

Abstract
Cascades of kinases and phosphatases are regulated by selective protein-protein interactions that are essential for signal transduction. Peptide modulators of these interactions have been used to dissect the function of individual components of the signaling cascade, without relying on either the over- or underexpression of proteins. Previously, we identified RACK1 as an endogenous substrate, binding partner and inhibitor of Src tyrosine kinases. Here we utilized cell-permeable peptides that selectively disrupt or enhance the interaction of RACK1 and Src to further examine the function of RACK1. Our results provide direct physiologic evidence that RACK1 regulates growth of NIH3T3 cells by suppressing the activity of Src and other cell cycle regulators in G1, and delaying entry into S phase. They also demonstrate the potential for using peptide modulators of Src activity as a tool for regulating cell growth, and for designing new strategies for cancer therapy that target specific protein-protein interactions.
Keywords: Src, RACK1, PKC, tyrosine kinases, G1/S transition, cell cycle regulation
RACK1 is the founding member of a family of intracellular receptors for activated C kinase (PKC) collectively called RACKs [reviewed in 1]. RACKs determine the specificity and function of PKC isozymes by selectively anchoring activated isozymes to specific subcellular sites, and near to specific substrates that mediate distinct cellular functions. For example, εRACK anchors activated εPKC, which protects the heart from ischemic and reperfusion damage, whereas RACK1 anchors activated βIIPKC, which mediates phenylepherine-induced cardiac hypertrophy [reviewed in 2].
Using the yeast two-hybrid assay, we identified RACK1 as a binding partner for the Src family of tyrosine kinases [3]. Following serum or platelet-derived growth factor (PDGF) stimulation or PKC activation, we observed that RACK1 co-localizes with Src at the plasma membrane and functions as a substrate and inhibitor of Src [3–6].
Peptide modulators of selective protein-protein interactions have proven invaluable in defining the function of individual components of signaling cascades, without relying on either the over- or underexpression of proteins [reviewed in 7]. For example, peptides that selectively activate or inhibit specific PKC isozymes have been used to identify the opposing roles of δ and εPKC in cardiac ischemia and reperfusion [reviewed in 2] and the δPKC inhibitor is now used in human clinical trials, in patients with acute myocardial infarction. Moreover, a selective activator peptide of εPKC confers cardioprotection in vivo, in part by reducing the incidence of lethal arrhythmias during ischemic-reperfusion injury [8]. Thus, peptides that activate εPKC may be useful treatment for patients with ischemic heart disease.
Here we used peptides that selectively disrupt or enhance the interaction of RACK1 with Src, via a βIIPKC-dependent mechanism, to define the role of RACK1 in regulating growth of cells. Our results provide direct physiologic evidence that RACK1 regulates growth of NIH 3T3 cells by suppressing the activity of Src and Src-mediated cell cycle regulators in G1 and delaying entry of cells into S phase. Our results also demonstrate the potential for using peptide modulators of Src activity as a tool for dissecting the function of Src in cells, regulating cell growth and designing new strategies for cancer therapy that target specific protein-protein interactions.
Materials and methods
Cell Culture and Cell Proliferation Assays
NIH3T3 cells were maintained in Dulbecco’s modified Eagle medium (Mediatech, Herndon, VA). Cells were supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), unless otherwise stated. Methods for peptide incubations were previously described [9]. For cell proliferation assays, cells were seeded in 96-well plates (5 × 103 cells/well). Peptides and 10% Alamar Blue were added and absorbance was measured hourly for 8 h at a wavelength of 562 and 595 nm using a Vmax kinetic microplate reader (Molecular Devices, Palo Alto, CA). Fresh peptide was added every 2 h. Reduction of alamarBlue was calculated from absorbance, according to the manufacturer’s protocol (Biosource, Camarillo, CA).
Peptides
Peptides βC2-4 (SLNPEWNET, corresponding to amino acids 218–226 in βPKC; 10), βIIV5-3 (QEVIRN, corresponding to amino acids 645–650 in βIIPKC; 11) and β agonist (pseudo-βRACK1 or ψβRACK1; SVEIWD, derived from the C2 regions of βPKC; 12) were conjugated to carrier peptide derived from the TAT protein (TAT47–57: YGRKKRRQRRR) for delivery into cells [13].
Immunoblot Analysis, Antibodies and In Vitro Protein-Kinase Assays
Immunoprecipitates or lysates of total cellular proteins were analyzed by protein immunoblotting (after SDS–PAGE) with the specified mouse monoclonal or rabbit polyclonal antibody, as described [3–6]. Primary antibodies included mouse monoclonal antibodies to Src (MAb 327; 14), RACK1, Stat3, phosphotyrosine (Py20), Kip1/p27 (Transduction Laboratories, Lexington, KY), c-Myc (9E10), and CDK2 (D-12), (Santa Cruz Biotechnology, Santa Cruz, CA). Other primary antibodies included rabbit polyclonal antisera to CDK4 (C-22), cyclin D1 (M-20), cyclin E (M-20), cyclin A (C-19), p16 (M-156), E2F1 (KH95) (Santa Cruz Biotechnology), phospho-Rb (Ser795 or Ser807/811) and p15 INK4B (Cell signaling, Beverley, MA). Methods to assess the phosphorylating activity of Src, CDK4 and CDK2 by kinase assays in vitro have been described [3–6].
Flow Cytometric Analysis for DNA Content and Ki-67 Expression
For analysis of G1/S progression, cells were synchronized in G0, released into G1 and analyzed for cellular DNA content as described [6]. For analysis of Ki-67 expression [15], 106 asynchronous cells were fixed in 70% ethanol, collected by low speed centrifugation, resuspended in PBS containing fluorescein isothiocyanate (FITC)-conjugated anti-Ki67 or mouse IgG1, and incubated for 30 min at room temperature in the dark according to the manufacturer’s protocol (BD Pharmingen) before staining with propidium iodide (PI; Sigma). Cells (104) were analyzed by FACScan cell sorting (Becton Dickinson, San Jose, CA). Dot plots were prepared using FlowJo software (Tree Star, San Carlos, CA).
Results
RACK1 regulates Src-mediated signaling pathways that culminate in induction of Myc
Previously, we suggested that RACK1 regulates NIH 3T3 cell growth by modulating Src activity at the G1 checkpoint [6]. This conclusion was based largely on indirect nonphysiologic evidence, namely, by enforced overexpression or depletion of RACK1 in cells. With the development of cell-permeable peptides that selectively disrupt or enhance the interaction of Src and RACK1 in a βIIPKC-dependent manner [9], we had the opportunity to test this hypothesis directly by using an approach that does not rely on either over- or underexpression of proteins. We first confirmed our previous finding that incubation of NIH 3T3 cells with either of two peptides that disrupt the RACK1-βIIPKC complex [antagonist βC2-4, which inhibits all the classical PKC isozymes (10) or βIIV5-3, which selectively inhibits βIIPKC and not any other member of the PKC family (11)] results in disruption of the Src-RACK1 complex and thus increases Src in vitro kinase activity (9, and Fig. 1, top two panels, compare lanes 2 and 3 with 1). In contrast, incubation of cells with a peptide that increases RACK1-βIIPKC binding (agonist βAg; 12) increases RACK1-Src binding and decreases Src in vitro kinase activity (compare lane 4 with 1).
Fig. 1.
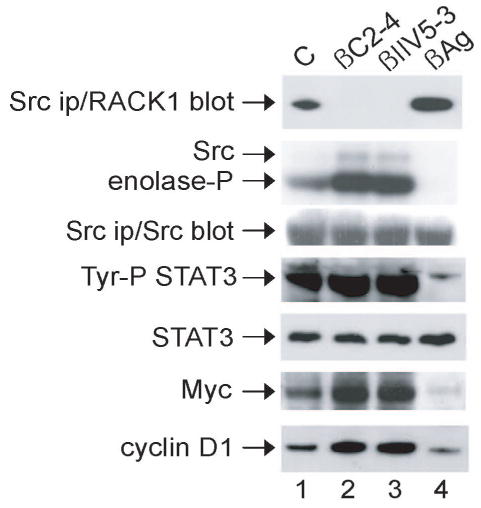
Effect of peptides on Src kinase activity and Src-mediated signaling in NIH 3T3 cells. Cells were incubated with peptide (1 μM) as described in the Methods. Proteins were immunoprecipitated with anti-Src (MAb 327) (panels 1–3) or phosphotyrosine (Py20) (panel 4) from lysate containing 500 μg of cellular protein and subjected to immunoblot analysis with anti-RACK1 (panel 1), Src (panel 3) or Stat3 (panel 4), or were assayed for in vitro protein-kinase activity by incubating with [γ-32P]ATP together with MnCl2 and enolase for 10 min at 30°C (panel 2). Otherwise, lysates containing 20 μg of total cellular protein were subjected to immunoblot analysis with anti-Stat3, Myc, or cyclin D1, as indicated (lower panels). C, Tat carrier peptide (control); βC2-4, βPKC antagonist peptide conjugated to Tat (an inhibitor of all the classical PKC isozymes); βIIV5-3, βIIPKC antagonist-selective peptide conjugated to Tat; βAg, pseudo-RACK1 agonist peptide conjugated to Tat (ψβRACK1). Data are representative of 2–3 independent experiments.
Myc and cyclin D1 are downstream effectors of Src and positive regulators of G1/S progression. Stat3 is a Src substrate and a cytoplasmic transcription factor that is required for Rac1 induction of Myc [6, 16]. Following activation by Src phosphorylation, Stat3 dimerizes, translocates to the nucleus and binds promoter elements of responsive genes involved in cell proliferation including myc and cyclin D1 [17, 18]. Thus, we next examined the influence of the peptides on Src-mediated signaling that culminates in induction of Myc and cyclin D1 (Fig. 1, lower panels). We found that incubation of cells with either antagonist peptide resulted in increased tyrosine phosphorylation of Stat3 and induction of Myc and cyclin D1 (compare lanes 2 and 3 with 1). The agonist peptide had the opposite effect (compare lane 4 with 1). These results provide direct evidence that RACK1 regulates Src-mediated signaling pathways that culminate in induction of Myc.
RACK1 Regulates Proliferation of NIH 3T3 Cells by Suppressing Src Activity
To assess the influence of the peptides on proliferation of NIH 3T3 cells, we first measured the ability of metabolically-active cells to reduce alamarblue (Fig. 2A). We observed that cells incubated with either antagonist peptide, which activate Src by disrupting its interaction with RACK1 (hence Src-activating peptides), were more metabolically active than cells incubated with carrier peptide or no peptide. In contrast, cells incubated with agonist peptide, which inhibits Src by enhancing its interaction with RACK1 (hence Src-inhibiting peptide), were less metabolically-active than control cells.
Fig. 2.
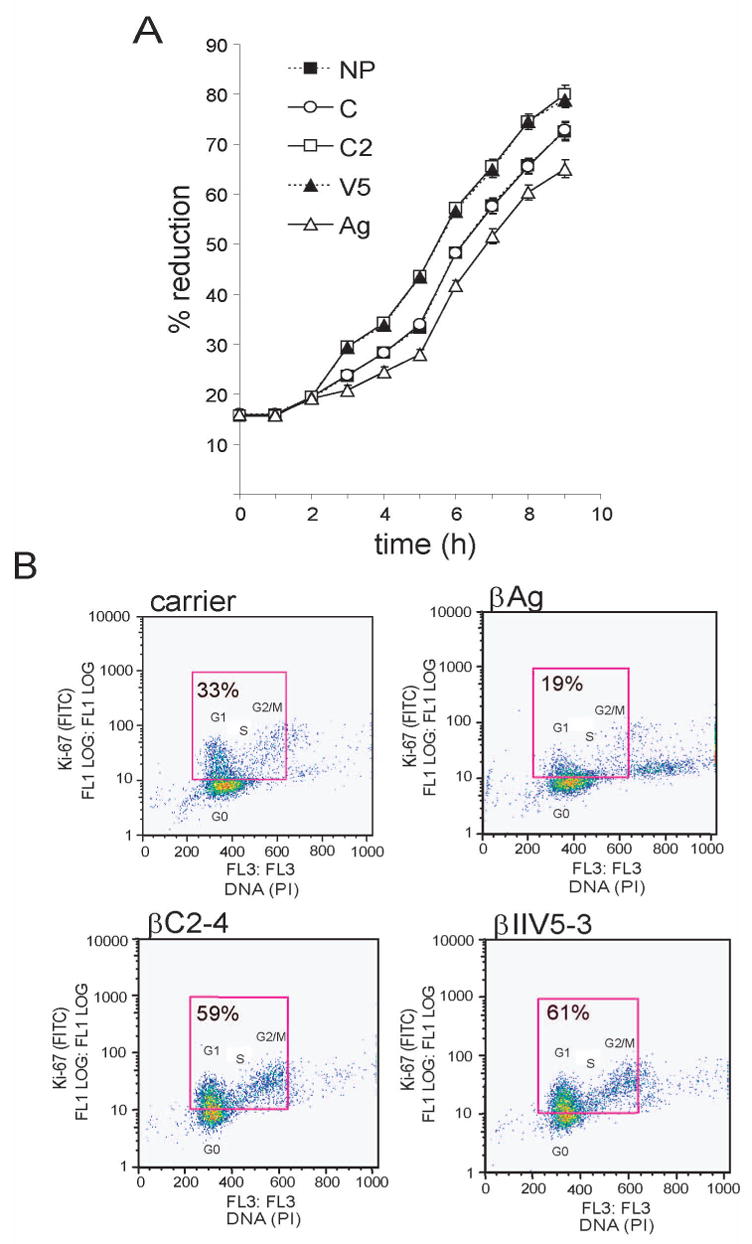
Effect of peptides on proliferation of NIH 3T3 cells. (A) Effect of peptides on the metabolic activity of cells. Cells (5 × 103) were incubated with peptide (1 μM) and alamarBlue (10%), and absorbance was measured hourly at a wavelength of 562 and 595 nm. Fresh peptide was added every 2h for 8 h. Reduction of alamarBlue was calculated from absorbance, according to the manufacturer’s protocol. x-axis, hours incubated; y-axis, relative reduction of alamarBlue. --■--, no peptide (NP); -○-, carrier peptide (C); -□-, βC2-4 peptide (C2); --▲--, βIIV5-3 peptide (V5); △, β agonist peptide (Ag). Data represent mean values +/− standard errors from 5 wells and are representative of 2 independent experiments. (B) Effect of peptides on Ki-67 expression and cell cycle distribution. Cells (2 × 106) were seeded onto 10-cm plates. 24 h later, cells were incubated with peptide for 30 min prior to fixation, stained with FITC-conjugated Ki-67 mAb and propidium iodide (PI), and analyzed by bivariate flow cytometry. x-axis, DNA content; y-axis, Ki-67-positive cell number. Ki-67-positive cells (in the G1, S or G2/M phase of the cycle) are located within the box. Ki-67-negative cells (in G0) are located below the box. %, percentage of cells that are Ki-67-positive. Data are representative of 2 independent experiments.
In a complementary proliferation assay, we used bivariate-flow-cytometric analysis to assess cells for expression of the nuclear proliferation antigen Ki-67 (which is expressed during G1, S, G2 and M, but not G0), and for phase of the cell cycle (Fig. 2B). Ki-67 expression was nearly twice as high in cells incubated with either of the Src-activating peptides (lower panels) than in cells incubated with carrier peptide (upper left panel). In contrast, Ki-67 expression was considerably lower in cells incubated with the Src-inhibiting peptide (upper right panel) than in control cells. Moreover, most of the Src-inhibited cells were in the G0/G1 phase of the cycle, whereas many more of the Src-activated cells were in S and G2/M. These results provide direct evidence that RACK1, in part via its interaction with Src, regulates growth of NIH 3T3 cells.
RACK1 Works Through Src to Regulate G1/S Progression
Results from the flow cytometry analyses indicated that enhanced binding of RACK1 to Src prolongs the G0/G1 phase of the cell cycle (Fig. 2B). This suggested that RACK1, via its interaction with Src, might regulate G1/S progression. To test this directly, we synchronized NIH 3T3 cells in G0 with serum withdrawal for 48 h, released them into G1 with the addition of serum, added peptide and analyzed cellular DNA content by flow cytometry (Fig. 3). After treatment of serum-starved cells for 6 h with 10% serum, approximately three quarter of cells incubated with carrier peptide or no peptide were in G1 (77 and 75%, respectively) and few had entered S phase (13 or 10%, respectively). In contrast, only half of the cells incubated with either of the Src-activating peptides, remained in G1 and a third had moved into S phase. Cells incubated with the Src-inhibiting peptide were arrested in G1 (90%). These results provide direct evidence that RACK1 works through Src to regulate G1/S progression.
Fig. 3.
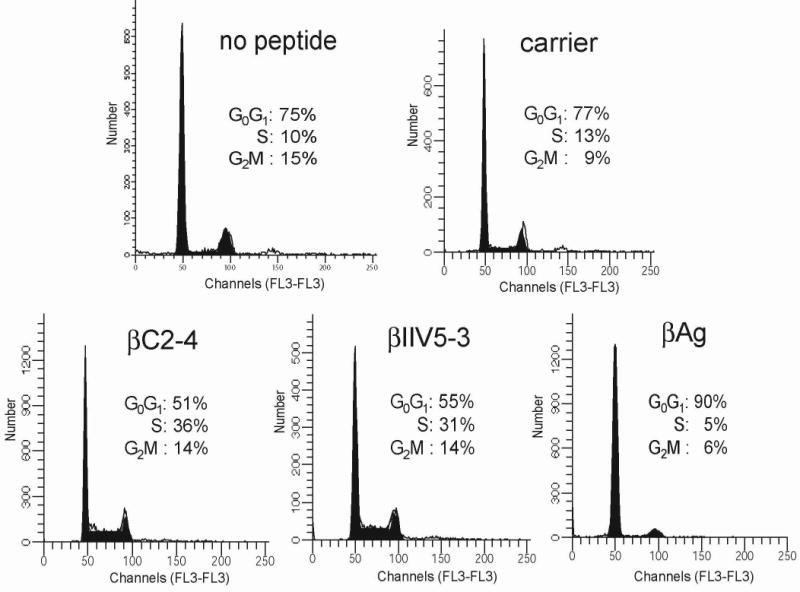
Effect of peptides on G1/S progression in NIH 3T3 cells. Cells were synchronized in G0 by serum starvation for 48 h and released into G1 with addition of 10% FBS for 6 h. Cells were incubated with peptide and analyzed for cellular DNA content by flow cytometry. Data are representative of 3 independent experiments.
By Suppressing Src Activity in G1, RACK1 Regulates Key Cell Cycle Proteins that Control G1/S Transition
To determine the mechanism whereby RACK1 regulates G1/S transition, we examined the influence of the peptides on the activities of Src and Src-mediated regulators of cell cycle progression at the G1/S boundary. G1/S transition is controlled by two key families of proteins: cyclin-dependent kinases (CDKs) and cyclins. Cyclins bind to, and activate CDKs [19, 20]. Another critical event in cell cycle progression at G1/S is hyperphosphorylation and inactivation of the retinoblastoma protein (pRb), which releases regulatory proteins such as E2F that stimulate transcription of target genes that are required for S phase entry and DNA replication. v-Src, the transforming homolog of c-Src, directly influences cell [21, 22, reviewed in 23]. For example, in cycle proteins that regulate G1/S transition fibroblasts, v-Src induces rapid transit through the G1 checkpoint and entry into S phase by simultaneously suppressing the CDK inhibitor p27 and inducing cyclins D1, E and A.
Thus, we examined the influence of the peptides on these Src-mediated regulators of G1/S progression (Fig. 4). Cells were synchronized in G0 by serum withdrawal for 48 h and released into G1 by the addition of serum for 6 h. Incubation of G1 cells with peptides that disrupt the Src-RACK1 complex resulted in increased in vitro kinase activities of Src, CDK4 and CDK2, induction of cyclins D1, E and A, and enhanced binding of CDKs to their activating cyclin partners (CDK4 to cyclin D1, and CDK2 to cyclin E and A) (Fig. 4A and 4B, compare lanes 2 and 3 with 1). Incubation of G1 cells with the peptide that enhances binding of RACK1 to Src had the opposite effect: inhibiting the kinase activities of Src, CDK4 and CDK2, the expression of cyclins and the binding of CDKs to their cyclin partners (compare lane 4 with 1).
Fig. 4.
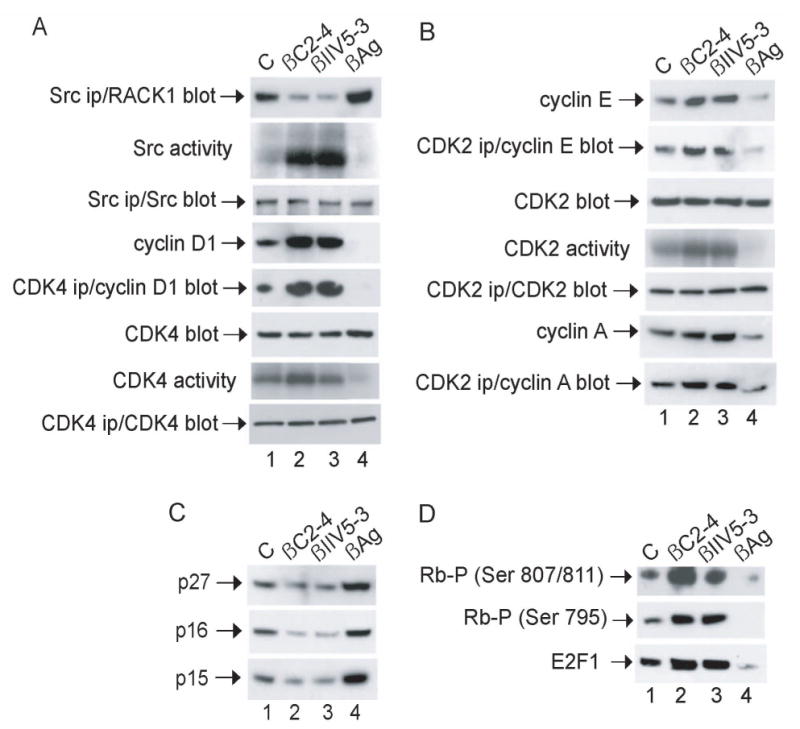
Effect of peptides on key regulators of G1/S transition in NIH 3T3 cells. Cells were synchronized in G0, released into G1 and incubated with peptide as described in the legend to Fig 3. (A and B) Proteins were precipitated with anti-Src, CDK4 or CDK2 from lysates containing 500 μg of cellular protein and analyzed for in vitro protein-kinase activity by incubating with [γ-32P]ATP together with MnCl2 and enolase (Src ips), MgCl2 and Rb peptide (CDK4 ips) or MgCl2 and histone H1 (CDK2 ips) for 10 min at 30°C, or subjected to immunoblot analysis with anti-Src, CDK4 or CDK2, respectively. Otherwise, proteins from lysates containing 20 μg of cellular protein were subjected to immunoblot analysis with anti-CDK4, CDK2 or cyclin D1, E or A, as indicated. (C and D) Proteins from lysates containing 20 μg of cellular protein were subjected to immunoblot analysis with anti-p16, p15, p27, phospho-Rb (Ser 795 or Ser 807/811) or E2F1, as indicated. Data are representative of 2–3 independent experiments.
We next assessed the influence of the peptides on CDK inhibitors (CDKIs), which play a key role in the function of the G1/S checkpoint by inhibiting G1-phase cyclin-CDK complexes [reviewed in 24]. There are two families of CDKIs, the INK4 inhibitors (p16, p15, p18 and p19) and the Cip/Kip inhibitors (p27, p21 and p57). The INK4 family inhibits CDK4 and 6 activity during G1 phase specifically, whereas the Cip/Kip family can inhibit CDK activity during all phases of the cell cycle. Both families of CDKIs have important roles in the G1/S checkpoint. For example, p16 mediates the p53-independent G1 arrest in response to DNA damage through abrogation of cyclin-D-CDK4 and CDK6-mediated phosphorylation of pRb [25]. We found that the Src-activating peptides inhibited expression of CDKIs p16, p15 and p27 in G1 (Fig. 4C, compare lanes 2 and 3 with 1). In contrast, the Src-inhibiting peptide induced expression of the CDKIs in G1 (compare lane 4 and 1).
Finally, we examined the influence of the peptides on phosphorylation of pRb and release of E2F1 (Fig. 4D). We observed that Src-activating peptides induced the hyperphosphorylation of pRb at multiple serine residues (795, 807 and 811), and induced the release of E2F1 (lanes 2 and 3). In contrast, the Src-inhibiting peptide induced the hypophosphorylation of pRb and the sequestration of E2F1 (lane 4). Together, these results provide direct evidence that RACK1, by suppressing Src activity in G1, regulates key cell cycle proteins that control G1/S transition.
Discussion
As part of our ongoing investigation of the function of RACK1 in cells, we sought a means of modulating RACK1’s influence on Src that did not rely on the non-physiologic approach of either over- or underexpressing proteins. Cell-permeable peptides that modulate specific protein-protein interactions in cells provide such a means. Previously, we used peptide modulators of RACK1’s interaction with βIIPKC and thus Src to define RACK1’s influence on Src-mediated Sam68 and p190RhoGAP signaling, Rho activity and actin cytoskeleton rearrangement [9]. Here, we used the peptides to define RACK1’s influence on cell growth. Our results provide direct evidence that RACK1 regulates growth of NIH 3T3 cells (Fig 2), at least in part, by suppressing the activity of Src and Src-mediated cell cycle regulators in G1, (Figs 1 and 4) and delaying entry of cells into S phase (Fig 3). These results confirm our previous indirect evidence obtained by over- or underexpressing RACK1 [6].
Peptide modulators of protein-protein interaction show great promise in treating patients with ischemic heart disease [8, 26, reviewed in 27]. We and others showed that the specific activity of Src is strikingly elevated in some human cancers [reviewed in 28]. Thus, with a long-term goal of exploring potential new strategies for treating cancer, we started by assessing the influence of the peptide modulators of Src activity on the growth of normal cells. We found that a peptide that enhances βIIPKC’s and thereby Src’s interaction with RACK1 [9], suppresses Src activity at the G1 checkpoint (Fig 4A) and slows the growth of NIH 3T3 cells (Figs 2 and 3). Peptides that disrupt βIIPKC-RACK1-Src complex [9] have the opposite effect. These results reveal a novel mechanism of cell cycle control in G1 of cells that works via an endogenous inhibitor of Src kinases. They also demonstrate the potential for using peptide modulators of βIIPKC and thereby Src activity for designing new strategies of cancer therapy that target specific protein-protein interactions. Although a peptide with intracellular activity has not yet been approved for use in clinical medicine, if its effect is validated in animal studies, it could justify the investment in developing a drug that affects the same protein-protein interaction. Moreover, if the current clinical trials with another PKC peptide regulator are positive, stabilizing the peptides and devising means for sustained delivery may remove at least some of the barriers to the use of the peptides themselves as a drug, because they appear to be very safe and selective even when delivered chronically at high dose via an Alzet pump [29].
Previously, we demonstrated that RACK1, βIIPKC and Src function in a trimolecular complex where βIIPKC’s association with RACK1 is required for Src phosphorylation of, and binding to RACK1, and for RACK1 inhibition of Src [9]. Thus, specificity of peptide effect on biological activities is determined by the selectivity of the peptides for the βIIPKC-RACK1 interaction. We have carried out numerous studies (in cell-based assays and in vivo) that support the selectivity of the peptides for the isozyme they were designed for: activation of only the right isozyme (e.g. βIIPKC) is affected; other isozymes present in the cells and affected by the same stimulus (e.g. βIPKC), are unaffected by the peptide modulators [reviewed in 1]. Furthermore, the peptide regulators for one isozyme have biological activities that are different and sometimes even opposing to peptide regulators of another isozyme [reviewed in 2]. Together, these results provide strong evidence that the peptides selectively and specifically affect biological activities involving the βIIPKC-RACK1 interaction.
Because the peptides modulate RACK1’s interaction with βIIPKC, and only indirectly the interaction of RACK1 with Src, their striking effects on G1/S progression (Fig. 3) and cell growth (Fig 2) could be due to their effect on the RACK1-βIIPKC interaction. However, ours results clearly show that the peptides modulate biological activities that are known to be Src-mediated. For example, the inhibitory effect of the peptide that enhances the βIIPKC-RACK1-Src interaction on: a) tyrosine phosphorylation of the Src-specific substrates Sam68 and Stat3 (Fig 1), b) induction of the Src effectors Myc and cyclin D1 (Fig 1), and c) on Src-mediated p190RhoGAP activity and actin-cytoskeleton rearrangement [9], is strikingly similar to the effect observed with: a) the Src-specific tyrosine kinase inhibitors SU6656, PP2 or PP1 [reviewed in 23, 30], b) dominant-negative mutants of Src [23, 30], or c) overexpression of the endogenous Src inhibitor RACK1 [6]. Conversely, the activating effects of the peptides that disrupt the βIIPKC-RACK1-Src interaction are similar to those observed with v-Src [23, 30, 31], constitutively-active Y527F c-Src [23, 30] or with depletion of RACK1 [6]. Thus, perturbing RACK1’s interaction with Src is a major mechanism by which the peptides function to regulate cell growth.
Recently, caveolae were shown to be important for Src function downstream of the PDGF receptor [32]. Moreover, Src activity appears to be regulated in caveolae [33], perhaps by RACK1 [34]. Thus, caveolae may serve an important function by recruiting signaling kinases and their regulators and substrates to growth factor receptor complexes.
Acknowledgments
We thank Jenny Cheng for assistance with some experiments and for critical review of the manuscript. This work was supported by a grant from the National Institutes of Health to C. A. C. (DK43743). L. D. M. was supported, in part, by NIH Digestive Disease Center grant DK65339.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Vidya Mamidipudi, Department of Medicine, Stanford University School of Medicine, Stanford, California 94305-5187.
Laura D. Miller, Department of Medicine, Stanford University School of Medicine, Stanford, California 94305-5187
Daria Mochly-Rosen, Department of Molecular Pharmacology, Stanford University School of Medicine, Stanford, California 94305-5187.
Christine A. Cartwright, Department of Medicine, Stanford University School of Medicine, Stanford, California 94305-5187
References
- 1.Dorn GW, Mochly-Rosen D. Intracellular transport mechanisms of signal transducers. Annu Rev Physiol. 2002;64:407–429. doi: 10.1146/annurev.physiol.64.081501.155903. [DOI] [PubMed] [Google Scholar]
- 2.Murriel CL, Mochly-Rosen D. Opposing roles of delta and epsilon PKC in cardiac ischemia and reperfusion: targeting the apoptotic machinery. Arch Biochem Biophys. 2003;420:246–254. doi: 10.1016/j.abb.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 3.Chang BY, Conroy KB, Machleder EM, Cartwright CA. RACK1, a receptor for activated C kinase and a homolog of the β subunit of G proteins, inhibits activity of Src tyrosine kinases and growth of NIH 3T3 cells. Mol Cell Biol. 1998;18:3245–3256. doi: 10.1128/mcb.18.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang BY, Chiang M, Cartwright CA. The interaction of Src and RACK1 is enhanced by activation of protein kinase C and tyrosine phosphorylation of RACK1. J Biol Chem. 2001;276:20346–20356. doi: 10.1074/jbc.M101375200. [DOI] [PubMed] [Google Scholar]
- 5.Chang BY, Harte RA, Cartwright CA. RACK1: a novel substrate for the Src protein-tyrosine kinase. Oncogene. 2002;21:7619–7629. doi: 10.1038/sj.onc.1206002. [DOI] [PubMed] [Google Scholar]
- 6.Mamidipudi V, Zhang J, Lee KC, Cartwright CA. RACK1 regulates G1/S progression by suppressing Src kinase activity. Mol Cell Biol. 2004;24:6788–6798. doi: 10.1128/MCB.24.15.6788-6798.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Souroujon MC, Mochly-Rosen D. Peptide modulators of protein-protein interactions in intracellular signaling. Nature Biotechnology. 1998;16:919–924. doi: 10.1038/nbt1098-919. [DOI] [PubMed] [Google Scholar]
- 8.Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation. 2005;111:44–50. doi: 10.1161/01.CIR.0000151614.22282.F1. [DOI] [PubMed] [Google Scholar]
- 9.Miller LD, Lee KC, Mochly-Rosen D, Cartwright CA. RACK1 regulates Src-mediated Sam68 and p190RhoGAP signaling. Oncogene. 2004;23:5682–5686. doi: 10.1038/sj.onc.1207735. [DOI] [PubMed] [Google Scholar]
- 10.Ron D, Luo J, Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of beta protein kinase C in vivo. J Biol Chem. 1995;270:24180–24187. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- 11.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. J Biol Chem. 2001;276:29644–29650. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 12.Ron D, Mochly-Rosen D. An autoregulatory region in protein kinase C: the pseudoanchoring site. Proc Natl Acad Sci USA. 1995;92:492–496. doi: 10.1073/pnas.92.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 14.Lipsich LA, Lewis AJ, Brugge JS. Isolation of monoclonal antibodies that recognize the transforming proteins of avian sarcoma viruses. J Virol. 1983;48:352–360. doi: 10.1128/jvi.48.2.352-360.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleury S, Rizzardi GP, Chapuis A, Tambussi G, Knabenhans C, Simeoni E, Meuwly JY, Corpataux JM, Lazzarin A, Miedema F, Pantaleo G. Long-term kinetics of T cell production in HIV-infected subjects treated with highly active antiretroviral therapy. Proc Natl Acad Sci USA. 2000;97:5393–5398. doi: 10.1073/pnas.97.10.5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000;290:144–147. doi: 10.1126/science.290.5489.144. [DOI] [PubMed] [Google Scholar]
- 17.Sinibaldi D, Wharton W, Turkson J, Bowman T, Pledger WJ, Jove R. Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene. 2000;19:5419–5427. doi: 10.1038/sj.onc.1203947. [DOI] [PubMed] [Google Scholar]
- 18.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA, Jove R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci USA. 2001;13:7319–7324. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donjerkovic D, Scott DW. Activation-induced cell death in B lymphocytes. Cell Res. 2000;1:1–16. doi: 10.1038/sj.cr.7290047. [DOI] [PubMed] [Google Scholar]
- 20.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 21.Riley D, Carragher NO, Frame MC, Wyke JA. The mechanism of cell cycle regulation by v-Src. Oncogene. 2001;20:5941–50. doi: 10.1038/sj.onc.1204826. [DOI] [PubMed] [Google Scholar]
- 22.Furstoss O, Manes G, Roche S. Cyclin E and cyclin A are likely targets of Src for PDGF-induced DNA synthesis in fibroblasts. FEBS Lett. 2002;526:82–86. doi: 10.1016/s0014-5793(02)03120-4. [DOI] [PubMed] [Google Scholar]
- 23.Thomas SM, Brugge JA. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 24.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 25.Shapiro GI, Edwards CD, Rollins BJ. The physiology of p16(INK4A)-mediated G1 proliferative arrest. Cell Biochem Biophys. 2000;33:189–197. doi: 10.1385/CBB:33:2:189. [DOI] [PubMed] [Google Scholar]
- 26.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–2307. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 27.Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. 2006;70:222–230. doi: 10.1016/j.cardiores.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Cartwright C. Intestinal cell growth control: role of Src tyrosine kinases. Gastroenterology. 1998;114:1335–1338. doi: 10.1016/s0016-5085(98)70443-3. [DOI] [PubMed] [Google Scholar]
- 29.Begley R, Liron T, Baryza J, Mochly-Rosen D. Biodistribution of intracellularly acting peptides conjugated reversibly to Tat. Biochem Biophys Res Commun. 2004;318:949–954. doi: 10.1016/j.bbrc.2004.04.121. [DOI] [PubMed] [Google Scholar]
- 30.Bromann PA, Korkaya J, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 31.Mamidipudi V, Chang BY, Harte RA, Lee KC, Cartwright CA. RACK1 inhibits the serum- and anchorage-independent growth of v-Src transformed cells. FEBS Lett. 2004;567:321–6. doi: 10.1016/j.febslet.2004.03.125. [DOI] [PubMed] [Google Scholar]
- 32.Veracini L, Franco M, Boureux A, Simon V, Roche S, Benistant C. Two distinct pools of Src family tyrosine kinases regulate PDGF-induced DNA synthesis and actin dorsal ruffles. J Cell Sci. 2006;119:2921–34. doi: 10.1242/jcs.03015. [DOI] [PubMed] [Google Scholar]
- 33.Li S, Couet J, Lisanti MP. Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J Biol Chem. 1996;271:29182–90. doi: 10.1074/jbc.271.46.29182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang JW, Chen CL, Chuang NN. Shift syndecan-2 from RACK1 to caveolin-2 upon transformation with oncogenic ras. Biochem Biophys Res Commun. 2005;329:855–62. doi: 10.1016/j.bbrc.2006.09.035. [DOI] [PubMed] [Google Scholar]