

Abstract
FLICE-like inhibitory protein (FLIP), a naturally occurring caspase-inhibitory protein that lacks the critical cysteine domain necessary for catalytic activity, is a negative regulator of Fas-induced apoptosis. Decreased FLIP levels sensitize tumor cells to Fas- and TRAIL-mediated apoptosis; however, the cellular mechanisms regulating FLIP expression have not been defined. Here, we examined the roles of the PKC and NF-κB pathway in the regulation of FLIP in human colon cancers. FLIP mRNA levels were increased in Caco-2 cells by treatment with PMA; actinomycin D completely inhibited the induction of FLIP by PMA, indicating transcriptional regulation. PKC inhibitors Gö6983 and Ro-31-8220, blocked PMA-stimulated FLIP expression. Pretreatment with the PKCδ selective inhibitor rottlerin or transfection with PKCδ siRNA inhibited PMA-induced FLIP expression, which identifies a role for PKCδ in FLIP induction. Treatment with the proteasome inhibitor, MG132, or the NF-κB inhibitor (eg, PDTC and gliotoxin), or overexpression of the superrepressor of IκB-α inhibited PMA-induced upregulation of FLIP. Moreover, PMA-induced NF-κB transactivation was blocked by GF109203x. In conclusion, our results demonstrate a critical role for PKCδ/NF-κB in the regulation of FLIP in human colon cancer cells.
Keywords: colon cancer, FLIP, PKCδ, NF-κB, signal transduction
INTRODUCTION
The caspase-8 homologue, FLICE-like inhibitory protein (FLIP), functions as a caspase-8 dominant negative by blocking apoptosis induced by the oligomerization of the adapter protein FADD/MORT-1.1, 2 At the transcription level, cellular FLIP appears to exist as multiple splice variants but only two endogenous forms, FLIPL and FLIPS, are detected.3-5 FLIP expression correlates with resistance to apoptosis induced by various members of the tumor necrosis factor (TNF) family such as TRAIL.6 High levels of FLIP are commonly found in tumors; forced expression of FLIP renders cells resistant to Fas-mediated apoptosis.1, 7, 8 Conversely, reduction in FLIP levels can sensitize normally resistant cancer cells to chemotherapy-induced apoptosis.9, 10 FLIP expression is dependent on the phosphatidylinositol-3 kinase (PI3-kinase) pathway in some tumor cell lines.2 Forced overexpression of protein kinase D (PKD) upregulates FLIP expression along with a reduction of CD95-mediated apoptosis in pancreatic carcinoma cells.11 Inducers of the transcription factor NF-κB induce FLIP expression.12, 13 However, the oncogenic signaling pathways that regulate FLIP expression in tumor cells are largely unknown.
The protein kinase C (PKC) family, some isoforms of which are stimulated by phorbol esters such as phorbol 12-myristate 13-acetate (PMA), is responsible for transducing many cellular signals important for mitogenesis, cellular metabolism, differentiation, tumor promotion, and apoptosis.14–16 Phorbol esters attenuate Fas-induced apoptosis.16 Moreover, in other cellular models of apoptosis, there is selective inhibition/activation of PKC isotypes, depending on cell type and apoptotic stimuli considered.17, 18 PKC functions in the cell death program to positively or negatively regulate apoptosis. For example, PKCδ acts as a pro-survival factor in human breast tumor cells.19 Activation of PKCδ/NF-κB increases expression of the inhibitor of apoptosis protein-2 (cIAP-2) 20, whereas, inhibition of PKCδ with rottlerin or transfection of a kinase-dead mutant of PKCδ increased apoptosis and potentiated chemotherapy-induced apoptosis in non-small cell lung cancer cells.21 In contrast, overexpression of PKCδ inhibited the proliferation of fibroblasts 22, induced monocytic differentiation of the myeloid progenitor cell line 23, and enhanced enterocyte-like differentiation of colon cancer cell line Caco-2.24 PKCδ has been reported to undergo tyrosine phosphorylation in response to various stimuli such as PMA, epidermal growth factor, and platelet-derived growth factor (PDGF).23, 25, 26 TNF-α has been shown to induce NF-κB activation through PKCδ in human neutrophils.27 Induction of NF-κB by PKC activation can prevent apoptosis in certain cells.28–30
Our laboratory is interested in mechanisms regulating the expression of the apoptotic-related genes in colorectal cancers.20, 31 Recently, we demonstrated that selective induction of TRAF1 in human colon cancer cells is through a Ca2+-dependent PKC/Raf-1/ERK/NF-κB-dependent pathway 31 and moreover, we found that the PKCδ/NF-κB pathway plays an important role in the regulation of the anti-apoptosis protein cIAP-2 in human colon cancer cells.20 It appears, however, that FLIP is a more efficient inhibitor of death ligand-induced apoptosis than TRAF1, TRAF-2, cIAP-1, and cIAP-2, which have been previously proposed to be responsible for the inhibition of death receptor-induced cell death.12, 32 Although the regulation of FLIP expression has been characterized to some degree in certain cancer cells 2, 33, the regulation of expression in colon cancers has not been defined. Therefore, the purpose of this present study was to analyze the mechanisms regulating FLIP gene expression in colorectal cancers. Phorbol esters, such as PMA, were originally described as tumor promoters but these agents can also modulate a variety of cell processes such as growth, differentiation, apoptosis and gene transcription through the PKC signaling pathway. PMA, which can substitute for diacylglycerol (the endogenous PKC activator), has been utilized as a model agent to analyze the potential mechanisms used by growth factors and hormones to modulate cell growth and differentiation.34–37 Here, we found that PMA induced FLIP expression in a dose- and time-dependent fashion. Moreover, we found that the induction of FLIP in human colon cancer cells is mediated through a PKCδ/NF-κB-dependent pathway.
MATERIALS AND METHODS
Materials
PMA, sodium butyrate (NaBT), hydrogen peroxide (H2O2), and anti-actin antibodies were purchased from Sigma Chemical Company (St. Louis, MO). Bis-indolylmaleimide I (GF109203x), PD98059, wortmannin, Gö6983, Gö6976, Ro-31-8220, Rottlerin, MG132, pyrrolidine dithiocarbamate (PDTC), and gliotoxin were from Calbiochem (San Diego, CA). The anti-c-FLIP mAb (NF6) (mouse IgG1) was a gift from Dr. Marcus E. Peter (University of Chicago, Chicago, IL). Adenovirus vector encoding hemagglutinin-tagged IκB-α superrepressor (Ad5IκB-AA) and its control vector (Ad5GFP) were gifts from Dr. Christian Jobin (University of North Carolina, Chapel Hill). PKCδ and control siRNA, purchased from Dharmacon, Inc. (Lafayette, CO), were used as we have described previously.38 The siRNAs for targeting PKCδ consist of four pooled SMARTselection-designed siRNAs and the sequences were PKCδ siRNA1 (5′-GAAAGAACGCUUCAACAUC-3′), PKCδ siRNA2 (5′-AGAAGAAGCCGACCAUGUA-3′), PKCδ siRNA3 (5′-GUUGAUGUCUGUUCAGUAU-3′), and PKCδ siRNA4 (5′-AGAAAUGCAUCGACAAGAU-3′). All siRNAs are synthesized with UU as 3′-overhangs on each strand, and 5′-phosphate on the antisense strand only. The pGEMT-FLIP construct was a gift from Dr. Alicia Algeciras-Schimnich (University of Chicago, Chicago, IL). The pNF-κB-luc reporter plasmid was from Clontech (Palo Alto, CA) and the pRL-Tk-luc reporter plasmid was purchased from Promega (Madison, WI). The luciferase reporter assay system was from Promega. The constitutively expressed glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was obtained from Ambion (Austin, TX) and used to ensure the integrity of the RNA samples analyzed by Northern blot. [γ-32P] ATP (3,000 Ci/mmol) was from Amersham Pharmacia Biotech (Piscataway, NY). Tissue culture media and reagents were obtained from GIBCO BRL (Grand Island, NY). Polyvinylidene difluoride (PVDF) membranes for Western blots were from Bio-Rad Laboratories (Hercules, CA). The enhanced chemiluminescence (ECL) system for Western immunoblot analysis was purchased form Amersham.
Cell culture
The human colon cancer cell lines Caco-2, HT29 and HCT116 were purchased from ATCC (Rockville, MA). The human colon cancer cell lines, KM20 and KM12C, were obtained from Dr. Isaiah Fidler (MD Anderson, Houston TX). Caco-2, KM20, and KM12C cells were incubated in MEM supplemented with either 15% (Caco-2,) or 10% (KM20, KM12C) fetal calf serum (FCS), respectively. HT29 and HCT116 cells were maintained in McCoy’s 5A supplemented with 10% FCS. PMA and inhibitors were initially dissolved in dimethyl sulfoxide (DMSO) and compared to cells treated with DMSO at the same final concentration. Cells were plated in 100 mm dishes and treated with PMA, the next day when ~80% confluent. Neither PMA nor the protein kinase inhibitors induced obvious cell death at the time points assessed in this study.
Transient transfections and luciferase assays
The PKCδ and control siRNA duplexes were introduced into cells by electroporation (Gene Pulser, Bio-Rad) as we have described previously.38. The pNF-κB-luc construct was co-transfected with 0.05 μg of pRL-Tk plasmid (Promega) using the calcium phosphate method.39 The pRL-Tk plasmid was co-transfected to normalize for variation in transfection efficiency. After 24 h, the cells were treated with various reagents for 16 h prior to luciferase assay. Luciferase activity was measured using the dual luciferase assay system (Promega) according to the manufacturer’s instructions and as we have previously described.40
RNA isolation and Northern blot analysis
Total RNA was isolated using the Ultraspectm RNA reagent. RNA extracts (30 μg) were run in 1.2% agarose/formaldehyde gels and transferred to supported nitrocellulose. Membranes were hybridized to random-primed 32P-labeled FLIP cDNA probe, containing nucleotides 383-1826 of the human FLIP cDNA sequence, overnight at 42°C and then washed three times at 68°C for 15 min with 2 × SSC and 0.5% SDS. After hybridization with a GAPDH probe, membranes were washed again and signals detected by autoradiography.
Protein preparation and Western immunoblot
Western immunoblot analyses were performed as described previously.41 Cells were lysed with TNN buffer (50 mM Tris HCl [pH 7.5], 150 mM NaCl, 0.5 mM Nonidet P-40, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM dithiothreitol [DTT], and 1 mM phenylmethylfuldonyl fluoride and 25 μg/m each of aprotinin, leupeptin and pepstatin A) at 4°C for 30 min. Lysates were clarified by centrifugation (10,000 g for 30 min at 4°C) and protein concentrations determined using the method of Bradford.42 Briefly, total protein (100 μg) was resolved on a 10% polyacrylamide gel and transferred to PVDF membranes. Membranes were incubated overnight at 4°C in blotting solution (Tris-buffer saline containing 5% nonfat dried milk and 0.1% Tween 20). FLIP and actin were detected with anti-FLIP (NF6) or anti-actin antibodies, respectively, following blotting with a horseradish peroxidase-conjugated secondary antibody and visualized using ECL detection.
DNA fragmentation assay
Cells were plated in 96-well plates 24 h before treatment. After treatment, DNA fragmentation was evaluated by examination of cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) using a Cell Death Detection ELISAPlus kit (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer’s instructions as we have described previously.43
Statistical analysis
Data in Fig. 6C and 6D was analyzed using analysis of variance for a two-factor experiment. Fisher’s least significant difference procedure was used for multiple comparisons with Bonferroni adjustment for the number of comparisons. Data in Fig. 5D was analyzed using the Kruskal-Wallis test.
Figure 6. PMA-induced FLIP expression acts through NF-κB activation.
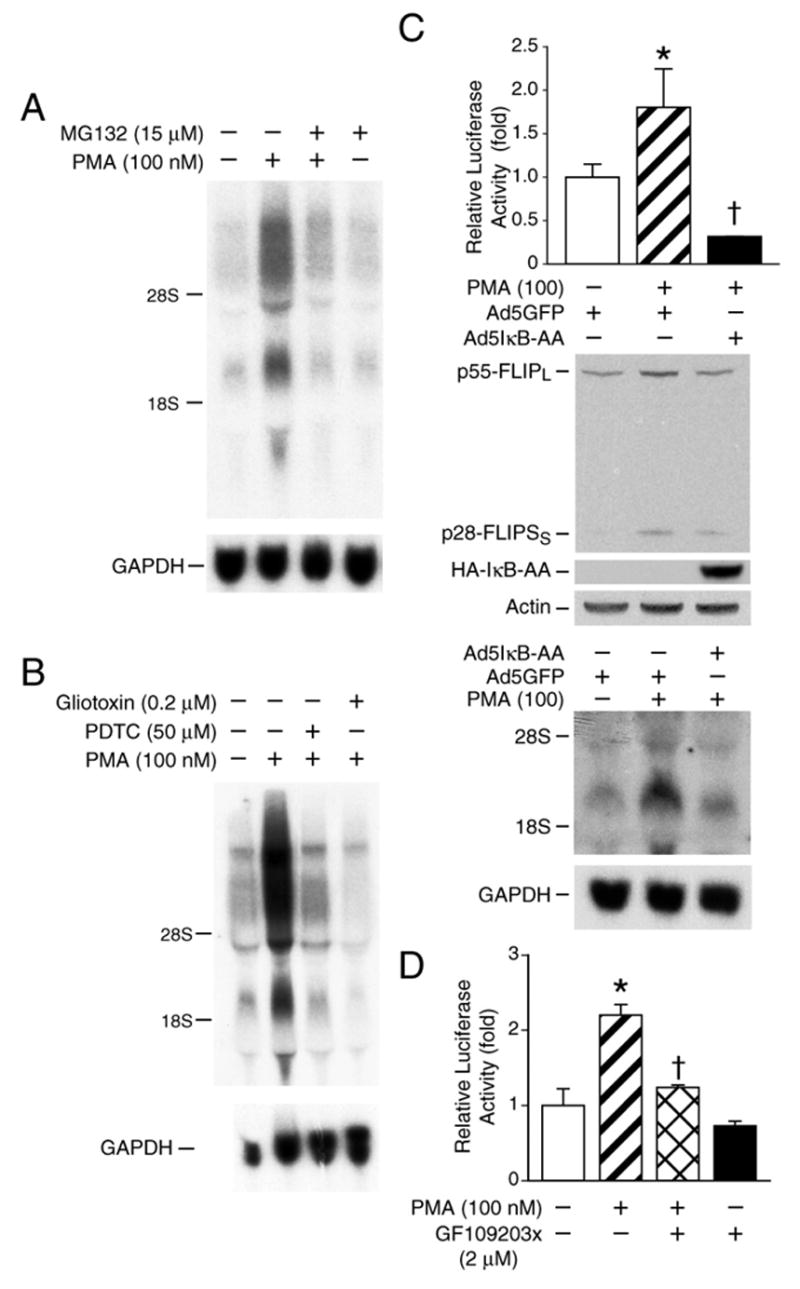
A. Caco-2 cells were preincubated for 30 min with the proteosome inhibitor MG132 (15 μM) and then treated with PMA (100 nM) for 8 h in the presence or absence of the inhibitor. Total RNA was isolated for Northern blot. B. Caco-2 cells were preincubated for 30 min with PDTC (50 μM) or gliotoxin (0.2 μM) and then treated with PMA (100 nM) for 8 h in the presence or absence of PDTC or gliotoxin. Total RNA was isolated for Northern blot. C. Top panel, Caco-2 cells were transfected with 0.3 μg of a plasmid containing the consensus NF-κB binding site linked to the luciferase reporter gene. The cells were then infected with a recombinant adenovirus encoding the Ad5IκB-AA or vector control encoding GFP. After 24 h, cells were treated with PMA or vehicle control for 16 h and then extracted and luciferase activity measured in the crude cell lysates as described in the “Materials and Methods.” Middle panel & bottom panel, Caco-2 cells were infected with a recombinant adenovirus encoding the Ad5IκB-AA or vector control encoding GFP. After 24 h, cells were treated with PMA (100 nM) or vehicle control for 8 h and then extracted for RNA and protein. Cell lysates (100 μg of protein) were fractionated by SDS-PAGE and blotted with anti-FLIP, anti-HA and anti-actin antibodies (middle panel). Total RNA was fractionated and probed with a labeled FLIP cDNA; blots were stripped and reprobed with GAPDH (bottom panel). D. Caco-2 cells were transfected with 0.3 μg of a plasmid containing the consensus NF-κB binding site linked to the luciferase reporter gene. Twenty-four hours after transfection, cells were treated with PMA with or without GF109203x (2 μM) for an additional 16 h and then extracted and luciferase activity measured in the crude cell lysates as described in the “Materials and Methods”. All results from the luciferase assay were normalized for transfection efficiency using the pRL-Tk-luc plasmid (Promega). Data are expressed as mean ±SD; * p < 0.05 compared with control; † p < 0.05 compared with PMA alone.
Figure 5. PKCδ contributes to PMA-induced FLIP expression.
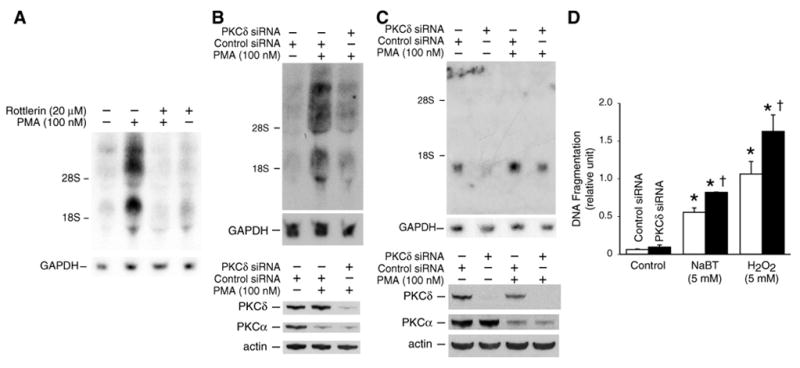
A. Caco-2 cells were starved for 24 h in serum-free medium. Cells were pretreated with or without the PKCδ inhibitor rottlerin (20 μM) for 30 min and then treated with PMA (100 nM) alone or in combination. RNA was isolated and analyzed by Northern blot. B. Caco-2 cells were transfected with control or PKCδ siRNA as described under “Materials and Methods” and then treated with PMA (100 nM) for 4 h. Total RNA was extracted for assessment of FLIP mRNA expression by Northern blot (upper panel). The whole cell lysates were extracted, resolved by SDS-PAGE (10% polyacrylamide) and assayed for PKCδ and PKCα expression by immunoblotting with anti-PKCδ or anti-PKCα antibodies (lower panel). C. HT29 cells were transfected with control or PKCδ siRNA as described in “Materials and Methods” and then treated with PMA (100 nM) for 4 h. Total RNA was extracted for assessment of FLIP mRNA expression by Northern blot (upper two panels). Cells from parallel dishes were harvested, and the lysates resolved by SDS-PAGE (10% polyacrylamide) and assayed for PKCδ and PKCα expression by immunoblotting with anti-PKCδ or anti-PKCα antibodies (lower two panels). D. After transfection with PKCδ siRNA to knockdown endogenous PKCδ or control siRNA, Caco-2 cells were treated with 5 mM NaBT or 5 mM H2O2. Apoptosis was estimated by an ELISA method as described in “Materials and Methods.” Columns, means of triplicate determinations; bar, SD. *, P < 0.05 compared with control; †, P < 0.05 compared with NaBT or H2O2 alone.
RESULTS
PMA induced FLIP mRNA expression in Caco-2 cells
PKC regulates expression of certain anti-apoptotic proteins.20, 34, 44, 45 For example, activation of PKCδ/NF-κB increases cIAP-2, inhibitor of apoptosis protein.20 Reduction of PKCα levels decreases Bcl-xL content and leads to increased sensitivity to apoptosis in hepatic epithelial cells.45 In this study, we examined the effect of PMA treatment on the levels of FLIP mRNA in human colon cancer cell line Caco-2. As shown in Fig. 1A, PMA treatment induced the expression of multiple splice variants FLIP mRNA in a time-dependent fashion. Induction of FLIP occurred by 2 h with maximal expression at 8 h. In addition, PMA induced FLIP expression in a dose-dependent fashion with concentrations as low as 5 nM resulting in an increase in expression (Fig. 1B).
Figure 1. PMA treatment increases FLIP mRNA level in Caco-2 cells.
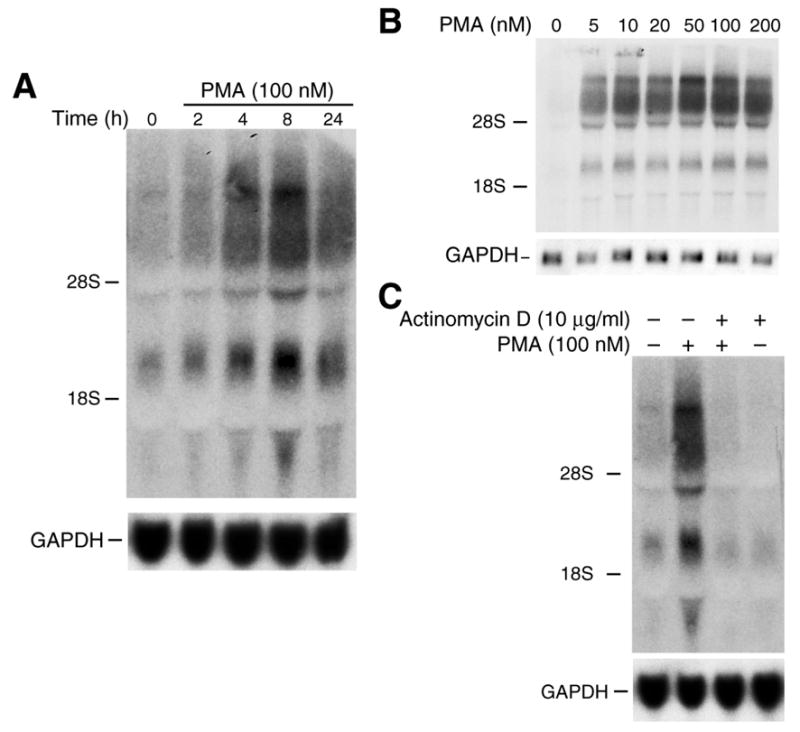
A. Northern blot of total RNA (40 μg) from Caco-2 cells treated with PMA (100 nM) for various times and hybridized to a 1.5 kb fragment of FLIP cDNA probe. The same membrane was reprobed with a human GAPDH probe as an internal loading control. B. To determine whether induction of FLIP mRNA by PMA occurs in a dose-dependent manner, Caco-2 cells were treated with various concentrations of PMA for 8 h; RNA was extracted and Northern blot performed as above. C. Cells were treated with 0 or 100 nM PMA and actinomycin D (10 μg/ml) for 8 h. Total cellular RNA was extracted, and Northern blot was performed as described above.
Steady-state levels of mRNAs may be modulated by transcriptional or post-transcriptional mechanisms. To determine the mechanisms for PMA-mediated FLIP induction, Caco-2 cells were exposed to PMA (100 nM) for 8 h in the presence or absence of actinomycin D (10 μg/ml), which inhibits transcription.46 Total cellular RNA was extracted and Northern analysis was performed (Fig. 1C). Actinomycin D alone slightly decreased FLIP mRNA levels which is consistent with findings of other investigators utilizing actinomycin D to assess expression of various genes.47–51 The increased expression of FLIP mRNA splice variants by PMA was completely blocked by co-incubation with actinomycin D, suggesting transcriptional regulation as the mechanism for FLIP induction by PMA (Fig. 1C).
Regulation of PMA-stimulated FLIP expression through the PKC pathway
PMA can stimulate downstream gene expression through the PKC, PI3-kinase or MAPK pathways, depending on the cell type.37, 52 Therefore, we examined which signaling pathway is involved in the PMA-induced FLIP expression.
Caco-2 cells were pretreated with the MEK/MAPK inhibitor PD98059 (10–50 μM) for 1 h followed by combination treatment with PMA (100 nM) for 2 h; activation of MEK/MAPK was assayed by the determination of ERK1/2 phosphorylation using anti-phospho-ERK1/2 antibody. Treatment with PMA induced ERK1/2 phosphorylation and this induction was attenuated by pretreatment with PD98059 (Fig. 2A). Treatment with PMA (100 nM) for 8 h increased FLIP mRNA expression detected by Northern blot; however, pretreatment with PD98059 (10–50 μM) did not affect PMA-mediated FLIP mRNA induction (Fig. 2B). To determine the effect of PMA on FLIP protein levels, whole cell protein was extracted and FLIP protein was analyzed by Western blot using a specific anti-human FLIP antibody. At least two splice variants of FLIP are expressed in a variety of cells: the full-length form, p55-FLIPL, and the shorter form, p28-FLIPS.8, 53–55 Both FLIPL and FLIPS are recruited into the death-inducing signaling complex (DISC) and block death receptor-mediated apoptosis.8, 54 As shown in Fig. 2C, PMA induced both FLIPL and FLIPS expression in Caco-2 cells. Consistently, pretreatment with PD98059 did not affect the PMA increased FLIP expression. Together, these results suggested that PMA-induced FLIP expression did not involve MEK/MAPK.
Figure 2. MEK/MAPK and PI3-kinase may not be involved in PMA-induced FLIP expression.
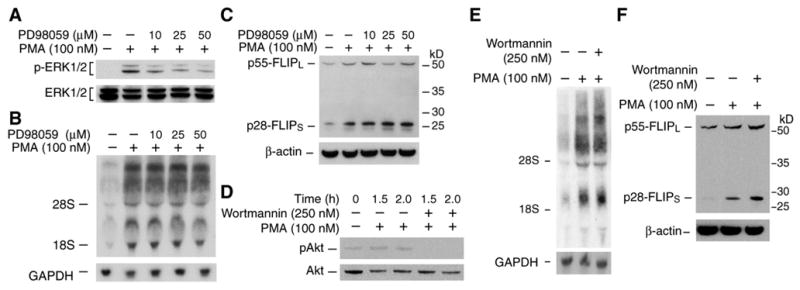
A. Caco-2 cells were pre-treated with the MEK1 inhibitor PD98059 (10–50 μM) for 1 h followed by treatment with PMA (100 nM) alone or together with PD98059 for 2 h. Whole cell protein was extracted and phospho-ERK1/2 level was determined by Western blot. B&C. Caco-2 cells were pre-treated with PD98059 (10–50 μM) for 1 h followed by treatment with PMA (100 nM) alone or together with PD98059. After incubation for 8 h, total RNA was extracted and FLIP mRNA level was determined by Northern blot (B); whole cell protein was extracted and FLIP level was determined by Western blot (C). D. Caco-2 cells were pre-treated with the PI3-kinase inhibitor wortmannin (250 nM) for 1 h followed by treatment with PMA (100 nM) alone or together with wortmannin for 1.5 or 2 h. Whole cell protein was extracted and phospho-Akt level was determined by Western blot. E&F. Caco-2 cells were pre-treated with wortmannin (250 nM) for 1 h followed by treatment with PMA (100 nM) alone or together with wortmannin. After incubation for 8 h, total RNA was extracted and FLIP mRNA level was determined by Northern blot (E); whole cell protein was extracted and FLIP expression was determined by Western blot (F).
To determine whether PI3-kinase plays a role in the FLIP induction by PMA, Caco-2 cells were pretreated with the PI3-kinase inhibitor wortmannin (250 nM) for 1 h followed by combination treatment with PMA (100 nM) for various times; activation of the PI3-kinase pathway was assayed by determining Akt phosphorylation, a downstream target of PI3-kinase, using anti-phospho-Akt antibody. Treatment with PMA increased Akt phosphorylation and this induction was completely blocked by pretreatment with wortmannin (Fig. 2D). In contrast, pretreatment with wortmannin (250 nM) did not affect either PMA-mediated FLIP mRNA (Fig. 2E) or protein induction (Fig. 2F), suggesting that the PI3-kinase pathway may not play a role in PMA-mediated FLIP induction.
To determine weather PKC is involved in FLIP inhibition, we next assessed the effect of various PKC inhibitors on PMA-induced FLIP mRNA expression in Caco-2 cells. Caco-2 cells were treated with PMA (100 nM) in the presence or absence of the PKC inhibitors Gö6983 (2 μM), Ro-31-8220 (2 μM), or Gö6976 (2 μM). Both Gö6983 and Ro-31-8220 completely attenuated PMA-induced FLIP mRNA expression (Fig. 3A) while the Ca2+-dependent PKC inhibitor Gö6976 has a minor effect. These results suggest a major role for Ca2+-independent novel PKC in PMA-mediated regulation of FLIP expression.
Figure 3. Effect of protein kinase C inhibitors on PMA-induced FLIP expression.
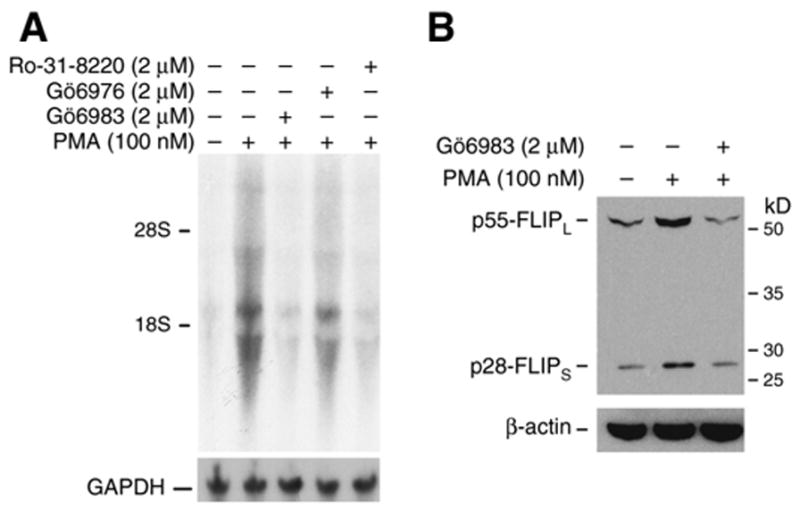
A. Caco-2 cells were treated with PMA (100 nM) alone or together with either Gö6983 (2 μM), Gö6976 (2 μM), or Ro-31-8220 (2 μM). After 8 h treatment, total RNA was extracted and FLIP mRNA level was determined by Northern blot. B. Caco-2 cells were treated with PMA (100 nM) alone or together with Gö6983 (2 μM). After 8 h treatment, whole cell protein was extracted and FLIP protein level was determined by Western blot.
We have shown induction of FLIP mRNA expression through a PKC-mediated mechanism. To determine the effect on FLIP protein levels, Caco-2 cells were treated with PMA (100 nM) with or without the PKC inhibitor Gö6983 (2 μM) for 8 h. PMA increased FLIP expression was blocked by Gö6983 (Fig. 3B). Taken together, our results identify regulation of both FLIP mRNA and protein by PKC.
PMA induces FLIP mRNA expression in KM12C, KM20, HT29, and HCT116 colon cancer cells
To determine whether PKC-mediated FLIP induction occurs in other colon cancer cell lines, FLIP expression was assessed in the human colon cancer cell lines KM12C, KM20, HT29, and HCT116 after treatment with PMA (100 nM) for 8 h with or without the PKC inhibitor Gö6983 (2 μM) (Fig. 4). PMA induced FLIP mRNA expression in all four cell lines compared with control (Fig. 4A). Interestingly, there was only one major splice variant which was expressed in these cell lines. Similar to Caco-2 cells, FLIP mRNA induction by PMA was blocked by Gö6883. Analysis of FLIP protein expression by Western blot also showed an induction of only p28-FLIPs in HT29 and KM20 cells; this induction was attenuated by Gö6883 (Fig. 4B). These results confirm the findings in the Caco-2 cell line and suggest a regulation of FLIP expression by the PKC pathway in human colon cancer cells
Figure 4. PMA induces FLIP mRNA expression in KM12C, KM20, HT29, and HCT116 colon cancer cells.
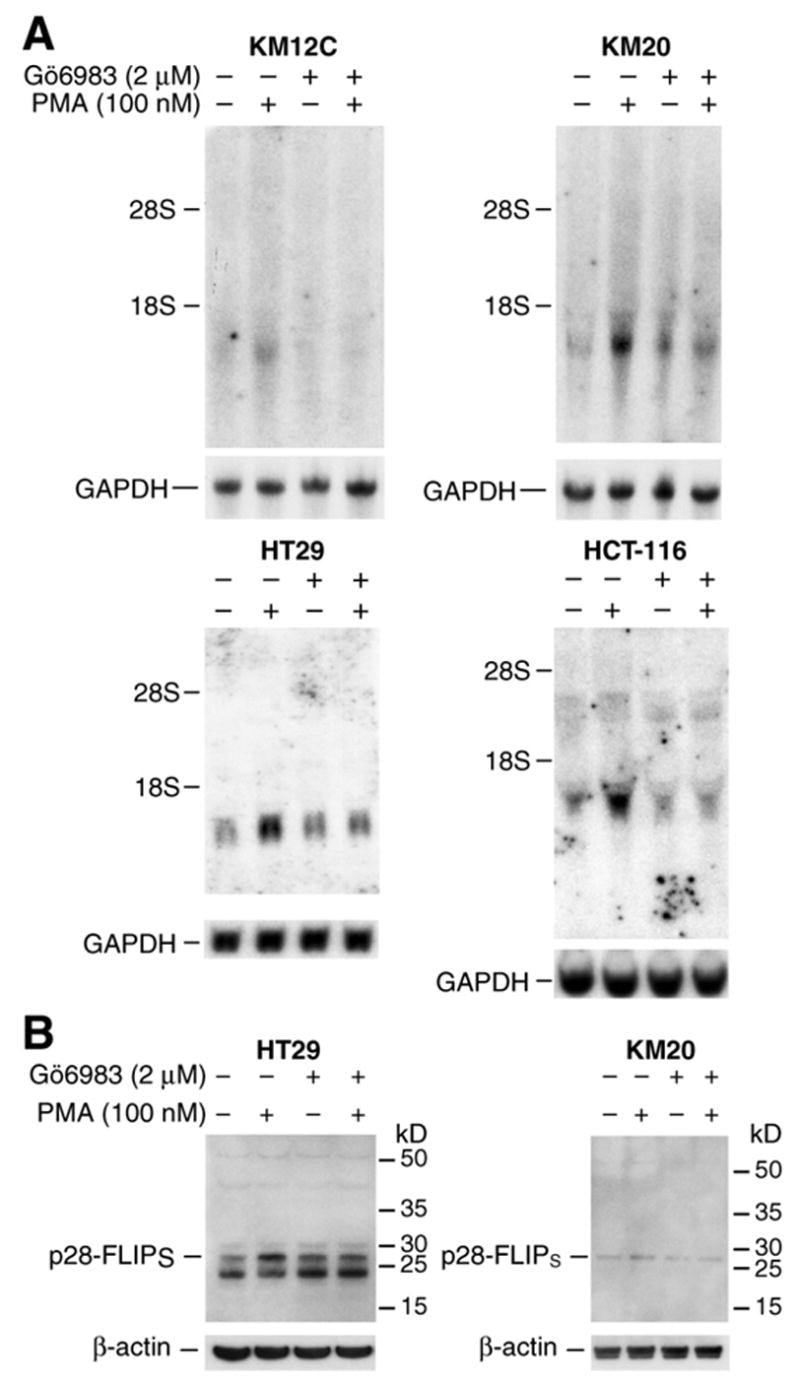
A. KM12C, KM20, HT29, and HCT116 colon cancer cells were treated with PMA (100 nM) alone or together with PKC inhibitor Gö6983 (2 μM) for 8 h. Total RNA was extracted and FLIP mRNA level was determined by Northern blot. B. HT29 and KM20 cells were treated with PMA (100 nM) alone or together with Gö6983 (2 μM) for 8 h. Whole cell protein was extracted and FLIP protein level was determined by Western blot.
PKCδ contributes to PMA-induced FLIP expression
The PKCδ isoform contributes to the regulation of various genes, such as ICAM-1 56, cyclin D1 57, GM-CSF and RANTES expression.58 Recently, we have found that PMA induces cIAP-2 expression through PKCδ activation in colon cancer cells.20 To determine whether PKCδ is involved in the induction of FLIP, Caco-2 cells were pretreated with the PKCδ-selective inhibitor rottlerin for 30 min prior to the addition of PMA; cells were harvested 4 h later and RNA analyzed by Northern blot (Fig. 5A). Pretreatment with rottlerin resulted in a complete inhibition of PMA-mediated FLIP induction in Caco-2 cells cultured in serum-free medium for 24 h prior to treatment. The inhibition of PMA-induced FLIP mRNA expression by rottlerin suggests that the PKCδ isoenzyme may play a role in PMA-mediated FLIP induction.
To further confirm the role of PKCδ in FLIP mRNA induction, Caco-2 cells were transfected with control siRNA or siRNA targeting PKCδ for 48 h followed by PMA treatment for 4 h. As shown in Fig. 5B (top), transfection with the PKCδ siRNA suppressed FLIP mRNA induction by PMA. PKCδ protein inhibition with PKCδ siRNA treatment was confirmed by Western blot; PKCα expression, which was decreased by PMA, was not affected by PKCδ siRNA, thus confirming selective inhibition of PKCδ by the PKCδ siRNA (Fig. 5B; bottom). In order to determine whether this regulation was limited to Caco-2 cells or occurs in other colon cancer cells, HT29 cells were transfected with control siRNA or siRNA targeting the PKCδ gene for 48 h followed by PMA treatment for 4 h; transfection with the PKCδ siRNA attenuated PMA-mediated FLIP induction with PMA (Fig. 5C). Taken together, these results, using complementary approaches (i.e., chemical inhibition of PKCδ and transfection with PKCδ siRNA) demonstrate a contributory role for PKCδ in PMA-mediated FLIP induction in human colon cancer cells.
We have shown that activation of PKCδ increased expression of the anti-apoptotic proteins, FLIP and cIAP-2 20, in colon cancer cells. To explore the biological consequences of these changes in protein expression levels, we investigated NaBT- or H2O2-induced apoptotic changes in Caco-2 cells that were transfected with siRNA to PKCδ. Treatment with either NaBT or H2O2 induced obvious cell death; this effect was further enhanced in the cells transfected with PKCδ siRNA (Fig. 5D). These results suggest a moderate anti-apoptotic role of PKCδ, which was associated with regulation of FLIP and cIAP-2 expression.
PMA-mediated FLIP induction is dependent on NF-κB activation
FLIP expression is preferentially up-regulated by stimuli that activate NF-κB.12, 13 To investigate whether NF-κB activation is involved in PKC-mediated FLIP induction in colon cancer cells, we used either MG132, a proteasome inhibitor that inhibits IκB degradation 59, or the antioxidant PDTC, which functions as an NF-κB inhibitor by blocking the dissociation of the NF-κB·IκB complex 60, or gliotoxin, a potent inhibitor of NF-κB.61 Caco-2 cells were preincubated 30 min, with or without MG132, PDTC or gliotoxin, followed by the addition of PMA (100 nM) in the presence or absence of the inhibitor; FLIP expression was assessed by Northern blot. Treatment with either MG132 (Fig. 6A), PDTC or gliotoxin (Fig. 6B) inhibited PMA-induced upregulation of FLIP.
To further confirm the involvement of NF-κB activation in PMA-induced FLIP expression, an adenovirus expressing the superrepressor of IκB-α (IκB-AA) was used to infect Caco-2 cells.62 First, we assessed the inhibitory role of IκB-AA on NF-κB transactivation by transfection of the NF-κB-luciferase plasmid, which contains four tandem copies of the NF-κB consensus sequence. Caco-2 cells were then either infected with the adenovirus encoding IκB-AA or the adenoviral control vector encoding GFP. Infection was carried out for 1 h followed by the replacement of fresh medium and an additional 24 h of incubation. Cells were then treated with PMA (100 nM) for 16 h. Treatment with PMA significantly increased NF-κB transactivation compared with control (Fig. 6C, top panel). Moreover, this induction was completely blocked by infection with adenovirus encoding IκB-AA. Next, we assessed the effect of IκB-AA on PMA-mediated FLIP expression. Caco-2 cells were infected with adenovirus encoding IκB-AA or the adenoviral control vector encoding GFP. Cells were treated with PMA or vehicle and protein and RNA extracted for Western and Northern blot analysis, respectively (Fig. 6C, middle panel & bottom panel). Consistent with the effect observed previously by chemical inhibitors, IκB-AA overexpression attenuated PMA-induced FLIP mRNA and protein expression. IκB-AA expression was confirmed by Western blot (Fig. 6C, middle panel). Notably, IκB-AA completely reversed the PMA-induced NF-κB reporter activity but did not completely reverse PMA-induced FLIP expression, suggesting NF-κB-dependent and NF-κB-independent regulation of FLIP expression by PKCδ.
Finally, we determined whether inhibition of PKC results in decreased NF-κB transactivation by transfection of the NF-κB-luciferase plasmid and treatment with PMA in the presence or absence of GF109203x. Treatment with PMA (100 nM) significantly increased NF-κB transactivation compared with control (Fig. 6D). Moreover, this induction was completely blocked by treatment with GF109203x. We have found that PKCδ significantly increased NF-κB transactivation in Caco-2 cells.20 Taken together, these findings suggest the regulation of FLIP induction in colon cancer cells through PKCδ-mediated NF-κB activation.
DISCUSSION
Previously, we demonstrated regulation of the anti-apoptotic cIAP-2 protein through a PKCδ/NF-κB-dependent pathway in human colon cancer cells.20 Here, we show that the PKCδ/NF-κB-dependent pathway also regulates expression of the anti-apoptotic FLIP gene. We demonstrate induction of FLIP expression by the phorbol ester, PMA, in human colon cancer cell lines. Inhibition of PKC by inhibitors markedly decreased PMA-mediated FLIP induction strongly suggesting that the PKC pathway plays a critical role in the regulation of FLIP expression levels in human colon cancer.
The PKC family consists of conventional PKC isoforms α, βI, βII and γ that are Ca2+-dependent and PMA-responsive, novel PKC isoforms δ, ε, η and θ that are Ca2+-independent and atypical PKC isoforms which are not responsive to PMA.34, 44 To delineate the PKC isoforms involved in PMA-mediated FLIP stimulation, we used a battery of PKC inhibitors. Isoform-selective PKC inhibitors Gö6983 and rottlerin, both of which inhibit PKCδ 63, blocked FLIP induction by PMA. The role of PKCδ in PMA-mediated FLIP expression was further confirmed using PKCδ siRNA. Gö6976, which inhibits PKCα, βI and μ but not PKCδ 63 had minor effect on PMA induction of FLIP. Although PKD, described originally as a novel PKC and named PKCμ, has been shown to upregulate FLIP expression in pancreatic adenocarcinoma cell lines 11, PKD does not appear to play a predominant role in the regulation of FLIP in human colon cancer as noted by the fact that treatment with Gö6976, which inhibits PKD (IC50 = 20 nM),64 had a minor effect on FLIP expression, whereas Gö6983, which does not inhibit PKD (IC50 of 20 μM) at the dosage used in this study,64 blocked PMA induction of FLIP.
The role of PKCδ in tumor cell lines is of particular interest, since contradictory reports exist in the literature regarding the role of PKCδ in cell survival and apoptosis.65 PKCδ has been implicated as a prosurvival factor in some tumor cells 19, 21 whereas other studies suggest a pro-apoptotic effect of PKCδ, thus demonstrating that the role of PKCδ is highly dependent on cell type and cellular context. For instance, PKCδ overexpression inhibits Sindbis virus-induced apoptosis but enhances etoposide-induced apoptosis in the same cell line.66, 67 A recent study showed that increased expression of the PKCδ isoform enhanced the rate of apoptosis in the Caco-2 human colon cancer cell line, which was further augmented by phorbol ester treatment.24 However, we demonstrate that activation of PKCδ increased expression of the anti-apoptotic proteins, FLIP and cIAP-2 20, levels in colon cancer cells, including Caco-2. In agreement with our results, PKCδ has been identified as a prosurvival factor in human breast tumor cell lines.19 PKCδ is commonly expressed in multiple myeloma cells and its downregulation by rottlerin results in apoptosis.68 These findings emphasize an important role of PKCδ in the regulation of anti-apoptotic molecules in certain cancer cells.
The transcription factor NF-κB controls transcription of numerous genes including genes fundamental for survival, such as TRAF1, TRAF-2, cIAP-1, cIAP-2, and FLIP.12, 32 Indeed, PKCδ/NF-κB plays a key role in the regulation of various genes, such as ICAM-1 56, cyclin D1 57, GM-CSF, and RANTES.58 We have shown that the presence of p65 RelA and p50 NF-κB1 in the PMA-induced DNA binding complex and overexpression of PKCδ significantly increased NF-κB transactivation.20 Consistent with these results, we found that NF-κB inhibition by either MG132 or PDTC abolished or significantly reduced PMA-induced FLIP expression, indicating that NF-κB is also involved in FLIP activation by PKCδ. Furthermore, the PKC inhibitor GF109203x abolished NF-κB transactivation by PMA and blocked PMA-induced FLIP expression. Collectively, these findings suggest a regulation of survival-associated gene expression through the PKCδ/NF-κB signaling pathway.
PI3-kinase and MAPK, which are critical regulators of cell proliferation and survival, have been reported to regulate the expression of FLIP in some cells.2, 69, 70 PMA can stimulate the MAPK/ERK and PI3-kinase pathways.37, 52 However, PI3-kinase and MAPK/ERK may not be involved in PMA-mediated FLIP induction in colon cancer cells, since either wortmannin, a PI3-kinase inhibitor, or PD98059, a MEK inhibitor, had no effect on PMA-induced FLIP expression in Caco-2 cells. Thus, FLIP induction noted in colon cancer cells by PMA was through PKC.
In conclusion, we demonstrate FLIP induction in human colon cancer cells through PKC activation by PMA. Importantly, this induction occurs through a PKCδ/NF-κB-dependent pathway. Together, our findings suggest that PKCδ plays a critical role in the regulation of NF-κB-dependent anti-apoptotic gene expression. The selective modulation of FLIP levels in certain cancers may be useful in sensitizing resistant cancers to the effects of agents which induce apoptosis through the Fas or TRAIL pathway.
Acknowledgments
The authors thank Tatsuo Uchida for statistical analysis, and Eileen Figueroa and Karen Martin for manuscript preparation. This work was supported by grants R01 DK48498 and P01 DK35608 from the National Institutes of Health.
Abbreviations
- FLIP
FLICE-like inhibitory protein
- TNF
tumor necrosis factor
- PI3-kinase
phosphatidylinositol-3 kinase
- PKD
protein kinase D
- PMA
phorbol 12-myristate 13-acetate
- PDGF
platelet-derived growth factor
- PKC
protein kinase C
References
- 1.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388(6638):190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 2.Panka DJ, Mano T, Suhara T, Walsh K, Mier JW. Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP expression in tumor cells. J Biol Chem. 2001;276(10):6893–6. doi: 10.1074/jbc.C000569200. [DOI] [PubMed] [Google Scholar]
- 3.Shu HB, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6(6):751–63. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 4.Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop BF, Peterson EP, et al. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5(4):271–88. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- 5.Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21(24):8247–54. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonsson G, Paulie S, Grandien A. High level of cFLIP correlates with resistance to death receptor-induced apoptosis in bladder carcinoma cells. Anticancer Res. 2003;23(2B):1213–8. [PubMed] [Google Scholar]
- 7.Tschopp J, Irmler M, Thome M. Inhibition of fas death signals by FLIPs. Curr Opin Immunol. 1998;10(5):552–8. doi: 10.1016/s0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- 8.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274(3):1541–8. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 9.Kim Y, Suh N, Sporn M, Reed JC. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem. 2002;277(25):22320–9. doi: 10.1074/jbc.M202458200. [DOI] [PubMed] [Google Scholar]
- 10.Hyer ML, Sudarshan S, Kim Y, Reed JC, Dong JY, Schwartz DA, Norris JS. Downregulation of c-FLIP sensitizes DU145 prostate cancer cells to Fas-mediated apoptosis. Cancer Biol Ther. 2002;1(4):401–6. doi: 10.4161/cbt.1.4.15. [DOI] [PubMed] [Google Scholar]
- 11.Trauzold A, Schmiedel S, Sipos B, Wermann H, Westphal S, Roder C, Klapper W, Arlt A, Lehnert L, Ungefroren H, Johannes FJ, Kalthoff H. PKCmu prevents CD95-mediated apoptosis and enhances proliferation in pancreatic tumour cells. Oncogene. 2003;22(55):8939–47. doi: 10.1038/sj.onc.1207001. [DOI] [PubMed] [Google Scholar]
- 12.Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21(16):5299–305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21(12):3964–73. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blobe GC, Obeid LM, Hannun YA. Regulation of protein kinase C and role in cancer biology. Cancer Metastasis Rev. 1994;13(3–4):411–31. doi: 10.1007/BF00666107. [DOI] [PubMed] [Google Scholar]
- 15.Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10(8):529–42. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 16.Meng XW, Heldebrant MP, Kaufmann SH. Phorbol 12-myristate 13-acetate inhibits death receptor-mediated apoptosis in Jurkat cells by disrupting recruitment of Fas-associated polypeptide with death domain. J Biol Chem. 2002;277(5):3776–83. doi: 10.1074/jbc.M107218200. [DOI] [PubMed] [Google Scholar]
- 17.Selbie LA, Schmitz-Peiffer C, Sheng Y, Biden TJ. Molecular cloning and characterization of PKC iota, an atypical isoform of protein kinase C derived from insulin-secreting cells. J Biol Chem. 1993;268(32):24296–302. [PubMed] [Google Scholar]
- 18.Mochly-Rosen D, Gordon AS. Anchoring proteins for protein kinase C: a means for isozyme selectivity. Faseb J. 1998;12(1):35–42. [PubMed] [Google Scholar]
- 19.McCracken MA, Miraglia LJ, McKay RA, Strobl JS. Protein kinase C delta is a prosurvival factor in human breast tumor cell lines. Mol Cancer Ther. 2003;2(3):273–81. [PubMed] [Google Scholar]
- 20.Wang Q, Wang X, Evers BM. Induction of cIAP-2 in human colon cancer cells through PKC delta/NF-kappa B. J Biol Chem. 2003;278(51):51091–9. doi: 10.1074/jbc.M306541200. [DOI] [PubMed] [Google Scholar]
- 21.Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003;63(4):780–6. [PubMed] [Google Scholar]
- 22.Kang Y, Park JS, Kim SH, Shin YJ, Kim W, Joo HJ, Chun JS, Kim HJ, Ha MJ. Overexpression of protein kinase C delta represses expression of proliferin in NIH3T3 cells that regulates cell proliferation. Mol Cell Biol Res Commun. 2000;4(3):181–7. doi: 10.1006/mcbr.2001.0276. [DOI] [PubMed] [Google Scholar]
- 23.Deszo EL, Brake DK, Cengel KA, Kelley KW, Freund GG. CD45 negatively regulates monocytic cell differentiation by inhibiting phorbol 12-myristate 13-acetate-dependent activation and tyrosine phosphorylation of protein kinase Cdelta. J Biol Chem. 2001;276(13):10212–7. doi: 10.1074/jbc.M010589200. [DOI] [PubMed] [Google Scholar]
- 24.Cerda SR, Bissonnette M, Scaglione-Sewell B, Lyons MR, Khare S, Mustafi R, Brasitus TA. PKC-delta inhibits anchorage-dependent and -independent growth, enhances differentiation, and increases apoptosis in CaCo-2 cells. Gastroenterology. 2001;120(7):1700–12. doi: 10.1053/gast.2001.24843. [DOI] [PubMed] [Google Scholar]
- 25.Denning MF, Dlugosz AA, Threadgill DW, Magnuson T, Yuspa SH. Activation of the epidermal growth factor receptor signal transduction pathway stimulates tyrosine phosphorylation of protein kinase C delta. J Biol Chem. 1996;271(10):5325–31. doi: 10.1074/jbc.271.10.5325. [DOI] [PubMed] [Google Scholar]
- 26.Kronfeld I, Kazimirsky G, Lorenzo PS, Garfield SH, Blumberg PM, Brodie C. Phosphorylation of protein kinase Cdelta on distinct tyrosine residues regulates specific cellular functions. J Biol Chem. 2000;275(45):35491–8. doi: 10.1074/jbc.M005991200. [DOI] [PubMed] [Google Scholar]
- 27.Vancurova I, Miskolci V, Davidson D. NF-kappa B activation in tumor necrosis factor alpha-stimulated neutrophils is mediated by protein kinase Cdelta. Correlation to nuclear Ikappa Balpha. J Biol Chem. 2001;276(23):19746–52. doi: 10.1074/jbc.M100234200. [DOI] [PubMed] [Google Scholar]
- 28.Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, Kato RM, Kang S, Patrone L, Wall R, Teitell M, Leitges M, et al. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. 2002;3(8):780–6. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 29.Storz P, Toker A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. Embo J. 2003;22(1):109–20. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilpatrick LE, Lee JY, Haines KM, Campbell DE, Sullivan KE, Korchak HM. A role for PKC-delta and PI 3-kinase in TNF-alpha-mediated antiapoptotic signaling in the human neutrophil. Am J Physiol Cell Physiol. 2002;283(1):C48–57. doi: 10.1152/ajpcell.00385.2001. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Wang Q, Hu W, Evers BM. Regulation of phorbol ester-mediated TRAF1 induction in human colon cancer cells through a PKC/RAF/ERK/NF-kappaB-dependent pathway. Oncogene. 2004;23(10):1885–95. doi: 10.1038/sj.onc.1207312. [DOI] [PubMed] [Google Scholar]
- 32.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281(5383):1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 33.Yang BF, Xiao C, Roa WH, Krammer PH, Hao C. Calcium/calmodulin-dependent protein kinase II regulation of c-FLIP expression and phosphorylation in modulation of Fas-mediated signaling in malignant glioma cells. J Biol Chem. 2003;278(9):7043–50. doi: 10.1074/jbc.M211278200. [DOI] [PubMed] [Google Scholar]
- 34.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. Faseb J. 1995;9(7):484–96. [PubMed] [Google Scholar]
- 35.Jaken S. Protein kinase C isozymes and substrates. Curr Opin Cell Biol. 1996;8(2):168–73. doi: 10.1016/s0955-0674(96)80062-7. [DOI] [PubMed] [Google Scholar]
- 36.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. Embo J. 2000;19(4):496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee HW, Ahn DH, Crawley SC, Li JD, Gum JR, Jr, Basbaum CB, Fan NQ, Szymkowski DE, Han SY, Lee BH, Sleisenger MH, Kim YS. Phorbol 12-myristate 13-acetate up-regulates the transcription of MUC2 intestinal mucin via Ras, ERK, and NF-kappa B. J Biol Chem. 2002;277(36):32624–31. doi: 10.1074/jbc.M200353200. [DOI] [PubMed] [Google Scholar]
- 38.Li J, O’Connor KL, Hellmich MR, Greeley GH, Jr, Townsend CM, Jr, Evers BM. The role of protein kinase D in neurotensin secretion mediated by protein kinase C-alpha/-delta and Rho/Rho kinase. J Biol Chem. 2004;279(27):28466–74. doi: 10.1074/jbc.M314307200. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q, Wang X, Hernandez A, Kim S, Evers BM. Inhibition of the phosphatidylinositol 3-kinase pathway contributes to HT29 and Caco-2 intestinal cell differentiation. Gastroenterology. 2001;120(6):1381–92. doi: 10.1053/gast.2001.24044. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Ji Y, Wang X, Evers BM. Isolation and molecular characterization of the 5′-upstream region of the human TRAIL gene. Biochem Biophys Res Commun. 2000;276(2):466–71. doi: 10.1006/bbrc.2000.3512. [DOI] [PubMed] [Google Scholar]
- 41.Wang Q, Ding Q, Dong Z, Ehlers RA, Evers BM. Downregulation of mitogen-activated protein kinases in human colon cancers. Anticancer Res. 2000;20(1A):75–83. [PubMed] [Google Scholar]
- 42.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 43.Wang Q, Li N, Wang X, Kim MM, Evers BM. Augmentation of sodium butyrate-induced apoptosis by phosphatidylinositol 3′-kinase inhibition in the KM20 human colon cancer cell line. Clin Cancer Res. 2002;8(6):1940–7. [PubMed] [Google Scholar]
- 44.Hug H, Sarre TF. Protein kinase C isoenzymes: divergence in signal transduction? Biochem J. 1993;291 (Pt 2):329–43. doi: 10.1042/bj2910329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsieh YC, Jao HC, Yang RC, Hsu HK, Hsu C. Suppression of protein kinase Calpha triggers apoptosis through down-regulation of Bcl-xL in a rat hepatic epithelial cell line. Shock. 2003;19(6):582–7. doi: 10.1097/01.shk.0000065705.84144.ed. [DOI] [PubMed] [Google Scholar]
- 46.Shi MM, Kugelman A, Iwamoto T, Tian L, Forman HJ. Quinone-induced oxidative stress elevates glutathione and induces gamma-glutamylcysteine synthetase activity in rat lung epithelial L2 cells. J Biol Chem. 1994;269(42):26512–7. [PubMed] [Google Scholar]
- 47.Abbas T, White D, Hui L, Yoshida K, Foster DA, Bargonetti J. Inhibition of human p53 basal transcription by down-regulation of protein kinase Cdelta. J Biol Chem. 2004;279(11):9970–7. doi: 10.1074/jbc.M306979200. [DOI] [PubMed] [Google Scholar]
- 48.Fang X, Yu S, Bast RC, Liu S, Xu HJ, Hu SX, LaPushin R, Claret FX, Aggarwal BB, Lu Y, Mills GB. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. J Biol Chem. 2004;279(10):9653–61. doi: 10.1074/jbc.M306662200. [DOI] [PubMed] [Google Scholar]
- 49.Jiang JX, Choi RC, Siow NL, Lee HH, Wan DC, Tsim KW. Muscle induces neuronal expression of acetylcholinesterase in neuron-muscle co-culture: transcriptional regulation mediated by cAMP-dependent signaling. J Biol Chem. 2003;278(46):45435–44. doi: 10.1074/jbc.M306320200. [DOI] [PubMed] [Google Scholar]
- 50.Shanmugam N, Kim YS, Lanting L, Natarajan R. Regulation of cyclooxygenase-2 expression in monocytes by ligation of the receptor for advanced glycation end products. J Biol Chem. 2003;278(37):34834–44. doi: 10.1074/jbc.M302828200. [DOI] [PubMed] [Google Scholar]
- 51.Wang Z, Malone MH, He H, McColl KS, Distelhorst CW. Microarray analysis uncovers the induction of the proapoptotic BH3-only protein Bim in multiple models of glucocorticoid-induced apoptosis. J Biol Chem. 2003;278(26):23861–7. doi: 10.1074/jbc.M301843200. [DOI] [PubMed] [Google Scholar]
- 52.Park SJ, Kang SY, Kim NS, Kim HM. Phosphatidylinositol 3-kinase regulates PMA-induced differentiation and superoxide production in HL-60 cells. Immunopharmacol Immunotoxicol. 2002;24(2):211–26. doi: 10.1081/iph-120003751. [DOI] [PubMed] [Google Scholar]
- 53.Kataoka T, Tschopp J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol. 2004;24(7):2627–36. doi: 10.1128/MCB.24.7.2627-2636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276(23):20633–40. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 55.Hennino A, Berard M, Casamayor-Palleja M, Krammer PH, Defrance T. Regulation of the Fas death pathway by FLICE-inhibitory protein in primary human B cells. J Immunol. 2000;165(6):3023–30. doi: 10.4049/jimmunol.165.6.3023. [DOI] [PubMed] [Google Scholar]
- 56.Rahman A, Anwar KN, Uddin S, Xu N, Ye RD, Platanias LC, Malik AB. Protein kinase C-delta regulates thrombin-induced ICAM-1 gene expression in endothelial cells via activation of p38 mitogen-activated protein kinase. Mol Cell Biol. 2001;21(16):5554–65. doi: 10.1128/MCB.21.16.5554-5565.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Page K, Li J, Corbit KC, Rumilla KM, Soh JW, Weinstein IB, Albanese C, Pestell RG, Rosner MR, Hershenson MB. Regulation of airway smooth muscle cyclin D1 transcription by protein kinase C-delta. Am J Respir Cell Mol Biol. 2002;27(2):204–13. doi: 10.1165/ajrcmb.27.2.20010016oc. [DOI] [PubMed] [Google Scholar]
- 58.Page K, Li J, Zhou L, Iasvovskaia S, Corbit KC, Soh JW, Weinstein IB, Brasier AR, Lin A, Hershenson MB. Regulation of airway epithelial cell NF-kappa B-dependent gene expression by protein kinase C delta. J Immunol. 2003;170(11):5681–9. doi: 10.4049/jimmunol.170.11.5681. [DOI] [PubMed] [Google Scholar]
- 59.Jiang B, Xu S, Hou X, Pimentel DR, Brecher P, Cohen RA. Temporal control of NF-kappaB activation by ERK differentially regulates interleukin-1beta-induced gene expression. J Biol Chem. 2004;279(2):1323–9. doi: 10.1074/jbc.M307521200. [DOI] [PubMed] [Google Scholar]
- 60.Snyder JG, Prewitt R, Campsen J, Britt LD. PDTC and Mg132, inhibitors of NF-kappaB, block endotoxin induced vasodilation of isolated rat skeletal muscle arterioles. Shock. 2002;17(4):304–7. doi: 10.1097/00024382-200204000-00011. [DOI] [PubMed] [Google Scholar]
- 61.Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, Haslett C, Rossi AG. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J Biol Chem. 1999;274(7):4309–18. doi: 10.1074/jbc.274.7.4309. [DOI] [PubMed] [Google Scholar]
- 62.Kim S, Domon-Dell C, Kang J, Chung DH, Freund JN, Evers BM. Down-regulation of the tumor suppressor PTEN by the tumor necrosis factor-alpha/nuclear factor-kappaB (NF-kappaB)-inducing kinase/NF-kappaB pathway is linked to a default IkappaB-alpha autoregulatory loop. J Biol Chem. 2004;279(6):4285–91. doi: 10.1074/jbc.M308383200. [DOI] [PubMed] [Google Scholar]
- 63.Shapiro BA, Ray S, Jung E, Allred WT, Bollag WB. Putative conventional protein kinase C inhibitor Godecke 6976 [12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)py rrolo(3,4-c)-carbazole] stimulates transglutaminase activity in primary mouse epidermal keratinocytes. J Pharmacol Exp Ther. 2002;302(1):352–8. doi: 10.1124/jpet.302.1.352. [DOI] [PubMed] [Google Scholar]
- 64.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392(2):77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 65.Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003;8(1):19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- 66.Zrachia A, Dobroslav M, Blass M, Kazimirsky G, Kronfeld I, Blumberg PM, Kobiler D, Lustig S, Brodie C. Infection of glioma cells with Sindbis virus induces selective activation and tyrosine phosphorylation of protein kinase C delta. Implications for Sindbis virus-induced apoptosis. J Biol Chem. 2002;277(26):23693–701. doi: 10.1074/jbc.M111658200. [DOI] [PubMed] [Google Scholar]
- 67.Blass M, Kronfeld I, Kazimirsky G, Blumberg PM, Brodie C. Tyrosine phosphorylation of protein kinase Cdelta is essential for its apoptotic effect in response to etoposide. Mol Cell Biol. 2002;22(1):182–95. doi: 10.1128/MCB.22.1.182-195.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ni H, Ergin M, Tibudan SS, Denning MF, Izban KF, Alkan S. Protein kinase C-delta is commonly expressed in multiple myeloma cells and its downregulation by rottlerin causes apoptosis. Br J Haematol. 2003;121(6):849–56. doi: 10.1046/j.1365-2141.2003.04368.x. [DOI] [PubMed] [Google Scholar]
- 69.Shiomi Y, Shinozaki A, Sugimoto K, Usukura J, Obuse C, Tsurimoto T. The reconstituted human Chl12-RFC complex functions as a second PCNA loader. Genes Cells. 2004;9(4):279–90. doi: 10.1111/j.1356-9597.2004.00724.x. [DOI] [PubMed] [Google Scholar]
- 70.Wang W, Prince CZ, Mou Y, Pollman MJ. Notch3 signaling in vascular smooth muscle cells induces c-FLIP expression via ERK/MAPK activation. Resistance to Fas ligand-induced apoptosis. J Biol Chem. 2002;277(24):21723–9. doi: 10.1074/jbc.M202224200. [DOI] [PubMed] [Google Scholar]