

Abstract
The ubiquity of mobile elements in mammalian genomes poses considerable challenges for the maintenance of genome integrity. The predisposition of mobile elements towards participation in genomic rearrangements is largely a consequence of their interspersed homologous nature. As tracts of nonallelic sequence homology, they have the potential to interact in a disruptive manner during both meiotic recombination and DNA repair processes, resulting in genomic alterations ranging from deletions and duplications to large-scale chromosomal rearrangements. Although the deleterious effects of transposable element (TE) insertion events have been extensively documented, it is arguably through post-insertion genomic instability that they pose the greatest hazard to their host genomes. Despite the periodic generation of important evolutionary innovations, genomic alterations involving TE sequences are far more frequently neutral or deleterious in nature. The potentially negative consequences of this instability are perhaps best illustrated by the 25 human genetic diseases that are attributable to TE-mediated rearrangements. Some of these rearrangements, such as those involving the MLL locus in leukemia and the LDL receptor in familial hypercholesterolemia, represent recurrent mutations that have independently arisen multiple times in human populations. While TE-instability has been a potent force in shaping eukaryotic genomes and a significant source of genetic disease, much concerning the mechanisms governing the frequency and variety of these events remains to be clarified. Here we survey the current state of knowledge regarding the mechanisms underlying mobile element-based genetic instability in mammals. Compared to simpler eukaryotic systems, mammalian cells appear to have several modifications to their DNA-repair ensemble that allow them to better cope with the large amount of interspersed homology that has been generated by TEs. In addition to the disruptive potential of nonallelic sequence homology, we also consider recent evidence suggesting that the endonuclease products of TEs may also play a key role in instigating mammalian genomic instability.
Introduction
Because of the high density of repetitive elements in mammalian genomes, it is not surprising that we should find them frequently serving as substrates for genomic rearrangements. With nearly 50% of the human genome occupied by recognizable repetitive sequences, chance alone would place transposable elements (TEs) in the vicinity of a great number of genetic alterations. Yet the biological basis for involvement of repetitive sequences in genomic instability extends beyond their abundance in genomes. As tracts of nonallelic homologous sequences, they possess the potential to disrupt essential DNA repair processes, often resulting in genomic alterations. The variety of post-insertion mutations involving TE substrates ranges from genomic insertion/deletion (indel) mutations to larger-scale, intra and inter-chromosomal rearrangements. While a number of TE-related rearrangement events have resulted in key innovations during evolution (e.g.[1,2]), it is nevertheless the case that these events are more frequently neutral or deleterious in terms of organismal fitness. The negative consequences of instability are perhaps best illustrated by the 25 distinct human genetic diseases that are attributable to TE- related rearrangements [3]. Some of these rearrangements have independently arisen multiple times in the human population, suggesting the TE sequences involved are particularly disposed to instability. Well known examples of this phenomenon include Alu-mediated deletions in the LDL receptor locus, resulting in familial hypocholestrolemia, and rearrangements involving the MLL locus, a frequent factor in acute myeloid leukemias [4].
Prior to the advent of large-scale sequencing projects associated and comparative genomic studies, our ability to observe the results of instability events involving TEs was effectively limited to those events that were associated with observable disease phenotypes (Reviewed in [3,4]) or those that were induced under controlled experimental systems. The abundance of sequence data now available from several taxa, as well as the growing list of TE-related rearrangement events, make the profound impact TE expansion and post-insertion instability have had on the evolution of eukaryotic genomes increasingly evident. Nevetheless, the full extent to which ongoing negative selection is occurring against both individual and groups of TE insertions due to their potential for ectopic recombination remains to be clarified. In support of the hypothesis that a significant historical selection pressure has existed against TE copy accumulation based on instability, several studies have demonstrated a tendency for TEs to accumulate in low recombining areas of the genome (discussed in [5]). Although these observations are also consistent with several alternative hypotheses (e.g. accumulation of deleterious insertions due to decreased selection efficiency within these regions) that do not necessitate ectopic recombination, there is increasing support from the drosophila model for significant selection against TE loci due to their potential for instability [5] Recent data from humans, showing stronger selection against full-length L1 elements than their truncated counterparts, is also highly suggestive of selection against ectopic recombination instability in our own lineage [6], particularly when taken together with an earlier genomic analysis of TE accumulation on human sex chromosomes which indicated that negative selection on L1s was related to L1 insertion size [7] as opposed to the full length vs. non-full length status of insertions. In the latter work, a statistically significant accumulation of >500bp but less-than-full-length L1 inserts on the human sex chromosomes vs. the autosomes [7].
Although modern sequencing efforts and advances in experimental techniques have considerably improved our understanding of the range and impact of TE instability, significant gaps in our knowledge remain due to our inability to observe those naturally occurring mutational events that are likely to be rapidly removed from the population via natural selection. In addition, another group of mutation events remain largely inaccessible on account of their occurring in somatic cell lineages or subcomponents of tumors that can not readily be assayed. It is therefore important for researchers to remain cognizant of how current observational and experimental constraints impact our overall picture of both the variety and frequency of TE mutations in organisms.
As a group, transposable elements can contribute to the disruption of genetic information in a variety of ways, many of which do not involve physical rearrangements of the genome. Such events include, but are not limited to, direct disruption of genetic structures through insertion [4], generation of aberrant mRNA splices [8–10], introduction of premature polyadenylation signals [11] and/or transcription disruption [12], and as disruptive epigenetic signals which can spread to nearby genes. This larger class of genetic perturbations, along with their evolutionary implications, has been reviewed elsewhere [8,9,13–16]. In this review, we focus on the molecular biology underlying that subset of mutagenic events in which TEs serve as substrates for physical genomic rearrangements.
1 Mechanisms of Post-Insertional Transposable Element Instability
1.1 Interspersed Repeats and DNA Repair Systems
The majority of instability events involving TEs are thought to be the byproduct of errant double-strand break (DSB)-repair processes. Several major DSB-repair mechanisms rely on sequence homology to conserve sequence information while repairing damaged chromosomes. DSBs are a particularly disruptive form of DNA damage that typically must be corrected by the cell prior to its proceeding through the cell cycle [17]. DSBs are known to instigate a variety of genetic alterations, many of which directly increase the risk of tumorigenesis and/or metastasis [18,19]. The sources of these breaks are multi-fold; they can arise from free-radical byproducts of cellular reactions, topoisomerases, ionizing radiation, as well as during the process of meiosis (reviewed in [19,20]). DSBs can also be generated from single-stranded nicks in DNA; this most frequently occurs when replication forks traverse the broken strand, leading to a collapse in the fork which creates a DSB [21]. As a consequence, the S-phase of the cell cycle appears to endure the bulk of DSBs. In addition to these canonical sources of DNA breaks, our laboratory has recently reported that the endonuclease produced by L1, an autonomous retrotransposon that is widespread throughout the mammalian lineage [22–24], also has the capability of generating DSBs [25]. As we discuss below, the generation of DSBs by L1 and related TE endonucleases may play a significant role in some genetic instability events.
Current estimates place the number of DSBs naturally occurring during each cell cycle at ~50 [20]. This figure will no doubt vary among cell types that differ in their respective exposures to endogenous free radicals and environmental stresses as well as the genomic replication demands placed upon them. Eukaryotic cells possess a robust set of mechanisms for the detection and repair of DSBs [26,27]. In the most extreme cases, the detection of rampant DSBs throughout the genome results in signal cascades leading to apoptosis [28]. More typically, however, DNA-repair machinery is recruited to the site of the DSB, and repair processes are initiated.
Eukaryotic repair pathways can be categorized based on the extent to which they rely on tracts of sequence homology as well as the accuracy with which they repair the defect. There are those mechanisms that are dependent upon tracts of sequence homology, referred to as homologous recombination repeair (HRR), and those pathways, such as non-homologous end joining (NHEJ), which require little or no sequence homology to operate. HRR pathways can also be further subdivided into two basic types. The first type consists of the conservative HRR pathways, which are those that demonstrate a high degree of preservation of sequence information, such as classic double-strand break repair (DSBR), synthesis-dependent strand annealing (SDSA), and break-induced replication (BIR). The second type, nonconservative HRR, presently consists of a single pathway, single strand annealing (SSA). NHEJ can also be further subdivided into “accurate” and “inaccurate” pathways, depending on the particular protein pathway invoked. A complex network of cellular proteins, many of which remain to be characterized, are necessary for the DNA repair process; these proteins and their interactions are detailed in several available reviews [19,29–31]. In the sections below, we discuss current models for DNA repair along with their potential interactions with interspersed homologous sequence.
1.1.1 Homologous Recombination Repair (HRR)
It is through the interaction with the homology-based DNA DSB repair systems that TEs appear to be most disruptive to genome integrity. Yet these very same repair pathways offer the greatest potential for the cell to repair DSB breaks with no associated loss of genetic information. In most DSB repair processes involving homology, an undamaged copy of the severed chromosome is used to provide the information necessary for repair. Sister chromatids, which are available from S-phase until cell division, are the preferred template, being far more frequently used than the homologous chromosome as source templates for the repair process [32].
Numerous models currently exist for the interactions among chromosomes and templates during the homologous repair process [26]. In the classic DSB repair scenario of Szostak et al. (DSBR), which extended an earlier proposal by Resnick [33] and Resnick and Martin [34], the 3’ end of each of the exposed chromosome ends invades the undamaged chromosome at the homologous (allelic) counterpart of the damaged locus [33,35] (Figure 1). The invading strand displaces one of the template strands, and DNA synthesis is subsequently primed from the invading 3’ end. Meanwhile, the second exposed 3' end anneals to the displaced template strand, also initiating synthesis and resulting in the formation of two separate Holiday junctions which are subsequently resolved (Figure 1). The locations of the two Holiday junctions are not fixed and may migrate to expand the heteroduplex region. The process replicates the chromosomal region in the vicinity of the breakpoint. In this scenario, the repair process culminates with, or without, a crossover event, depending on how the Holiday junctions are resolved (Figure 1). Ideally, if an allelic template on the sister chromatid is used, no genetic information is lost. When the homologous chromosome is used, gene conversion can occur where hybrid strands anneal, potentially resulting in loss of heterozygosity (LOH) at the locus. Experimental evidence suggests, however, that extensive LOH is suppressed during DSB repair in mammalian cells [36].
Figure 1. Classic Double-strand Break Repair (DBSR) Model.
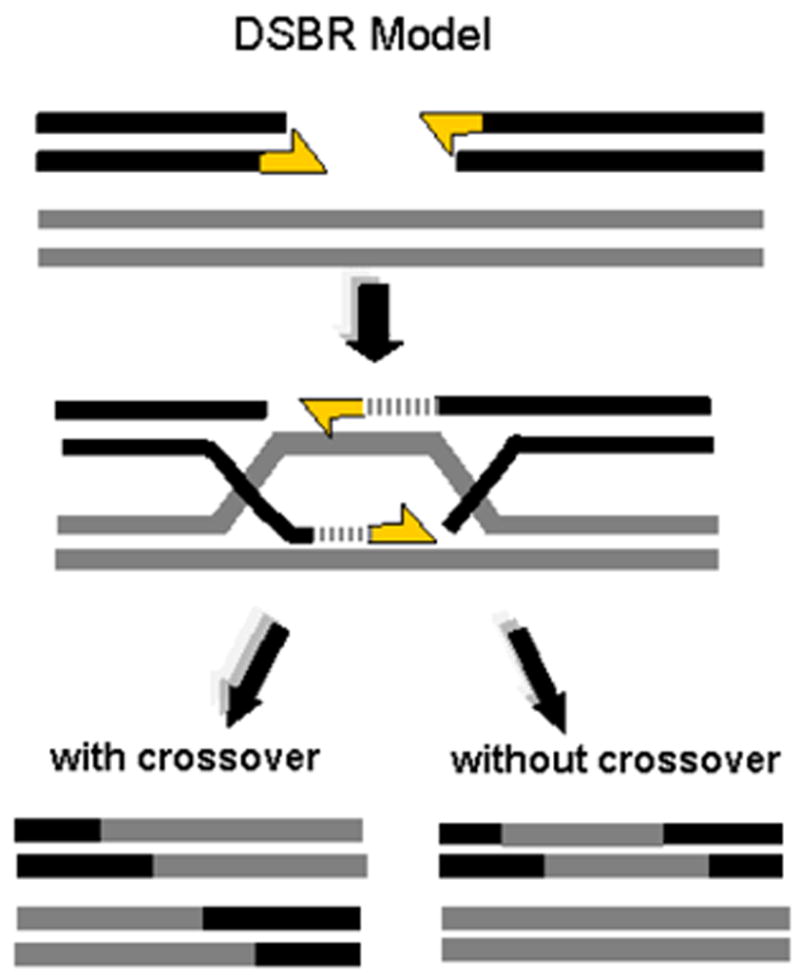
In the classic model for recombination-mediated DSB repair, invasion of the undamaged template by the exposed 3' ends of both sides of the break junction (indicated by yellow arrows), primes the synthesis of the nascent DNA used for repair and forms a pair of holiday junctions. As indicated in the lower portion of the figure, depending on how these Holiday junctions are resolved, the DBSR model predicts crossover and noncrossover (“gene conversion”) events will occur with approximately equal frequency.
Interspersed TE sequences can severely disrupt this classic homologous repair pathway by offering alternative nonallelic tracts of homology with which the invading strand can anneal (Figure 2). Several possible alleles can be generated from the resolution of these events, depending upon the relative locations of the nonallelic sequences involved. A subset of these possibilities are depicted in Figure 2. If the targeted sequence is a nonallelic homologous element located on the sister chromatid or homologous chromosome, two types of alleles are generated: one containing a deletion and one containing a duplication of the region (Figure 2). Such misrouted recombination events, collectively referred to as nonallelic homologous recombinations (NAHR), are thought to be a source of many TE-related instability events, particularly those resulting in duplications. If the targeted sequence is on the same chromosomal strand as the repair site, the intervening sequence can be inverted (Figure 2). As discussed below, most of the Alu-mediated deletion events in humans are consistent with either NAHR or SSA mechanisms [37].
Figure 2. Nonallelic Homologous Recombination Events Among TEs Generate Multiple Rearrangments.
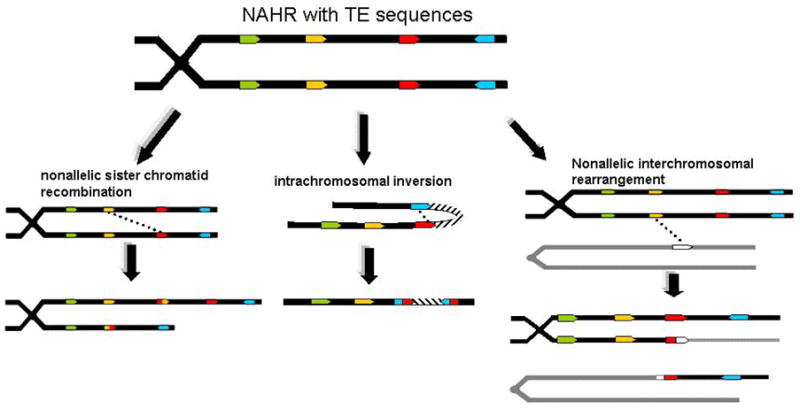
Three of several possible rearrangments resulting from nonallelic recombination are depicted. In the first instance, misallignment of sister chromatids during recombination or DNA repair yield insertion and deletion mutations. In the second instance, recombination between two homolgous TEs in direct orientation on the same physical chromsome results in an inversion of the intervening sequence. In the third image, alignment with a nonhomologous chromosome results in the translocation of chromosome arms.
NAHR involving TEs represents a viable mechanism for large-scale chromosomal translocation mutations. Yet although confirmed instances exist (e.g. [38]), there are surprisingly few authentic examples of TE-mediated translocations resulting in human diseases [39]. Under experimental conditions, it has been demonstrated that homologous Alu elements are capable of mediating such rearrangements at appreciable levels [39]. In addition, it has also been demonstrated that genomic L1 copies throughout the genome can serve as templates to repair DSBs that are experimentally induced within an L1 copy [40]. These data demonstrate that repair-related physical interactions do occur among homologous TE copies interspersed throughout the genome. While the rarity of TE-mediated translocation mutations no doubt partially stems from the lethality of these mutations relative to smaller-scale rearrangements, as we discuss further below, this observation is likely also related to a general reliance in mammals on non-crossover mechanisms of DNA repair such as SDSA and SSA.
While there is extensive support for the DSBR model described above for meiotic cells, a number of inconsistencies with experimental results were observed for mitotically dividing systems [32,37,41]. As a result, several alternative conservative HRR models have been put forward to accommodate the experimental data, including a SDSA and BIR (Reviewed in [26,41]).
1.1.2 Synthesis-Dependent Strand Annealing (SDSA)
In eukaryotic mitotic systems, gene conversion is frequently observed to occur without the associated crossover events predicted by the classic DSBR model described above [19]. In addition, the transfer of sequence information between chromosomes via gene conversion in several experimental systems appeared primarily unidirectional [26], an observation that is also inconsistent with predictions of the DSBR model. Experimental data in mammals further suggests that homologous repair using the sister chromatid without associated crossover is the predominant pathway in mammalian cells [32]. To address these and other inconsistencies with DSBR, several related models, collectively referred to as synthesis-dependent strand-annealing (SDSA), have been proposed (Reviewed in detail in [26]). The common theme of this family of models consists of the return of the newly synthesized strands to the original damaged chromosome. Three variations of SDSA are depicted in (Figures 3a–c). In the first scenario, as in the classic DSBR model described above, both free 3′ ends invade the homologous locus and initiate synthesis. Instead of forming holiday junctions, however, the nascent strands anneal with one another, resulting in no associated crossover and restriction of conversion events to the invading (non-template) strand (Figure 3a). The second scenario is similar to the first, with the exception that only one strand invades and primes synthesis; the newly synthesized sequence subsequently anneals to homologous sequence on the damaged chromosome (Figure 3b). A third scenario, which has received experimental support in mammals, occurs when newly synthesized strands are deprived of homology and NHEJ is employed to rejoin the strands to the original chromosome [42]. A variation of this process is depicted in Figure 3c, wherein invasion at a nonallelic homologous TE locus results in synthesis of nonhomologous sequence that relies on NHEJ to be rejoined to the source locus. Such hybrid HRR-NHEJ processes provide a mechanism for interspersed repetitive elements to serve as vehicles for replicating unique regions of sequence throughout the genome [42]. While the homology-associated pathways are believed to be the primary repair mechanisms involved in TE-related rearrangements, NHEJ likely plays an important role in finalizing some TE-related instability events. Additional SDSA pathways, not discussed here, have been proposed that also include the possibility of crossover events [26].
Figure 3. Three Variations of Synthesis-Dependent Strand Annealing (SDSA).

a) invasion of the undamaged template chromosome simultaneously by both 3' strands (indicated by yellow arrows) of the broken chromosome primes the synthesis of new DNA. Unlike the DBSR model, however, the newly synthesized strands anneal together and result in no crossover. b) In this model, only one strand invades the target template. Following synthesis, the new DNA strand reattaches to the originally damaged chromosome at a homologous location, resulting in a gene conversion repair without crossover. c) In this scenario, the DSB occurs adjacent to a TE insertion. The 3' end of the broken DNA strand invades the template strand at a nonallelic yet homologous TE locus. Synthesis occurs, copying both the homologous TE locus as well as the nonhomologous sequence flanking it. NHEJ is then used to reunite the newly synthesized sequence to the original damaged chromosome.
During SDSA repair, there are several avenues by which interspersed repetitive elements could serve to derail proper resolution of the DSB. Re-annealment and subsequent attachment of the newly synthesized strands with nonallelic homologous TEs (e.g. Figure 2c) could lead to a variety of rearrangements, several of which being difficult to distinguish from both DSBR and SSA pathways.
1.1.3 Single-strand Annealing (SSA)
Another HRR repair process, SSA, is fundamentally different from the above-mentioned repair methods in that it is invariably mutagenic and hence nonconservative. SSA can occur when a DSB occurs either within a pair of directly repeated sequences or somewhere between the repeating sequences. During SSA, sequences on both sides of the DSB are resected or “chewed back,” allowing homologous sequences adjacent to the exposed ends to associate and ligate [26] (Figure 4). Ideally, the homologous sequence would be located within the immediate vicinity of the break, thereby minimizing the inevitable genetic loss. Such nearby homology is typically only available for TEs, giving SSA a particularly important role in TE-related DNA repair events. Recent data from Drosophila indicate that SSA is the preferred mechanism of repair for repeat-related DSBs in that organism [43]. In addition, a large number of the observed smaller-scale deletion events in humans are consistent with the SSA pathway [4], and the use of SSA in preference to recombination based mechanisms would be consistent with the general tendency of mammalian lineages, as opposed to yeast, to suppress crossover during repair. Although the relatively short average size of Alu-Alu mediated deletions in humans (~806 bp) [37] is suggestive of an SSA model where limited resection is involved, the somewhat larger size of disease rearrangements involving Alu [3] indicates that larger events that are occurring may be rapidly purged from the genome. As a consequence, we should be cautious about extrapolating much from the characteristics of instability events that have already been heavily filtered by natural selection.
Figure 4. Single Strand Annealing (SSA).

In the SSA model, the 5' ends surrounding the breakage region are “chewed back” or resected, exposing adjacent homology. The homologous strands anneal with each other and are ligated, resulted in the deletion of intervening sequence.
While SSA also has the potential to produce larger intra and inter-chromosomal rearrangements, its overall contribution to these large-scale events is generally considered minimum, as such larger events would require either a) simultaneous DSBs at distant regions of the genome or b) that the resection (“chewing back”) process extends an exceptionally long distance. As is the case with NHEJ, SSA does not require a sister chromatid and remains available throughout the cell cycle. It remains unclear what determines when and where SSA will be invoked in preference to other available repair processes. With the availability of NHEJ—which theoretically necessitates less sequence loss on average than SSA—throughout the cell cycle, invocation of SSA would seldom seem desirable for the cell. However, experimental evidence increasingly points to a competition between multiple repair pathways whose outcome may be determined by a number of cell-state-dependent host factors [43,44].
1.1.4 Suppression of Promiscuous Recombination
Given the ubiquity of TEs in mammalian genomes, and the incredible potential for disruption which exists as a result, it may seem a wonder that the overall integrity of the genome is able to be maintained at all. As a consequence of the hazards such interspersed sequences pose, eukaryotic organisms have evolved a number of mechanisms to mitigate post-insertional TE instability. Although homologous interspersed repeats exist throughout mammalian genomes, many of these copies exhibit varying degrees of divergence from one another. Such between-copy diversity is primarily the result of the accumulation of neutral mutations in the sequences. One mechanism employed to avoid NAHR appears to be the discrimination of the level of sequence identity used as a repair template [45]. Experimental evidence from several systems suggests that interspersed repeats having greater levels of divergence are less likely to be used as templates [45,46]. This aspect of recombination surveillance is thought to be carried out, in part, by proteins involved in the mismatch repair system [47]. In yeast, a rapid drop-off in recombination frequency vs. percent divergence is observed for repetitive elements, wherein the first few mismatches have a more suppressive effect on recombination than the addition of further mismatches [48]. As a consequence of the relationship between recombination and divergence, the particular TE ecology of a host genome, including the number and age of copy numbers, will have a significant impact on the potential for ectopic instability. At the same time, the host organism's ability to suppress such rearrangements will in turn have a considerable impact on the manner in which TEs may expand—or not expand—in a given taxa.
In addition to sequence identity, the length of homology of sequences also appears to be a factor in template selection [45]. Data from multiple systems indicate that the longer the available tract of sequence homology, the more likely recombination-based instability events will occur [46,49]. Given that longer tracts of homology increase the likelihood of nonallelic recombination occurring, it may initially seem counterintuitive that most known recombination instability events leading to genetic disease in humans appear to be Alu-mediated [3,4]. This is the case despite the presence of 500,000 copies of considerably longer (up to 6kb) L1 sequence that together comprise a larger fraction of the total genomic sequence than any other repeat family (roughly 17% vs. Alu’s 10%) [50]. There may be multiple factors contributing to this observation. Foremost among these is that the lower L1 copy numbers result in greater inter-copy distances between L1 sequences [49,51,52], rendering recombinations between L1 elements far more likely to be large, mutagenic, and lethal. The distance between Alu elements, by comparison, is smaller, both due to their higher copy number (~1.5 million) and their tendency to cluster in GC-rich regions, often in the proximity of genes. The tendency of Alu sequences to cluster in and around genes also serves to increase the probability of Alu-mediated deletions that knock out components of individual genes without resulting in larger multi-genic lethal mutations. The frequency of meiotic recombination is also observed to vary considerably across the genome, and is generally found to be higher in GC-rich genic regions [51,52] where Alu elements reside. All of these factors likely contribute to some degree to Alu’s predominance in human disease-causing rearrangements.
1.2 Insertion Mediated Deletions
There is a class of genetic rearrangements which appear to occur in concert with the TE insertion process itself. Insertion-related deletions and other rearrangements were first observed at newly inserted human L1 retrotransposition loci recovered from cell culture experiments [53,54]. Rearrangements were heterogeneous in size, with deletions ranging from a few base pairs to several thousand base pairs. Numerous products recovered from these insertions appeared to be fusions between the novel L1 copy and existing endogenous elements, suggesting the involvement of SSA or other homology dependent pathways for resolution of the insertion. Bioinformatic analyses, complemented with molecular validations, have subsequently confirmed that, for both L1 and Alu inserts, these events occur in the germline, and their products are readily observable in the public human genome sequence ([55,56]). The precise mechanism(s) resulting in these rearrangements remain uncertain, but may indicate the rather fragile nature of the exposed chromosome sites during the insertion process. The hallmark of DNA repair activity associated with these insertion events may indicate a competitive struggle occurring, at the time of the TE insertion process, wherein the host repair machinery attempts to repair the nascent insertion location and derail the retrotransposon insertion [53]. The generation of DSBs by L1 products, which are used by L1, Alu, and SVA insertions, probably alerts the cell DSB signaling system and recruits DNA repair machinery to the insertion loci. This raises the possibility that heterogeneity in pathway preference, both at the level of cell cycle phase [57] and across tissue types and developmental stages [44], could greatly impact the relative efficiency of retrotransposon insertions.
1.3 Transcription and Recombination
Increased transcriptional activity in certain contexts is believed to be highly recombinogenic (reviewed in [58]). A common explanation for this phenomenon is that interactions between replication forks and transcriptional machinery can increase the likelihood of DSBs [58]. By introducing potentially active promoters throughout the genome, TEs may increase the overall level of transcription-related stress, inflating the average number of DSBs per cell cycle. This phenomenon is no doubt largely mitigated in normal cells due to the repression of many TE promoters by methylation and associated chromatin structures [59–61]. However, some TE promoters evidently do manage—if only temporarily—to escape these repression mechanisms as evidenced by their continued survival in the vast majority of taxa surveyed. It is known, for example, that early developmental stages in mammalian species include a period of general hypomethylation across the genome [62]. Additionally, several forms of cancer are associated with a widespread hypomethylation [63]. Such hypomethylation may increase the transcriptional activity of these elements, thereby increasing the impact of transcriptional stress related DSBs. The increased “transcriptional stress,” by introducing additional DSBs and associated rearrangements, may be an important contributor to the genetic instability that is associated with many cancers as well as the rampant instability associated with loss of methyltransferases in humans and mice[64–66].
1.4 Segmental Duplications
Segmental duplications of chromosomal regions are a common occurrence in mammalian genomes and appear to be particularly enriched in human genomes [67]. Such segmental duplications have played substantial roles both in the evolution of genomes and in human disease. Segmental duplications in chromosomes are conducive to additional rearrangements on account of their very extensive nonallelic homology. A systematic analysis of evolutionarily recent segmental duplications in humans revealed a significantly increased density of Alu sequences at, or near, the duplication junctions, specifically those duplications that were separated by a span of unique sequence [68]. For the human lineage, it was hypothesized that the explosion of Alu retrotransposition ~30–40 mya effectively primed the human genome to be particularly subject to segmental duplications [68]. The precise mechanism by which Alu and other repetitive sequences are involved in generating some of these duplications remains unclear, although several of models have been proposed. While the aforementioned homologous repair pathways could readily generate duplication events in which the replicons are tandemly arranged, it is not obvious what mechanism generates the large number of segmental duplications that are physically separated by >1Mb of unique sequence. However, the increased density of Alu elements at the junction of these events, along with tendency for these segmental duplications to involve younger, presumably less-diverged, elements is highly suggestive of a prominent role for homology in the underlying mechanism [68]. In addition to the human data, similar observations have been made regarding the involvement of the B1 and B2 SINE lineages in segmental duplications within the mouse genome [69]. Together these data suggest that TE-driven segmental duplication may be a common theme in mammalian genome evolution.
1.5 Inverted Repeats and Instability
Multiple studies have demonstrated the particularly unstable nature of nearby TE copies that are in inverted orientation relative to one another [45,70,71]. Closely located, inverted Alu elements are extremely rare in the human genome [66]. In the case of Alu elements in the human genome, these inverted repeats appear to yield DSBs due to secondary structures formed during DNA replication. Although some degree of insertion preference exists for the direct orientation of immediately adjacent Alu insertions [72], it is likely that the inherent instability of the inverted insertions is the primary reason for their almost complete exclusion from the human genome [71]. Despite the infrequent nature of these inverted loci in humans, the fact that Alu insertions are still occurring in the human population at an appreciable rate [73] indicates that the potential for the de novo generation of inverted loci remains present in each new generation and possibly even in somatic cell lineages; the latter being increasingly likely given recent in vivo evidence demonstrating that L1 can mobilize in the soma [74] as well as ex vivo (cell culture) evidence for the possibility of somatic Alu mobilization [75].
1.6 Seeding Microsatellites
Several studies have demonstrated the ability of some TE sequences to seed the formation of unstable microsatellite repeats [76–78]. Because the structure of many TE insertions includes a poly-A rich tail, mutations in the A-tracts can readily create the initial repeats of a microsatellite sequence. These large tracts of repetitive sequence can subsequently become unstable [79–81]. Depending upon their location, such instabilities can range from innocuous to serious. Severe expansion of microsatellite sequence within introns can influence gene transcripts, such as in the case of an Alu-generated microsatellite in Friedrich's Ataxia [82].
1.7 TEs Alter Epigenetic Regulatory Structures
As the critical role of epigenetic regulation of the mammalian genome becomes increasingly evident, it is important to recognize the potential epigenetic ramifications of TE insertions, along with their mutagenic effects on the primary DNA sequence. These epigenetic alterations can be considered physical alterations in the genome’s information repository. Because of their potential to disrupt normal biological function during proliferation, a number of mammalian taxa have employed epigenetic modifications, many of which serve to constrain TE activity [60,83–86]. As a consequence, the insertion of TEs at particular regions of the genome can result in the recruitment of chromatin modulating factors, typically via CpG methylation and subsequent histone deacetylation, to the region containing the inserted element. In some cases, the introduction of these heterochromatin-inducing factors can not be effectively confined to the immediate vicinity of the TE element. The resulting spread of heterochromatin has the potential to negatively impact surrounding gene expression [87].
The ability for the genome to restrict chromatin modulation to the repeat location appears to vary among repeats. In humans, LTR and L1 repeats appear to generate larger chromatin modulating effects than Alu elements, which can be methylated in a more focused manner [88]. The relationship between chromatin state and the propensity for regions to accumulate genetic alterations remain unclear. It is conceivable that, by modulating the epigenetic structures associated with a particular region in the genome, TE insertions may exert an influence on the overall mutation rate.
2 Revisitation of Endonuclease Sites
2.1 L1 Endonuclease Generates DSBs
While our discussion of TE-related instability largely focuses on the disruptive aspects of their homology, the activity of mobile elements has also been implicated in instability. In order to facilitate transposition, TEs produce a variety of protein products, including reverse transcriptases and endonucleases. While the precise biochemical steps in the insertion process for many TEs remain unclear, it was recently reported from our laboratory that the endonuclease products of L1, an autonomous retrotransposon found throughout the mammalian order, is capable of generating DSBs far in excess of insertions in mammalian cell lines [25]. This capacity of the L1 endonuclease was experimentally demonstrated by showing an increase in H2AX foci following L1 transfection, a phenomena strongly associated with DSB formation [89,90]. The extent to which this represents an important source of natural endogenous DSBs remains unknown, as the levels of L1 expression used in these experiments were significantly greater than those expected in the normal cellular environment. Nevertheless, the formation of DSBs, which are highly recombinogenic and associated with numerous genetic alterations, has several possible implications for both genome stability and TE evolution in general. For those TEs that do produce DSBs during their insertion, the existence of a number of cellular detection mechanisms for DSBs and their associated repair, suggest a possible avenue for the cell to resist new insertion events. On the other hand, data showing ATM, an important protein in many DNA repair signaling processes, is required for L1 retrotransposition [25] may indicate that the L1 retrotransposon capitalizes on DNA host repair proteins for its own purposes [25]. Both the reliance on the DNA repair process by the cell to thwart new insertion attempts and the manipulation of the repair process by the TE for its own ends need not be mutually exclusive possibilities.
2.2 Endonuclease Targets remain Adjacent to Interspersed Repeats
Many varieties of TEs, including mammalian LINEs and SINEs, employ a mechanism termed target-primed reverse transcription (TPRT) as their means of integration into the genome [91]. In TPRT, the initial target site for endonuclease cleavage and integration is replicated, resulting in a pair of target site duplications (TSDs) that flank the new insert (Figure 5). This allows for possible further cleavage of these target sites by subsequent endonuclease visits (Figure 4). This “revisitation” of target sites by L1 endonucleases, as well as its potential consequences for instability, was initially proposed in conjunction with Alu-mediated low-copy repeat shuffling on chromosome 22q11 [92]. Further support was provided through subsequent bioinformatic analyses, which indicated that in both the human and rodent lineages, SINE elements flanked by perfect (i.e. nonmutated) L1 endonuclease consensus sites were more rapidly lost from the genome [92]. Although it should be noted that a similar analysis for dog SINEs failed to show this effect; there are, however, indications that SINE/LINE dynamics in the dog differ significantly in key ways from those of humans and rodents [93]. Taken together with recent experimental evidence for the ability of endonucleases to generate DSBs at endonuclease target sites [25], a connection between TSDs and post-insertion genomic instability involving TEs appears highly likely. Endonuclease-generated DSBs at TSD locations would have an enormous potential for genomic instability because they represent the “worst-case-scenario” conjunction of DSBs and adjacent interspersed homology. As described above, the interaction of interspersed homologous sequence and DSB repair pathways is a recipe for genetic rearrangements.
Figure 5. Enonuclease Target Site Duplications (TSDs) Adjacent to SINEs and LINEs Invite DSB Instability.

Insertion of SINEs and LINEs by the TPRT mechanism results in the duplication of the pre-insertion endonuclease cleavage site. The persistance of these sights adjacent to the element allows for subsequent cleavage by other L1 endonucleases. The DSBs generated by these L1 endonucleases, coupled with the adjacency of the TSDs to sequences with interspersed homologous copies, gives them a high potential for generating rearrangement events. Orange arrows indicate endonuclease target sites, which are part of the TSDs following TE insertion. Purple shapes represent L1 endonuclease molecules.
2.3 Alu “chi-like” Sequence and Chromosome Rearrangements
Several studies have reported the involvement of a short 26 bp “core” sequence 5'-CCTGTAATCCCAGCACTTTGGGAGGC-3'within the Alu left monomer in a number of rearrangement events [38,94]. Most recently, an examination of over 400 Alu mediated deletion events showed that the most frequent locations of deletion junctions did indeed occur within this Alu segment, with the previously described 26bp region showing a modest, though clear, two-fold increase in junction participation [37].
The presence within this 26 bp region of 5 bp of the prokaryotic chi sequence has served to fuel further speculation about a possible recombinogenic role for this region of the Alu left monomer. In E. coli, the short 8 bp chi site is the target site for RecA recombinase, the enzyme which initiates recombination [26]. However, the fact that the prokaryotic chi target site does not appear to be conserved in any other eukaryotic system examined to date makes it highly unlikely that a RecA-”chi-like” system would have been uniquely preserved in the primate lineage—particularly one maintaining target site identity at the nucleotide level. In addition, as others have noted previously [94], the 26 bp core Alu fragment represents the most highly conserved stretch of Alu sequence among genomic Alu copies. This elevated sequence conservation is likely attributable to both its location in the polIII promoter region as well as its complete lack of highly mutagenic CpG dinucleotides, which are present throughout the remainder of the Alu consensus. It is therefore plausible that the reason this segment of Alu is frequently represented at Alu-mediated rearrangement junctions is simply due to its sequence conservation [94]; as indicated above, two sequences with little divergence are more likely to engage in a recombination events. While sequence homology no doubt plays a key role in the phenomenon, the possible involvement of topoisomerases has also been suggested [94]. Due to its location within the polIII promoter, there is also a distinct possibility that DSBs related to transcriptional stress [58] contributes to the number of rearrangements involving this region. In addition to the factors listed above, we further speculate, based on the endonuclease revisitation, that the proximity of the 26bp sequence to the endonuclease target site 5’ of the Alu may play an important role in its involvement in rearrangement junctions. DSBs generated at the 5' end of the Alu sequence could frequently be involved in DSB repair, resulting in an increased appearance of the Alu sequences at both homologous and nonhomologous rearrangement junctions.
Conclusion
The of DNA repair systems in mammalian lineages to avoid NAHR related instability may have been, in part, a response to increased interspersed TE homology across their genomes [41]. As a result of the host organisms’s repair responses, the evolutionary landscape for TEs was likely significantly altered by the reduced risk of ectopic recombination. If, as has been suggested previously, ectopic recombination does play a major role in limiting total TE copy numbers [95], it follows that increased suppression of ectopic recombination in mammals and other taxa may have ultimately provided for more “safe” niche space within mammalian genomes for the expansion of larger, more similar, TE families [49]. Thus the repeat-driven expansion of many genomes, including our own, may in part represent the consequence of increased vigilance against TE-related instability.
The capacity for L1 to generate DSBs, along with interspersed homology, serves to further increase the culpability of TEs in mammalian genetic instability. From an epidemiological standpoint, L1-generated DSBs also suggest the possibility that variability in the number of actively expressing TE sequences in the human population could lead to differences between individuals in the risk for genetic alterations, both somatic and germinal. As a consequence, TE activity levels may contribute to heterogeneity among individuals for the onset of cancer, reproductive deficiencies, as well as the generation of de novo disease alleles in offspring.
Acknowledgments
We are very grateful to Louisiana State University in Baton Rouge, and specifically Dr. Mark Batzer, for hosting us in his laboratory during our evacuation for hurricane Katrina. We thank all members of Dr. Batzer’s laboratory for their kindness and help. This research was supported by grants from the National Institutes of Health, R01GM45668 (PLD), National Science Foundation, EPS-0346411 (PLD), and the State of Louisiana Board of Regents Support Fund (PLD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cordaux R, Udit S, Batzer MA, Feschotte C. Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc Natl Acad Sci U S A. 2006;103:8101–8106. doi: 10.1073/pnas.0601161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chou HH, Hayakawa T, Diaz S, Krings M, Indriati E, Leakey M, Paabo S, Satta Y, Takahata N, Varki A. Inactivation of CMP-N-acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc Natl Acad Sci U S A. 2002;99:11736–11741. doi: 10.1073/pnas.182257399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Callinan PA, Batzer MA. In: Retrotransposable Elemenets and Human Disease. J-N V, editor. Genome Dynamics; Karger, Basel: 2006. pp. 104–115. [DOI] [PubMed] [Google Scholar]
- 4.Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 5.Petrov DA, Aminetzach YT, Davis JC, Bensasson D, Hirsh AE. Size matters: non-LTR retrotransposable elements and ectopic recombination in Drosophila. Mol Biol Evol. 2003;20:880–892. doi: 10.1093/molbev/msg102. [DOI] [PubMed] [Google Scholar]
- 6.Boissinot S, Davis J, Entezam A, Petrov D, Furano AV. Fitness cost of LINE-1 (L1) activity in humans. Proc Natl Acad Sci U S A. 2006;103:9590–9594. doi: 10.1073/pnas.0603334103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boissinot S, Entezam A, Furano AV. Selection against deleterious LINE-1-containing loci in the human lineage. Mol Biol Evol. 2001;18:926–935. doi: 10.1093/oxfordjournals.molbev.a003893. [DOI] [PubMed] [Google Scholar]
- 8.Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res. 2002;12:1060–1067. doi: 10.1101/gr.229302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sorek R, Lev-Maor G, Reznik M, Dagan T, Belinky F, Graur D, Ast G. Minimal conditions for exonization of intronic sequences: 5' splice site formation in alu exons. Mol Cell. 2004;14:221–231. doi: 10.1016/s1097-2765(04)00181-9. [DOI] [PubMed] [Google Scholar]
- 10.Belancio VP, Hedges DJ, Deininger P. LINE-1 RNA splicing and influences on mammalian gene expression. Nucleic Acids Res. 2006;34:1512–1521. doi: 10.1093/nar/gkl027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perepelitsa-Belancio V, Deininger P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat Genet. 2003;35:363–366. doi: 10.1038/ng1269. [DOI] [PubMed] [Google Scholar]
- 12.Wheelan SJ, Aizawa Y, Han JS, Boeke JD. Gene-breaking: a new paradigm for human retrotransposon-mediated gene evolution. Genome Res. 2005;15:1073–1078. doi: 10.1101/gr.3688905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 14.Dagan T, Sorek R, Sharon E, Ast G, Graur D. AluGene: a database of Alu elements incorporated within protein-coding genes. Nucleic Acids Res. 2004;32:D489–492. doi: 10.1093/nar/gkh132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 16.Hedges DJ, Batzer MA. From the margins of the genome: mobile elements shape primate evolution. Bioessays. 2005;27:785–794. doi: 10.1002/bies.20268. [DOI] [PubMed] [Google Scholar]
- 17.Friedberg EC. Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol. 2005;6:943–953. doi: 10.1038/nrm1781. [DOI] [PubMed] [Google Scholar]
- 18.van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- 19.Pfeiffer P, Goedecke W, Obe G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis. 2000;15:289–302. doi: 10.1093/mutage/15.4.289. [DOI] [PubMed] [Google Scholar]
- 20.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. Embo J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- 23.Boissinot S, Furano AV. The recent evolution of human L1 retrotransposons. Cytogenet Genome Res. 2005;110:402–406. doi: 10.1159/000084972. [DOI] [PubMed] [Google Scholar]
- 24.Furano AV, Duvernell DD, Boissinot S. L1 (LINE-1) retrotransposon diversity differs dramatically between mammals and fish. Trends Genet. 2004;20:9–14. doi: 10.1016/j.tig.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol. 2006;357:1383–1393. doi: 10.1016/j.jmb.2006.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 28.Bree RT, Neary C, Samali A, Lowndes NF. The switch from survival responses to apoptosis after chromosomal breaks. DNA Repair (Amst) 2004;3:989–995. doi: 10.1016/j.dnarep.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 29.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 30.Thacker J, Zdzienicka MZ. The XRCC genes: expanding roles in DNA double-strand break repair. DNA Repair (Amst) 2004;3:1081–1090. doi: 10.1016/j.dnarep.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:909–918. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 32.Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. Embo J. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Resnick MA. The repair of double-strand breaks in DNA; a model involving recombination. J Theor Biol. 1976;59:97–106. doi: 10.1016/s0022-5193(76)80025-2. [DOI] [PubMed] [Google Scholar]
- 34.Resnick MA, Martin P. The repair of double-strand breaks in the nuclear DNA of Saccharomyces cerevisiae and its genetic control. Mol Gen Genet. 1976;143:119–129. doi: 10.1007/BF00266917. [DOI] [PubMed] [Google Scholar]
- 35.Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 36.Stark JM, Jasin M. Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol Cell Biol. 2003;23:733–743. doi: 10.1128/MCB.23.2.733-743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sen SK, Han K, Wang J, Lee J, Wang H, Callinan PA, Dyer M, Cordaux R, Liang P, Batzer MA. Human Genomic Deletions Mediated by Recombination between Alu Elements. Am J Hum Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hill AS, Foot NJ, Chaplin TL, Young BD. The most frequent constitutional translocation in humans, the t(11;22)(q23;q11) is due to a highly specific alu-mediated recombination. Hum Mol Genet. 2000;9:1525–1532. doi: 10.1093/hmg/9.10.1525. [DOI] [PubMed] [Google Scholar]
- 39.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 40.Tremblay A, Jasin M, Chartrand P. A double-strand break in a chromosomal LINE element can be repaired by gene conversion with various endogenous LINE elements in mouse cells. Mol Cell Biol. 2000;20:54–60. doi: 10.1128/mcb.20.1.54-60.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson RD, Jasin M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 2001;29:196–201. doi: 10.1042/0300-5127:0290196. [DOI] [PubMed] [Google Scholar]
- 42.Couedel C, Mills KD, Barchi M, Shen L, Olshen A, Johnson RD, Nussenzweig A, Essers J, Kanaar R, Li GC, Alt FW, Jasin M. Collaboration of homologous recombination and nonhomologous end-joining factors for the survival and integrity of mice and cells. Genes Dev. 2004;18:1293–1304. doi: 10.1101/gad.1209204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Preston CR, Engels W, Flores C. Efficient repair of DNA breaks in Drosophila: evidence for single-strand annealing and competition with other repair pathways. Genetics. 2002;161:711–720. doi: 10.1093/genetics/161.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Preston CR, Flores CC, Engels WR. Differential usage of alternative pathways of double-strand break repair in Drosophila. Genetics. 2006;172:1055–1068. doi: 10.1534/genetics.105.050138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gebow D, Miselis N, Liber HL. Homologous and nonhomologous recombination resulting in deletion: effects of p53 status, microhomology, and repetitive DNA length and orientation. Mol Cell Biol. 2000;20:4028–4035. doi: 10.1128/mcb.20.11.4028-4035.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Opperman R, Emmanuel E, Levy AA. The effect of sequence divergence on recombination between direct repeats in Arabidopsis. Genetics. 2004;168:2207–2215. doi: 10.1534/genetics.104.032896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- 48.Datta A, Hendrix M, Lipsitch M, Jinks-Robertson S. Dual roles for DNA sequence identity and the mismatch repair system in the regulation of mitotic crossing-over in yeast. Proc Natl Acad Sci U S A. 1997;94:9757–9762. doi: 10.1073/pnas.94.18.9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deininger PL, Roy-Engel AE. Mobile DNA in Animals and Plants. In: Craig NL, editor. Mobile DNA 2. ASM Press; Washington: 2002. pp. 1074–1092. [Google Scholar]
- 50.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 51.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 52.McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 53.Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 54.Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- 55.Callinan PA, Wang J, Herke SW, Garber RK, Liang P, Batzer MA. Alu retrotransposition-mediated deletion. J Mol Biol. 2005;348:791–800. doi: 10.1016/j.jmb.2005.02.043. [DOI] [PubMed] [Google Scholar]
- 56.Han K, Sen SK, Wang J, Callinan PA, Lee J, Cordaux R, Liang P, Batzer MA. Genomic rearrangements by LINE-1 insertion-mediated deletion in the human and chimpanzee lineages. Nucleic Acids Res. 2005;33:4040–4052. doi: 10.1093/nar/gki718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004;32:3683–3688. doi: 10.1093/nar/gkh703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aguilera A. The connection between transcription and genomic instability. Embo J. 2002;21:195–201. doi: 10.1093/emboj/21.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boccaccio C, Deschatrette J, Meunier-Rotival M. The human episome HALF1: structure of its genomic counterpart. Genetica. 1994;93:217–224. doi: 10.1007/BF01435253. [DOI] [PubMed] [Google Scholar]
- 60.Lavie L, Kitova M, Maldener E, Meese E, Mayer J. CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2) J Virol. 2005;79:876–883. doi: 10.1128/JVI.79.2.876-883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bestor TH. The host defence function of genomic methylation patterns. Novartis Found Symp. 1998;214:187–195. doi: 10.1002/9780470515501.ch11. discussion 195–189, 228–132. [DOI] [PubMed] [Google Scholar]
- 62.Allegrucci C, Thurston A, Lucas E, Young L. Epigenetics and the germline. Reproduction. 2005;129:137–149. doi: 10.1530/rep.1.00360. [DOI] [PubMed] [Google Scholar]
- 63.Hoffmann MJ, Schulz WA. Causes and consequences of DNA hypomethylation in human cancer. Biochem Cell Biol. 2005;83:296–321. doi: 10.1139/o05-036. [DOI] [PubMed] [Google Scholar]
- 64.Bourc'his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 65.Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005;65:8635–8639. doi: 10.1158/0008-5472.CAN-05-1961. [DOI] [PubMed] [Google Scholar]
- 66.Dodge JE, Okano M, Dick F, Tsujimoto N, Chen T, Wang S, Ueda Y, Dyson N, Li E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J Biol Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- 67.Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2002;3:65–72. doi: 10.1038/nrg705. [DOI] [PubMed] [Google Scholar]
- 68.Bailey JA, Liu G, Eichler EE. An Alu transposition model for the origin and expansion of human segmental duplications. Am J Hum Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jurka J, Kohany O, Pavlicek A, Kapitonov VV, Jurka MV. Clustering, duplication and chromosomal distribution of mouse SINE retrotransposons. Cytogenet Genome Res. 2005;110:117–123. doi: 10.1159/000084943. [DOI] [PubMed] [Google Scholar]
- 70.Stenger JE, Lobachev KS, Gordenin D, Darden TA, Jurka J, Resnick MA. Biased distribution of inverted and direct Alus in the human genome: implications for insertion, exclusion, and genome stability. Genome Res. 2001;11:12–27. doi: 10.1101/gr.158801. [DOI] [PubMed] [Google Scholar]
- 71.Lobachev KS, Stenger JE, Kozyreva OG, Jurka J, Gordenin DA, Resnick MA. Inverted Alu repeats unstable in yeast are excluded from the human genome. Embo J. 2000;19:3822–3830. doi: 10.1093/emboj/19.14.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.El-Sawy M, Deininger P. Tandem insertions of Alu elements. Cytogenet Genome Res. 2005;108:58–62. doi: 10.1159/000080802. [DOI] [PubMed] [Google Scholar]
- 73.Cordaux R, Hedges DJ, Herke SW, Batzer MA. Estimating the retrotransposition rate of human Alu elements. Gene. 2006;373:134–137. doi: 10.1016/j.gene.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 74.Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 75.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 76.Wilder J, Hollocher H. Mobile elements and the genesis of microsatellites in dipterans. Mol Biol Evol. 2001;18:384–392. doi: 10.1093/oxfordjournals.molbev.a003814. [DOI] [PubMed] [Google Scholar]
- 77.Chauhan C, Dash D, Grover D, Rajamani J, Mukerji M. Origin and instability of GAA repeats: insights from Alu elements. J Biomol Struct Dyn. 2002;20:253–263. doi: 10.1080/07391102.2002.10506841. [DOI] [PubMed] [Google Scholar]
- 78.Arcot SS, Wang Z, Weber JL, Deininger PL, Batzer MA. Alu repeats: a source for the genesis of primate microsatellites. Genomics. 1995;29:136–144. doi: 10.1006/geno.1995.1224. [DOI] [PubMed] [Google Scholar]
- 79.Hebert ML, Wells RD. Roles of double-strand breaks, nicks, and gaps in stimulating deletions of CTG.CAG repeats by intramolecular DNA repair. J Mol Biol. 2005;353:961–979. doi: 10.1016/j.jmb.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 80.Li HR, Shagisultanova EI, Yamashita K, Piao Z, Perucho M, Malkhosyan SR. Hypersensitivity of tumor cell lines with microsatellite instability to DNA double strand break producing chemotherapeutic agent bleomycin. Cancer Res. 2004;64:4760–4767. doi: 10.1158/0008-5472.CAN-04-0975. [DOI] [PubMed] [Google Scholar]
- 81.Richard GF, Dujon B, Haber JE. Double-strand break repair can lead to high frequencies of deletions within short CAG/CTG trinucleotide repeats. Mol Gen Genet. 1999;261:871–882. doi: 10.1007/s004380050031. [DOI] [PubMed] [Google Scholar]
- 82.Montermini L, Andermann E, Labuda M, Richter A, Pandolfo M, Cavalcanti F, Pianese L, Iodice L, Farina G, Monticelli A, Turano M, Filla A, De Michele G, Cocozza S. The Friedreich ataxia GAA triplet repeat: premutation and normal alleles. Hum Mol Genet. 1997;6:1261–1266. doi: 10.1093/hmg/6.8.1261. [DOI] [PubMed] [Google Scholar]
- 83.Yu F, Zingler N, Schumann G, Stratling WH. Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 2001;29:4493–4501. doi: 10.1093/nar/29.21.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matzke MA, Mette MF, Aufsatz W, Jakowitsch J, Matzke AJ. Host defenses to parasitic sequences and the evolution of epigenetic control mechanisms. Genetica. 1999;107:271–287. [PubMed] [Google Scholar]
- 85.Hata K, Sakaki Y. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene. 1997;189:227–234. doi: 10.1016/s0378-1119(96)00856-6. [DOI] [PubMed] [Google Scholar]
- 86.Lamb BT, Satyamoorthy K, Li L, Solter D, Howe CC. CpG methylation of an endogenous retroviral enhancer inhibits transcription factor binding and activity. Gene Expr. 1991;1:185–196. [PMC free article] [PubMed] [Google Scholar]
- 87.Turker MS. Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene. 2002;21:5388–5393. doi: 10.1038/sj.onc.1205599. [DOI] [PubMed] [Google Scholar]
- 88.Kang MI, Rhyu MG, Kim YH, Jung YC, Hong SJ, Cho CS, Kim HS. The length of CpG islands is associated with the distribution of Alu and L1 retroelements. Genomics. 2006 doi: 10.1016/j.ygeno.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 89.Andegeko Y, Moyal L, Mittelman L, Tsarfaty I, Shiloh Y, Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem. 2001;276:38224–38230. doi: 10.1074/jbc.M102986200. [DOI] [PubMed] [Google Scholar]
- 90.Modesti M, Kanaar R. DNA repair: spot(light)s on chromatin. Curr Biol. 2001;11:R229–232. doi: 10.1016/s0960-9822(01)00112-9. [DOI] [PubMed] [Google Scholar]
- 91.Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. Embo J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Babcock M, Pavlicek A, Spiteri E, Kashork CD, Ioshikhes I, Shaffer LG, Jurka J, Morrow BE. Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res. 2003;13:2519–2532. doi: 10.1101/gr.1549503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gentles AJ, Kohany O, Jurka J. Evolutionary diversity and potential recombinogenic role of integration targets of Non-LTR retrotransposons. Mol Biol Evol. 2005;22:1983–1991. doi: 10.1093/molbev/msi188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rudiger NS, Gregersen N, Kielland-Brandt MC. One short well conserved region of Alu-sequences is involved in human gene rearrangements and has homology with prokaryotic chi. Nucleic Acids Res. 1995;23:256–260. doi: 10.1093/nar/23.2.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bartolome C, Maside X, Charlesworth B. On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol. 2002;19:926–937. doi: 10.1093/oxfordjournals.molbev.a004150. [DOI] [PubMed] [Google Scholar]