε/φ stem-bulge sequence (Tang and McLachlan, 2002a). The location of the HBV polymerase with the three covalently attached nucleotides after translocation from ε to DR1 is indicated. The locations of the M14φ to M17φ mutations are also indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (B) RNA (Northern) filter hybridization analysis of HBV transcripts. Cells were transiently transfected with the wild type HBV DNA (4.1kbp) construct (lane 1) and the mutant HBV DNA (4.1kbp) constructs, X- (lane 2), PC-(lane 3), X-PC- (lane 4), M14φ (lane 5), M15φ (lane 6), M16φ (lane 7) and M17φ (lane 8). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (C) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA. (D) Quantitative analysis of the 3.5-kb HBV RNA and HBV DNA replication intermediates. The levels of the 3.5-kb HBV RNA and HBV DNA replication intermediates are reported relative to the HBV DNA (4.1kbp) X-PC- construct (lane 4) which are designated as having a relative activity of 1.0. The mean RNA and DNA levels plus standard deviations from two independent analyses are shown. The lower DNA/RNA ratios observed with the φ sequence mutant constructs that are statistically different by Student’s t test (p<0.05) from the DNA/RNA ratios observed with the constructs lacking φ sequence mutants are indicated with asterisks (*).
Effects of compensatory mutations in the ε and φ sequences on the efficiency of HBV replication
Since the M15φ mutation reduced replication to the greatest extent (Fig. 3), the effect of introducing the corresponding compensatory mutations into the 5′-half of ε was examined (Fig. 4). The compensatory mutation, M15ε (Fig. 1), introduced into the 5′-half of ε has been characterized previously and shown to support encapsidation of pregenomic RNA at a level two- to three- times lower than the wild-type RNA (Pollack and Ganem, 1993). Consequently, it was assumed that the M15ε mutant pregenomic RNA would be encapsidated and potentially support viral replication. The M15ε mutation introduced into the ε sequence at the 5′-end of the pregenomic RNA (M15 5′ε) reduced the level of viral replication per 3.5-kb viral transcript approximately five-fold without affecting pregenomic RNA synthesis (Fig. 4). The reduction in viral replication presumably reflects contributions from either or both the reduced efficiency of pregenomic RNA encapsidation and pregenomic RNA rearrangement during the formation of the ε/φ structure. To examine these possibilities, the level of replication supported by the double M15 5′εφ mutation was compared to the individual mutations (Fig. 4). The double M15 5′εφ mutation supported a higher level of replication per 3.5-kb viral transcript than either single mutation alone (M15 5′ε and M15φ) and the level of viral replication observed was approximately half that observed with the HBV DNA (4.1kbp) X-PC- construct whereas 3.5-kb RNA synthesis was essentially unaffected (Fig. 4). These observations suggest that the M15 5′ε mutation reduces encapsidation approximately two-fold as previously described (Pollack and Ganem, 1993) and the introduction of the compensating mutation M15φ into the φ sequence restores both the base pairing in the ε/φ structure and the replication efficiency of the encapsidated pregenomic RNA (Fig. 4). However, the possibility that the replication efficiency, in addition to the encapsidation efficiency, is affected to some degree by the sequence of the M15 mutations cannot be rigorously excluded. Introduction of the same combinations of mutations into the HBV DNA (4.1kbp) X-PC- construct carrying the M15ε mutation in the 3′ε sequence produced very similar results (Fig. 4). The M15 3′ε mutation did not affect HBV RNA or DNA synthesis (Fig. 4). The M15 5′ε3′ε and M15 φ3′ε double mutations did not affect pregenomic RNA synthesis but the level of viral replication per 3.5-kb viral transcript was reduced approximately three-fold. When the M15 5′εφ3′ε mutations were combined the level of viral replication per 3.5-kb viral transcript was approximately twice the level observed with the M15 5′ε3′ε and M15 φ3′ε double mutations again supporting the contention that the M15 5′ε and the M15φ mutations compensate for each other functionally (Fig. 4). These observations support the suggestion that the ε/φ structure is an important intermediate in the initial steps of HBV minus-strand DNA synthesis.
The M14φ mutation reduced replication to a rather modest extent without affecting pregenomic RNA synthesis (Figs. 3 and 5). Despite the limited effect of the M14φ mutation, the effect of introducing the corresponding compensatory mutations into ε was examined (Fig. 5). The compensatory mutation, M14ε (Figs. 1 and 5), introduced into the upper stem of ε has been characterized previously and shown to support encapsidation of pregenomic RNA at a level similar to the wild-type RNA (Pollack and Ganem, 1993). Consequently, it was assumed that the M14ε mutation would support viral replication. However, the M14ε mutation introduced into the ε sequence at the 5′-end of the pregenomic RNA (M14 5′ε) completely failed to support viral replication despite having no affect on pregenomic RNA synthesis (Fig. 5). The level of replication supported by the double M14 5′εφ mutation was examined but like the M14 5′ε mutation it failed to support any viral DNA synthesis (Fig. 5). These results indicate that certain mutations in ε may support encapsidation under certain circumstances but fail to support the complete viral replication cycle. This suggests there may be sequence specific requirements for the ε sequence which are necessary for viral DNA synthesis but not for encapsidation. Alternatively, the M14 5′ε mutation may simply fail to support encapsidation under the current conditions. In addition, these observations indicate how difficult it can be to generate functional ε mutations which might be used to test the hypothetical base pairing between the 5′-half of the ε sequence and the φ sequence for their roles in the initiation of minus-strand DNA synthesis (Tang and McLachlan, 2002a).
Figure 5.
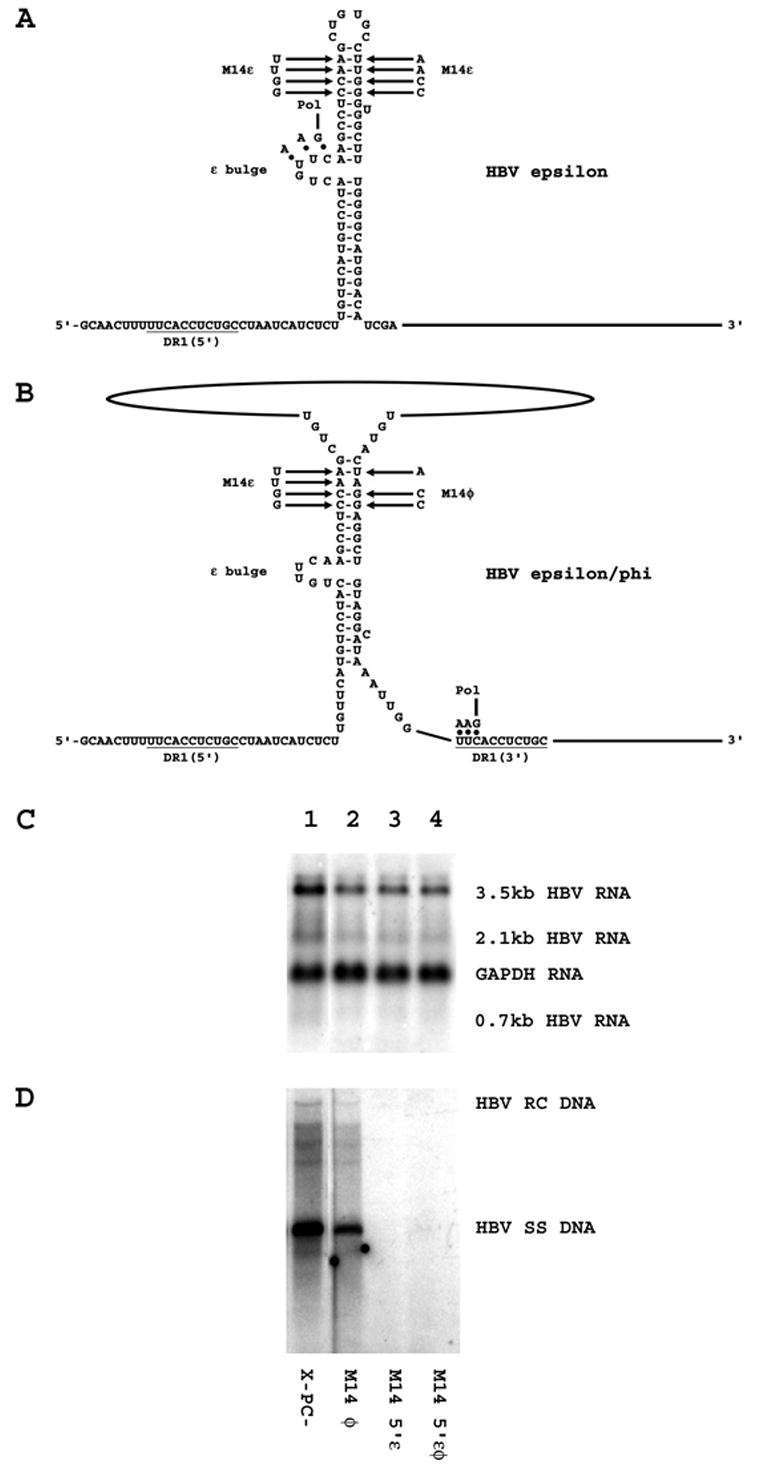
Effect of additional mutations in the ε and φ sequences on HBV transcription and replication in HepG2 cells. (A) Structure of the HBV ε stem-loop sequence (Junker-Niepmann et al., 1990). The location of the HBV polymerase with the three covalently attached nucleotides prior to translocation from ε to DR1 is indicated. The location of the M14ε mutation is also indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (B) Structure of the putative HBV ε/φ stem-bulge sequence (Tang and McLachlan, 2002a). The location of the HBV polymerase with the three covalently attached nucleotides after translocation from ε to DR1 is indicated. The location of the M14ε and M14φ mutations are also indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (C) RNA (Northern) filter hybridization analysis of HBV transcripts. Cells were transiently transfected with the mutant HBV DNA (4.1kbp) constructs, X-PC- (lane 1), M14 φ (lane 2), M14 5′ε (lane 3) and M14 5′εφ (lane 4). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (D) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA.
The φ sequence is not functionally equivalent to the ε sequence for HBV replication
From these studies, it is apparent that it is not trivial to find mutations in the ε sequence which can support pregenomic RNA encapsidation and viral replication. However, it is apparent that mutations can be introduced into the 3′-half of ε to convert this sequence to the φ sequence without altering the coding potential of the HBV DNA (4.1kbp) X-PC- construct (Figs. 1 and 6). Creating an HBV genome where the 3′-half of ε is converted to the φ sequence permits the potential functional properties of the putative ε/φ base pairing to be examined. Introduction of the φ sequence into the ε sequence at the 5′-end of the pregenomic RNA does not affect viral transcription but fails to support any viral replication. This indicates that the ε/φ base paired structure, if formed, is not functionally equivalent to the ε sequence in the initiation of viral replication even when this putative structure is introduced into the 5′-end of the pregenomic RNA at the same location as the ε sequence. Therefore this analysis is unable to determine if the ε/φ structure can be bound by the HBV polymerase as predicted by the proposed role of the φ sequence in the early steps of viral replication. This may reflect the inability of polymerase to recognize the ε/φ structure until the initial three nucleotides of minus-strand DNA are synthesized. The observation that the ε to φ mutation can be introduced into the ε sequence at the 3′-end of the pregenomic RNA (Mεtoφ 3′ε) indicates that this mutation does not have a dominant negative effect on viral replication (Fig. 6).
Figure 6.
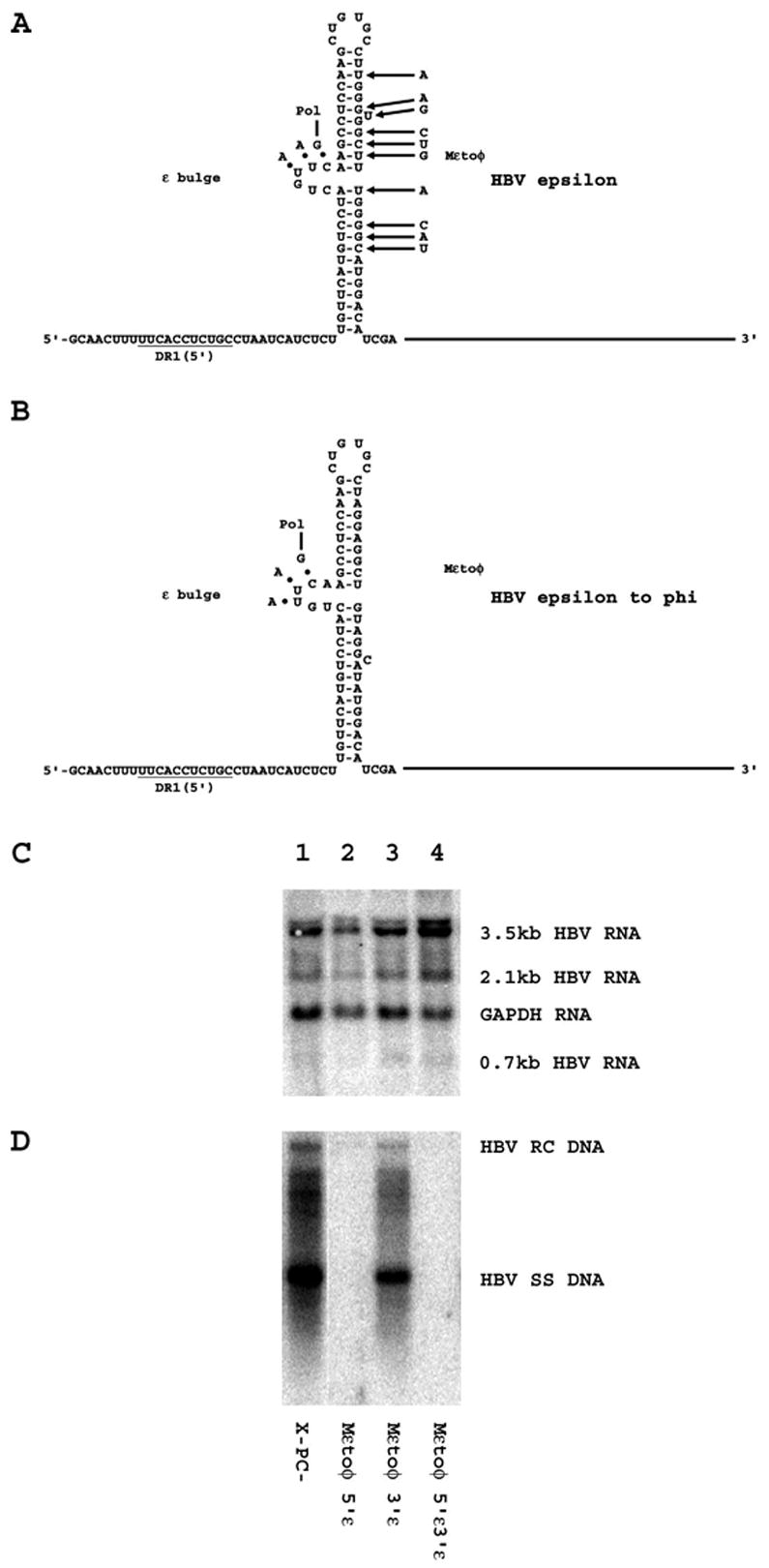
Effect of mutating the 3′-half of the ε sequence to the φ sequence on HBV transcription and replication in HepG2 cells. (A) Structure of the HBV ε stem-loop sequence (Junker-Niepmann et al., 1990). The location of the HBV polymerase with the three covalently attached nucleotides prior to translocation from ε to DR1 is indicated. The location of the Mεtoφ mutation is also indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (B) Putative structure of the HBV ε/φ stem-loop sequence produced by the Mεtoφ mutation. The presumed location of the HBV polymerase with the three covalently attached nucleotides prior to translocation from putative mutant HBV ε/φ stem-loop structure to DR1 is indicated. The pregenomic RNA sequence is derived from the HBVayw genome (Galibert et al., 1979). (C) RNA (Northern) filter hybridization analysis of HBV transcripts. Cells were transiently transfected with the mutant HBV DNA (4.1kbp) constructs, X-PC- (lane 1), Mεtoφ 5′ε (lane 2), Mεtoφ 3′ε (lane 3) and Mεtoφ 5′ε3′ε (lane 4). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript was used as an internal control for RNA loading per lane. (D) DNA (Southern) filter hybridization analysis of HBV replication intermediates. HBV RC DNA, HBV relaxed circular DNA; HBV SS DNA, HBV single-stranded DNA.
DISCUSSION
In our previous studies, the φ sequence was identified as a 19 nucleotide regulatory element located 32 nucleotides upstream of the DR1 element at the 3′-end of the HBV 3.5-kb pregenomic RNA (Tang and McLachlan, 2002a). This element did not affect the level of pregenomic RNA synthesis but was necessary for efficient viral DNA synthesis (Tang and McLachlan, 2002a). In addition, the φ sequence was complementary to the 5′-half of the ε sequence and phylogenetically conserved between the woodchuck and human hepatitis B viruses (Tang and McLachlan, 2002a). These observations led to the suggestion that the role of the φ sequence in viral replication is probably at an early step in the replication cycle and is most likely involved in the process of the translocation of the HBV polymerase from ε at the 5′-end of the pregenomic RNA to the DR1 sequence at the 3′-end of the pregenomic RNA (Tang and McLachlan, 2002a). The basic model involves the HBV polymerase recognizing and binding to ε. Inside the immature capsid, the polymerase uses the bulge region of ε as a template and the tyrosine reside in the terminal-protein domain of the polymerase as a primer to synthesize the first three nucleotides of the HBV minus-strand DNA. During or subsequent to this synthesis, the polymerase and/or ε undergoes a conformational change resulting in the displacement of the 3′-half of ε and its replacement with the φ sequence generating a polymerase plus ε/φ complex with a similar structure to the polymerase plus ε structure except the loop in the RNA stem/loop structure is now approximately 3-kb rather than six nucleotides as found in the ε structure (Tang and McLachlan, 2002a). This strand displacement reaction generates a complex where the original three nucleotides reverse transcribed from the loop of ε are now in close proximity to their target sequence at the 5′-end of the DR1 sequence (Tang and McLachlan, 2002a). Presumable the close proximity of this tri-nucleotide priming sequence to DR1 promotes their interaction and the initiation of the extension reaction necessary for complete minus-strand synthesis using the pregenomic RNA as a template. The subsequent synthesis of the plus-strand HBV DNA would complete the production of the viral genome and generate the mature capsid found in the virus particles (Ganem and Schneider, 2001; Raney and McLachlan, 1991).
Based on this model for the role of the φ sequence in the initiation of minus-strand DNA synthesis, it is predicted that for the φ sequence to facilitate HBV replication it must be able to base-pair with the 5′-half of the ε sequence. To functionally examine this possibility, it is necessary to identify mutations in the φ sequence that reduce viral replication efficiency and can be compensated for by the complementary mutations in the ε sequence. Although this is straightforward in principal, it is an extremely difficult challenge to identify the appropriate mutations given the functional importance of boththe ε sequence and structure to its role in polymerase recognition and the initiation of minus-strand DNA synthesis (Pollack and Ganem, 1993; Hu and Boyer, 2006).
In an initial attempt to address this issue, a comprehensive mutational analysis of the φ sequence was performed to determine in detail the region of this element that was most important for viral replication (Figs. 2 and 3). Based on mutations M11φ, M12φ and M15φ, it appears that the 5′-half of the φ sequence which is proposed to form the upper stem structure with the 5′-half of the ε sequence is the most important functional region of the φ sequence. Based on mutations M13φ, M16φ and M17φ, it appears that the 3′-half of the φ sequence which is proposed to form the lower stem structure with the 5′-half of the ε sequence is a less important functional region of the φ sequence. However this mutational analysis supports the suggestion that the whole φ sequence contributes to the function of this element in governing the efficiency of viral replication. In addition, the M10φ mutation suggests that up to six additional nucleotides located directly upstream of the φ sequence may also contribute to the efficiency of viral replication (Fig. 2). Previous analysis had also suggested that this region upstream of the φ sequence might contribute to the efficiency of viral replication (Tang and McLachlan, 2002a).
The mutagenesis analysis of the φ sequence suggested that the 5′-end of the φ sequence might be a suitable target region for designing mutations which might be compensated for by introducing the complementary mutations into the ε sequence. Based on previous studies (Pollack and Ganem, 1993), it appeared that the complementary mutation to M15φ within the ε sequence, M15ε, should support encapsidation of pregenomic RNA. Indeed, the M15ε and the M15φ mutations were able to support viral replication although at a significantly reduced level per 3.5-kb viral transcript compared with the HBV DNA (4.1kb) X-PC- construct (Fig. 4). When both the M15ε and the M15φ mutations were introduced into the pregenomic RNA the level of replication per 3.5-kb viral transcript was increased to approximately half the level observed with the HBV DNA (4.1kb) X-PC- construct (Fig. 4). This modest reduction in replication efficiency may be attributed to the reduced encapsidation efficiency of the M15ε mutation (Pollack and Ganem, 1993) or, alternatively, may reflect slightly reduced replication efficiency during the initial steps in minus-strand DNA synthesis. In general these findings support the suggestion base-pairing between the 5′-half of the ε sequence and the φ sequence contribute to efficient viral replication, presumable during the initial steps of minus-strand DNA synthesis.
In an attempt to find additional mutations in the ε sequence that could compensate for mutations in the φ sequence, the effect of the M14ε mutation was examined (Fig. 5). This mutation would permit base-pairing with the corresponding M14φ mutation in the φ sequence (Fig. 5) which we have demonstrated to modestly influence the efficiency of viral replication (Fig. 4). The M14ε mutation has been shown previously to permit essentially wild-type levels of encapsidation of pregenomic RNA (Pollack and Ganem, 1993) and therefore appeared to represent a suitable candidate for this analysis. However, the M14ε mutation failed to support any viral replication and the compensating M14φ mutation did not restore any HBV DNA synthesis (Fig. 5). This result suggests that the sequence in addition to the structure of ε is important in determining viral replication and illustrates the difficulty in identifying suitable mutations in ε for analysis of compensating mutations in the φ sequence.
In an attempt to directly establish the functional significance of the ε/φ structure for viral replication, the sequence of the 3′-half of ε was changed to the φ sequence (Fig. 6). If this mutant pregenomic RNA was capable of supporting any level of viral replication, it would demonstrate that the HBV polymerase was capable of functionally interacting with the ε/φ structure. However the Mεtoφ mutation did not support any viral replication (Fig. 6). This demonstrates that the ε/φ structure is not functionally equivalent to the ε stem/loop structure. In addition, these analyses indicate that the function of the ε stem/loop structure and the putative ε/φ structure depend both on their primary sequences as well as their secondary and potentially tertiary structure. Recently, similar conclusions were reached by others (Abraham and Loeb, 2006) supporting the suggestion that base-pairing between ε and φ sequence contribute to but are not the only determinant governing to the efficiency of viral replication.
MATERIALS AND METHODS
Plasmid constructions
The steps in the cloning of the plasmid constructs used in the transfection experiments were performed by standard techniques (Sambrook et al., 1989). HBV DNA sequences in these constructions were derived from the plasmid pCP10, which contains two copies of the HBV genome (subtype ayw) cloned into the EcoRI site of pBR322 (Dubois et al., 1980). The HBV DNA (4.1kbp) construct that contains 1.3 copies of the HBV genome includes the viral sequence from nucleotide coordinates 1072 to 3182 plus 1 to 1990. This plasmid was constructed by cloning the NsiI/BglII HBV DNA fragment (nucleotide coordinates 1072 to 1990) into pUC13, generating pHBV(1072–1990). Subsequently, a complete copy of the 3.2kbp viral genome linearized at the NcoI site (nucleotide coordinates 1375 to 3182 plus 1 to 1374) was cloned into the unique NcoI site (HBV nucleotide coordinate 1374) of pHBV(1072–1990), generating the HBV DNA (4.1kbp) construct (Fig. 1A).
The HBV DNA (4.1kbp) X-, PC-, and X-PC- constructs were derived by introducing the A1376G, A1816G and both nucleotide substitutions, respectively, into the X-gene and the precore coding regions in the HBV DNA (4.1kbp) construct using the QickChange II site-directed mutagenesis kit (Stratagene Cloning Systems, La Jolla, CA) according to the manufacturer's instructions. The A1376G and A1816G nucleotide substitutions converted the X-gene and precore translation initiation codons from ATG to GTG preventing the initiation of translation of the X-gene and precore polypeptides, respectively. The nucleotide substitutions introduced into the X-gene and precore initiation codons were verified by dideoxynucleotide sequencing (Sanger et al., 1977). The X-gene and precore initiation codons at both the 5′- and 3′-ends of these terminally redundant HBV constructs were mutated for this analysis.
The plasmids, M9φ, M10φ, M11φ, M12φ, M13φ, M14φ, M15φ, M16φ and M17φ, were derived by introducing the nucleotide substitutions indicated in Figure 1 into the φ region between coordinates 1763 and 1792 located immediately upstream of the 3′ DR1 element in the HBV DNA (4.1kbp) X-PC- construct. The plasmids, M14ε, M15ε and Mεtoφ, were derived by introducing the nucleotide substitutions indicated in Figure 1 into the ε sequence between coordinates 1869 and 1902 located at either or both the 5′- and 3′-ends of the HBV DNA (4.1kbp) X-PC- construct. The QickChange II site-directed mutagenesis kit (Stratagene Cloning Systems, La Jolla, CA) was using according to the manufacturer's instructions to generate these mutated HBV DNA (4.1kbp) X-PC-constructs. The nucleotide substitutions introduced into the ε and φ regions of these constructs were verified by dideoxynucleotide sequencing (Sanger et al., 1977).
Cells and transfections
The human hepatoma cell line HepG2 were grown in RPMI-1640 medium and 10% fetal bovine serum at 37°C in 5% CO2/air. Transfections for viral RNA and DNA analysis were performed as previously described (McLachlan et al., 1987) using 10 cm plates, containing approximately 1 X 106 cells. Cells were transfected with 15μg of HBV DNA (4.1kbp) constructs and DNA and RNA isolation was performed 3 days post transfection (Tang and McLachlan, 2001).
Characterization of HBV transcripts and viral replication intermediates
Transfected cells from a single plate were divided equally and used for the preparation of total cellular RNA and viral DNA replication intermediates as described previously (Summers et al., 1991) with minor modifications (Tang and McLachlan, 2001). For RNA isolation (Chomczynski and Sacchi, 1987), the cells were lysed in 1.8 ml of 25 mM sodium citrate, pH 7.0, 4 M guanidinium isothiocyanate, 0.5% (v/v) sarcosyl, 0.1 M 2-mercaptoethanol. After addition of 0.18 ml of 2M sodium acetate, pH 4.0, the lysate was extracted with 1.8 ml of water-saturated phenol plus 0.36 ml of chloroform-isoamyl alcohol (49:1). After centrifugation for 30 min. at 3,000 rpm in a Sorval RT6000, the aqueous layer was precipitated with 1.8 ml of isopropanol. The precipitate was resuspended in 0.3 ml of 25 mM sodium citrate, pH 7.0, 4 M guanidinium isothiocyanate, 0.5% (v/v) sarcosyl, 0.1 M 2-mercaptoethanol and precipitated with 0.6 ml of ethanol. After centrifugation for 20 min. at 14,000 rpm in an Eppendorf 5417C microcentifuge, the precipitate was resuspended in 0.3 ml of 10 mM Tris hydrochloride, pH 8.0, 5 mM EDTA, 0.1% (w/v) sodium lauryl sulfate and precipitated with 45 μl of 2 M sodium acetate plus 0.7 ml of ethanol.
For the isolation of viral DNA replication intermediates, the cells were lysed in 0.4 ml of 100 mM Tris hydrochloride, pH 8.0, 0.2% (v/v) NP40. The lysate was centrifuged for 1 min. at 14,000 rpm in an Eppendorf 5417C microcentrifuge to pellet the nuclei. The supernatant was adjusted to 6.75 mM magnesium acetate plus 200 ug/ml DNase I and incubated for 1 hr at 37°C to remove the transfected plasmid DNA. The supernatant was readjusted to 100 mM NaCl, 10 mM EDTA, 0.8% (w/v) sodium lauryl sulfate, 1.6 mg/ml pronase and incubated for an additional 1 hr at 37°C. The supernatant was extracted twice with phenol, precipitated with two volumes of ethanol and resuspended in 100 μl of 10 mM Tris hydrochloride, pH8.0, 1 mM EDTA.
RNA (Northern) and DNA (Southern) filter hybridization analysis were performed using 10 μg of total cellular RNA and 30 μl of viral DNA replication intermediates, respectively, as described (Sambrook et al., 1989). Transcription initiation sites for the HBV 3.5-kb transcripts were examined by primer extension analysis using 4 units of avian myeloblastosis virus reverse transcriptase (Roche), 7 ng 32P-labeled HBV oligonucleotide probe, GGAAAGAAGTCAGAAGGCAAAAACGAGAGTAACTCC (HBV coordinates 1976 to 1941), and 10 μg of total cellular RNA as described by the manufacturer. Filter hybridization and primer extension analyses were quantitated by phosphorimaging using a Molecular Dynamics Storm 860 phosphoimager.
Acknowledgments
We thank Philip Matsumura for many helpful suggestions and discussions. This work was supported by a Public Health Service grant AI30070 and AI55548 from the National Institutes of Health.
Contributor Information
Claudia E. Oropeza, Email: coropeza@uic.edu.
Alan McLachlan, Email: mclach@uic.edu.
References
- Abraham TM, Loeb DD. Base pairing between the 5′ half of ε and a cis-acting sequence, φ, makes a contribution to the synthesis of minus-strand DNA for human hepatitis B virus. J Virol. 2006;80:4380–4387. doi: 10.1128/JVI.80.9.4380-4387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, Junker-Niepmann M, Schaller H. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J Virol. 1990;64:5324–5332. doi: 10.1128/jvi.64.11.5324-5332.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, Schaller H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992;11:3413–3420. doi: 10.1002/j.1460-2075.1992.tb05420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck J, Nassal M. Formation of a functional hepatitis B virus replication initiation complex involves a major structural alteration in the RNA template. Mol Cell Biol. 1998;18:6265–6272. doi: 10.1128/mcb.18.11.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo R, Will H, Schaller H. Hepatitis B virus transcription in the infected liver. EMBO J. 1984;3:2191–2196. doi: 10.1002/j.1460-2075.1984.tb02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dubois MF, Pourcel C, Rousset S, Chany C, Tiollais P. Excretion of hepatitis B surface antigen particles from mouse cells transformed with cloned viral DNA. Proc Natl Acad Sci USA. 1980;77:4549–4553. doi: 10.1073/pnas.77.8.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallows DA, Goff SP. Mutations in the ε sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J Virol. 1995;69:3067–3073. doi: 10.1128/jvi.69.5.3067-3073.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galibert F, Mandart E, Fitoussi F, Tiollais P, Charnay P. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature. 1979;281:646–650. doi: 10.1038/281646a0. [DOI] [PubMed] [Google Scholar]
- Ganem D, Schneider R. Hepadnaviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Field's Virology. Lippincott, Williams and Wilkins; New York: 2001. pp. 2923–2970. [Google Scholar]
- Gerlich WH, Robinson WS. Hepatitis B virus contains protein attached to the 5′ terminus of its complete DNA strand. Cell. 1980;21:801–809. doi: 10.1016/0092-8674(80)90443-2. [DOI] [PubMed] [Google Scholar]
- Havert MB, Loeb DD. cis-acting sequences in addition to donor and acceptor sites are required for template switching during synthesis of plus-strand DNA for duck hepatitis B virus. J Virol. 1997;71:5336–5344. doi: 10.1128/jvi.71.7.5336-5344.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch RC, Lavine JE, Chang LJ, Varmus HE, Ganem D. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature. 1990;344:552–555. doi: 10.1038/344552a0. [DOI] [PubMed] [Google Scholar]
- Hu J, Boyer M. Hepatitis B virus reverse transcriptase and ε RNA sequences required for specific interaction in vitro. J Virol. 2006;80:2141–2150. doi: 10.1128/JVI.80.5.2141-2150.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imazeki F, Yaginuma K, Omata M, Okuda K, Kobayashi M, Koike K. RNA transcripts of hepatitis B virus in hepatocellular carcinoma. Hepatology. 1987;7:753–757. doi: 10.1002/hep.1840070423. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Loeb DD. Insertions within epsilon affect synthesis of minus-strand DNA before the template switch for duck hepatitis B virus. J Virol. 1997;71:5345–5354. doi: 10.1128/jvi.71.7.5345-5354.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker-Niepmann M, Bartenschlager R, Schaller H. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990;9:3389–3396. doi: 10.1002/j.1460-2075.1990.tb07540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus T, Nassal M. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res. 1993;21:3967–3975. doi: 10.1093/nar/21.17.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Notvall L, Beames B. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J Virol. 1995;69:4431–4439. doi: 10.1128/jvi.69.7.4431-4439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Notvall L, Lee H, Beames B. Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J Virol. 1997;71:2996–3004. doi: 10.1128/jvi.71.4.2996-3004.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien JM, Aldrich CE, Mason WS. Evidence that a capped oligoribonucleotide is the primer for duck hepatitis B virus plus-strand DNA synthesis. J Virol. 1986;57:229–236. doi: 10.1128/jvi.57.1.229-236.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien JM, Petcu DJ, Aldrich CE, Mason WS. Initiation and termination of duck hepatitis B virus DNA synthesis during virus maturation. J Virol. 1987;61:3832–3840. doi: 10.1128/jvi.61.12.3832-3840.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb DD, Gulya KJ, Tian R. Sequence identity of the terminal redundancies on the minus- strand DNA template is necessary but not sufficient for the template switch during hepadnavirus plus-strand DNA synthesis. J Virol. 1997;71:152–160. doi: 10.1128/jvi.71.1.152-160.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb DD, Hirsch RC, Ganem D. Sequence-independent RNA cleavages generate the primers for plus strand DNA synthesis in hepatitis B viruses: Implications for other reverse transcribing elements. EMBO J. 1991;10:3533–3540. doi: 10.1002/j.1460-2075.1991.tb04917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb DD, Tian R. Transfer of the minus strand of DNA during hepadnavirus replication is not invariable but prefers a specific location. J Virol. 1995;69:6886–6891. doi: 10.1128/jvi.69.11.6886-6891.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb DD, Tian R, Gulya KJ. Mutations within DR2 independently reduce the amount of both minus- and plus-strand DNA synthesized during duck hepatitis B virus replication. J Virol. 1996;70:8684–8690. doi: 10.1128/jvi.70.12.8684-8690.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan A. Molecular biology of the hepatitis B virus. CRC Press; Boca Raton, Florida: 1991. [Google Scholar]
- McLachlan A, Milich DR, Raney AK, Riggs MG, Hughes JL, Sorge J, Chisari FV. Expression of hepatitis B virus surface and core antigens: Influences of pre-s and precore sequences. J Virol. 1987;61:683–692. doi: 10.1128/jvi.61.3.683-692.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassal M, Rieger A. Bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J Virol. 1996;70:2764–2773. doi: 10.1128/jvi.70.5.2764-2773.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou JH, Bao H, Shih C, Tahara SM. Preferred translation of human hepatitis B virus polymerase from core protein- but not from precore protein-specific transcript. J Virol. 1990;64:4578–4581. doi: 10.1128/jvi.64.9.4578-4581.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack JR, Ganem D. Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J Virol. 1994;68:5579–5587. doi: 10.1128/jvi.68.9.5579-5587.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack JR, Ganem D. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J Virol. 1993;67:3254–3263. doi: 10.1128/jvi.67.6.3254-3263.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney AK, McLachlan A. The biology of hepatitis B virus. In: McLachlan A, editor. Molecular biology of the hepatitis B virus. CRC Press; Boca Raton, Florida: 1991. pp. 1–37. [Google Scholar]
- Russnak R, Ganem D. Sequences 5′ to the polyadenylation signal mediate differential poly(A) site use in hepatitis B viruses. Genes Dev. 1990;4:764–776. doi: 10.1101/gad.4.5.764. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; New York: 1989. [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Ganem D, Varmus HE. Biochemical and genetic evidence for the hepatitis B virus replication strategy. Science. 1986;232:447–484. doi: 10.1126/science.3961490. [DOI] [PubMed] [Google Scholar]
- Seeger C, Maragos J. Identification of a signal necessary for initiation of reverse transcription of the hepadnavirus genome. J Virol. 1991;65:5190–5195. doi: 10.1128/jvi.65.10.5190-5195.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Maragos J. Molecular analysis of the function of direct repeats and a polypurine tract for plus-strand DNA priming in woodchuck hepatitis virus. J Virol. 1989;63:1907–1915. doi: 10.1128/jvi.63.5.1907-1915.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staprans S, Loeb DD, Ganem D. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J Virol. 1991;65:1255–1262. doi: 10.1128/jvi.65.3.1255-1262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T, Lui WY, Lin LH, Han S, P'eng FK. Analysis of hepatitis B virus transcripts in infected human livers. Hepatology. 1989;9:180–185. doi: 10.1002/hep.1840090203. [DOI] [PubMed] [Google Scholar]
- Summers J, Smith PM, Huang M, Yu M. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J Virol. 1991;65:1310–1317. doi: 10.1128/jvi.65.3.1310-1317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, McLachlan A. A pregenomic RNA sequence adjacent to DR1 and complementary to epsilon influences hepatitis B virus replication efficiency. Virology. 2002a;303:199–210. doi: 10.1006/viro.2002.1645. [DOI] [PubMed] [Google Scholar]
- Tang H, McLachlan A. Mechanisms of inhibition of nuclear hormone receptor dependent hepatitis B virus replication by hepatocyte nuclear factor 3β. J Virol. 2002b;76:8572–8581. doi: 10.1128/JVI.76.17.8572-8581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, McLachlan A. Transcriptional regulation of hepatitis B virus by nuclear hormone receptors is a critical determinant of viral tropism. Proc Natl Acad Sci USA. 2001;98:1841–1846. doi: 10.1073/pnas.041479698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Ganem D. RNA sequences controlling the initiation and transfer of duck hepatitis B virus minus-strand DNA. J Virol. 1995;69:4283–4291. doi: 10.1128/jvi.69.7.4283-4291.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Ganem D. Evidence for activation of the hepatitis B virus polymerase by binding of its RNA template. J Virol. 1996;70:5741–5750. doi: 10.1128/jvi.70.9.5741-5750.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Massey B, Gong YH. The duck hepatitis B virus polymerase is activated by its RNA packaging signal, ε. J Virol. 1998;72:5789–5796. doi: 10.1128/jvi.72.7.5789-5796.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Perri S, Ganem D. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J Virol. 1994;68:3536–3543. doi: 10.1128/jvi.68.6.3536-3543.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- Wang G-H, Seeger C. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell. 1992;71:663–670. doi: 10.1016/0092-8674(92)90599-8. [DOI] [PubMed] [Google Scholar]
- Wang G-H, Seeger C. Novel mechanism for reverse transcription in hepatitis B viruses. J Virol. 1993;67:6507–6512. doi: 10.1128/jvi.67.11.6507-6512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-H, Zoulim F, Leber EH, Kitson J, Seeger C. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J Virol. 1994;68:8437–8442. doi: 10.1128/jvi.68.12.8437-8442.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Bronsema V, Bartos H, Bosserhoff A, Bartenschlager R, Schaller H. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J Virol. 1994;68:2994–2999. doi: 10.1128/jvi.68.5.2994-2999.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will H, Reiser W, Weimer T, Pfaff E, Buscher M, Sprengle R, Cattaneo R, Schaller H. Replication strategy of human hepatitis B virus. J Virol. 1987;61:904–911. doi: 10.1128/jvi.61.3.904-911.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka O, Omata M, Imazeki F, Ito Y, Okuda K. Hepatitis B virus RNA transcripts and DNA in chronic liver disease. N Engl J Med. 1986;315:1187–1192. doi: 10.1056/NEJM198611063151903. [DOI] [PubMed] [Google Scholar]
- Zoulim F, Seeger C. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J Virol. 1994;68:6–13. doi: 10.1128/jvi.68.1.6-13.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]