

Summary
Mad2 is an essential component of the spindle checkpoint that blocks activation of separase and dissolution of sister chromatids until microtubule attachment to kinetochores is complete. We show here that overexpression of Mad2 in transgenic mice leads to a wide variety of neoplasias, appearance of broken chromosomes, anaphase bridges and whole chromosome gains and losses, as well as acceleration of myc-induced lymphomagenesis. Moreover, continued overexpression of Mad2 is not required for tumor maintenance unlike the majority of oncogenes studied to date. These results demonstrate that transient Mad2 overexpression and chromosome instability can be an important stimulus in the initiation and progression of different cancer subtypes.
Significance
Genetic instability and aneuploidy are classical features of adult tumors which are usually associated with poor patient prognosis. Their actual contribution to oncogenic transformation, however, remains unclear. Elevated expression of the mitotic checkpoint gene Mad2, observed in a number of human cancers, promotes aneuploidy in vitro, but its role in tumor initiation or progression in mammals has not been established. We demonstrate here that overexpression of Mad2 in mice leads to tumor initiation most likely through the acquisition of a chromosomal instability (CIN) phenotype. In addition, once neoplastic transformation has occurred, Mad2 overexpression is no longer required to promote tumor progression, indicating that CIN could be an early and transient oncogenic event.
Introduction
The spindle assembly checkpoint is a signal transduction pathway which ensures that sister chromatids aligned at the metaphase plate do not separate prior to the bipolar attachment of all duplicated chromosomes to the mitotic spindle (Bharadwaj and Yu, 2004; Kops et al., 2005; Wassmann and Benezra, 2001). This pathway serves to restrain the protease separase which cleaves the cohesin proteins holding the sisters together at the metaphase-to-anaphase transition. Cyclin B/cdk1 phosphorylation of separase also negatively regulates its activity (Stemmann et al., 2001). Mad2 is a central component of this pathway since it is essential for inhibiting the E3 ubiquitin ligase cdc20-APC (or anaphase promoting complex) (Fang et al., 1998; Li et al., 1997) which itself targets securin (Visintin et al., 1997), a negative regulator of separase (Ciosk et al., 1998; Cohen-Fix et al., 1996), as well as cyclin B for degradation (Wasch and Cross, 2002; Yamamoto et al., 2005). Unoccupied kinetochores serve as loading machines for Mad2 onto cdc20-APC. This loading is thought to involve the association of a closed conformer of Mad2 (bound to Mad1 anchored at the kinetochore) with an open conformer capable of binding cdc20 and inhibiting APC activity (De Antoni et al., 2005). Once the last kinetochore is occupied with microtubules and the Mad1/Mad2 complex is displaced, closed conformers are capped by the presence of p31comet (Habu et al., 2002; Xia et al., 2004) and cdc20-APC is liberated. Securin is then ubiquitinated and degraded and after the loss of the inhibitory phosphorylation, separase is free to cleave the cohesins.
As anticipated from such a model, partial loss of Mad2 function by genetic manipulation leads to premature degradation of securin and separation of the sister chromatids (Michel et al., 2001). In cell lines and in animal models, this leads to a high rate of aneuploidy and polyploidy. Mad2 heterozygous animals develop lung tumors with very long latencies. Similar tumor predisposition or acceleration occurs in animal models in which other components of the mitotic checkpoint are partially inactivated and the animals exposed to chemical tumor promoters or other oncogenic stimuli (Babu et al., 2003; Baker et al., 2004; Dai et al., 2004; Rao et al., 2005). Complete loss of Mad2 or other mitotic checkpoint components, on the other hand, leads to early embryonic lethality and associated chromosome missegregation events (Babu et al., 2003; Dobles et al., 2000; Wang et al., 2004). In siRNA knockdown experiments in normal human fibroblasts and cell lines, near complete loss of Mad2 activity leads to massive chromosome missegregation and catastrophic cell death (Kops et al., 2004; Michel et al., 2004). The severity of this phenotype may be attributable to the concomitant loss of securin and cyclin B (Michel et al., 2004) and thereby complete loss of restraint on separase as well as reported microtubule disorganization. Indeed, to date tumor cells displaying complete loss of Mad2 function have not been found.
Mad2 overexpression on the other hand is a common event seen in many human cancers (Alizadeh et al., 2000; Chen et al., 2002; Garber et al., 2001) and is associated with poor prognosis (Hernando et al., 2004; Li et al., 2003; Tanaka et al., 2001; van ‘t Veer et al., 2002). Mad2 is an E2F target gene and is therefore expressed at high levels in tumors that are functionally or explicitly null for Rb activity (Hernando et al., 2004). Mad2 overexpression in human fibroblasts and cell lines can stabilize securin and cyclin B, delay exit from mitosis and increase nondisjunction events and aneuploidy. Whether such events contribute to tumorigenesis has not yet been explored. In the present study we conditionally overexpressed the Mad2 protein in mice using tetracycline inducible and repressible systems (Ewald et al., 1996) in which both high and intermediate levels of Mad2 overexpression are achieved. In addition, Mad2 was constitutively expressed in the Eμ-myc model of lymphomagenesis. Our results suggest that a hyperactive mitotic checkpoint plays a causal role in cancer initiation and progression and support the notion that enhanced chromosome instability, perhaps even transiently, contributes to the transformation process.
Results
Generation of mice carrying an inducible Mad2 gene
To generate transgenic mice containing a regulatable mouse Mad2 coding sequence, we constructed a 1.5 kb fragment of DNA (Figure 1A) consisting of seven direct repeats of the tet operator sequence (tetO7), a murine Mad2 cDNA and the SV40 polyadenylation site. To facilitate transgene detection, this construct encodes an HA epitope tag upstream of the Mad2 coding sequences (Figure 1A). Such a tag does not interfere with Mad2 activity (Wassmann et al., 2003). Injection of this construct into fertilized F2 eggs obtained from mating of (C57BL/6J x CBA/J) F1 mice produced 95 pups that were analyzed for the presence of the transgene by Southern blot analysis using a probe of 446 base pair specific for the exogenous Mad2 (Figure 1A). In order to regulate the expression of murine Mad2 we took advantage of two lines of transgenic mice, CMV-tTA (Tet-Off) (Furth et al., 1994) and CMV-rtTA (Tet-On) (kindly provided by H.Varmus and F.Cong). In the tTA (Tet-Off) system, the tetracycline analog doxycycline inhibits the activity of the transactivator (TA) and in the rtTA (Tet-On) system doxycycline stimulates the TA. In both systems the expression of the Mad2 responsive transgene is regulated in all tissues due to the expression of the TA from the human early cytomegalovirus promoter (PhCMV). The CMV-tTA (Furth et al., 1994) and CMV-rtTA mice (F.Cong, personal communication) are viable, fertile, and display no overt phenotype.
Figure 1.
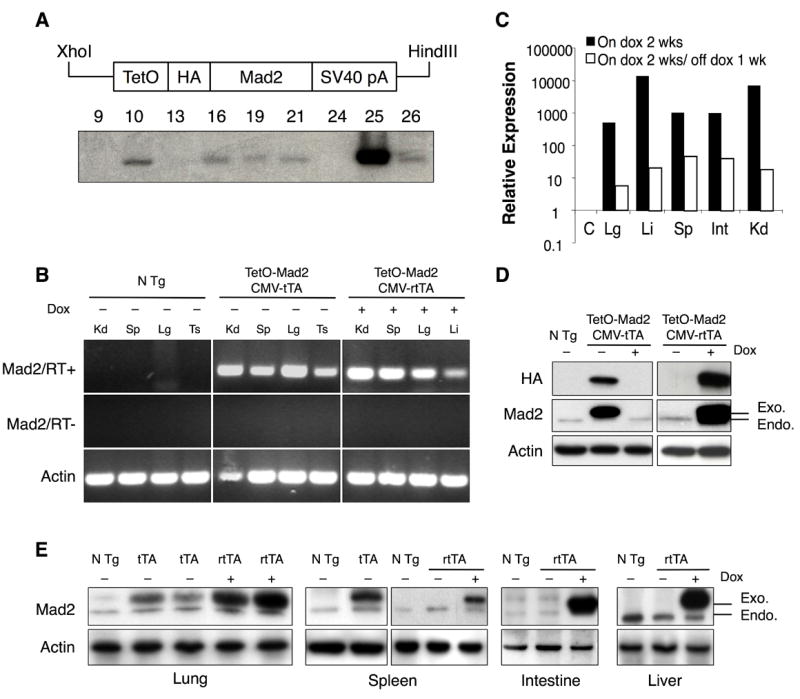
Mad2 transgene and expression pattern. (A) Construct used to generate the tetracycline operator-regulated Mad2 (TetO-Mad2) responder mice. TetO, tetracycline operator; HA, hemaglutinin; SV40 pA, SV40 gene polyadenylation sequence (upper panel). Southern blot of genomic DNA from different founders (lower panel). (B) RT-PCR from different tissues of non-transgenic, TetO-Mad2/CMV-tTA and TetO-Mad2/CMV-rtTA mice, the last ones exposed to doxycycline in the feed from 4 weeks to harvest at 8 weeks. PCR reactions were carried out in the presence (top) and absence (middle) of RT and products were visualized after electrophoresis in a 2% agarose gel. Amplification of actin mRNA by RT-PCR confirmed the presence of RNA in all samples. (C) Quantitative RT-PCR analysis of transgene expression in bitransgenic (Tet-On) mice on doxycycline and after doxycycline withdrawal (C= control, Kd=Kidney, Sp=spleen, Li=Liver, Lg=lung, Ts=Testis Int=intestine). (D) Western blot analysis for HA tag and Mad2 of non transgenic (N Tg) and TetO-Mad2/CMV-tTA MEFs (left panel) and TetO-Mad2/CMV-rtTA MEFs (right panel). Cells were maintained with (+) or without (−) doxycycline for 24h. (E) Western blot analysis showing Mad2 levels in different tissues from TetO-Mad2/CMV-tTA mice (lung and spleen) and TetO-Mad2/CMV-rtTA mice with and without doxycycline treatment (lung, spleen, intestine and liver). N Tg: non transgenic mice, tTA: is TetO-Mad2/CMV-tTA mice and rtTA: TetO-Mad2/CMV-rtTA mice. Anti-actin blots are shown as a loading control.
Five TetO-Mad2 positive founders (10, 16, 19, 21, 25 from Figure 1A) were crossed with both the CMV-tTA and CMV-rtTA mice. To induce Mad2 expression, we placed bitransgenic offspring from the rtTA system on a diet containing doxycycline after weaning, whereas in the tTA system food without doxycycline was administered at all times. After 4 weeks, progeny were assayed for transgene expression using a reverse transcriptase PCR (RT-PCR) assay specific for the transgene. As shown in Figure 1B, bitransgenic mice derived from TetO-Mad2 founder #25 expressed the transgene in both the Tet-On and Tet-Off system in all tissues tested. Similar results were observed with founder #10 (not shown) and both strains were used in subsequent analyses. Next, to determine whether expression of the transgene could be turned off, we performed quantitative RT-PCR on RNA samples derived from different tissues of TetO-Mad2/CMV-rtTA (Tet-On) bitransgenic mice fed doxycycline and after doxycycline withdrawal. Transgene expression was upregulated 500–10000-fold in tissues of TetO-Mad2/CMV-rtTA mice upon administration of doxycycline for 2 weeks and was reduced to 5–50 fold above background upon doxycycline withdrawal for 1 week (Figure 1C). Similar repressibility was observed with TetO-Mad2/CMV-tTA mice using the reverse doxycycline administration protocol (data not shown).
To measure induction of Mad2 protein encoded by the transgene, we performed western blots on cell extracts of murine embryonic fibroblasts (MEFs) maintained in normal media or in media containing doxycycline. We used antibodies against the HA tag (exogenous Mad2) as well as total Mad2 protein. As shown in figure 1D, the level of expression of exogenous Mad2 relative to endogenous in the Tet-Off system (left panel) was lower than in the Tet-On system (right panel) when doxycycline was added to the media. We also confirmed Mad2 protein overexpression by western blot analysis on tissues derived from TetO-Mad2/CMV-tTA mice maintained in normal food as well as on tissues from TetO-Mad2/CMV-rtTA mice on both normal diet and doxycycline-containing food (Figure 1E). Overall, these results demonstrate that we have generated transgenic lines in which we can achieve Mad2 overexpression in different tissues and that this can be turned on and off both in vivo and in vitro.
High level of Mad2 overexpression in MEFs leads to accumulation of mitotic cells and tetraploidy
Mouse embryonic fibroblasts (MEFs) obtained from TetO-Mad2/CMV-tTA (Tet-Off) mice express moderate levels of exogenous Mad2 compared to the endogenous levels of the protein (Figure 1D). Primary (P2) TetO-Mad2/CMV-tTA MEFs grow well in culture and do not display significant proliferative differences when compared to non-transgenic embryos (not shown). In contrast, MEFs obtained from TetO-Mad2/CMV-rtTA (Tet-On) mice express higher levels of exogenous Mad2 compared to the endogenous levels of the protein when exposed to doxycycline. When maintained in culture in the presence of doxycycline, these cells proliferate much more slowly than non transgenic cells or cells maintained in normal media (Figure 2A). These Mad2 overexpressing cells also form very few colonies when seeded at low density in the presence of doxycycline (Figure 2B).
Figure 2.
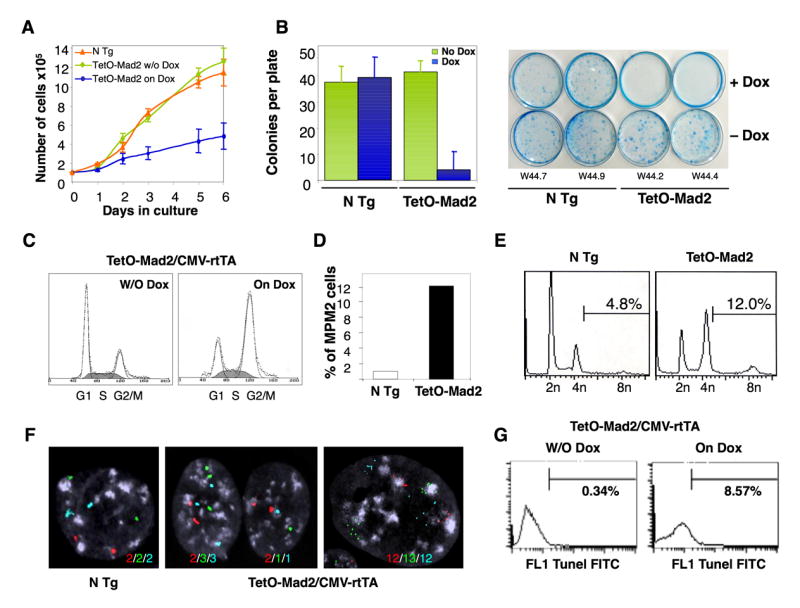
Growth properties of TetO-Mad2/CMV-rtTA MEFs. (A) Proliferation of early passage MEFs with or without the addition of doxycycline. (B) Plating efficiency of Mad2 wild type and TetO-Mad2/CMV-rtTA MEFs with and without doxycycline treatment. (C) Cell cycle profile of asynchronous cultures at passage 2. (D) Percentage of cells positive for MPM2 as a marker of mitosis. (E) FACS analysis of DNA content profile of asynchronously growing non transgenic and TetO-Mad2/CMV-rtTA MEFs on doxycycline media. (F) FISH analysis on MEFs using centromeric probes for chromosomes 12 (red), 16 (green) and 17 (blue) showing a non transgenic cell (left panel), a binucleated cell (middle panel) and a mononucleated cell with more than 4N (right panel). (G) TUNEL assay of TetO-Mad2/CMV-rtTA MEFs with and without doxycycline for 48 hours.
To better understand why MEFs overexpressing high levels of Mad2 stop proliferating we performed FACS analysis on asynchronously growing cultures with and without the addition of doxycycline. As shown in figure 2C, cells arrest in G2/M when Mad2 is overexpressed. MPM2 staining reveals that this is indeed a partial mitotic block (Figure 2D), a result consistent with our previously published data on IMR90 primary fibroblasts (Hernando et al., 2004). It was also shown previously that Mad2 overexpression does not lead to a permanent block but rather “escape” into the next cycle generating cells with >4N DNA content. In order to determine if this was also true in the tetracycline inducible system, FACS analysis was performed on growing populations 48 hours after Mad2 induction. Indeed, there were significantly more cells with >4N DNA content in Mad2 overexpressing MEFs than in non transgenic controls (12% versus 4.8%, Figure 2E). Analogous results were obtained using fluorescent in situ hybridization (FISH) to track specific chromosomes. We observed that overexpression of Mad2 in MEFs leads to both generation of binucleated cells and mononuclear cells with abnormal chromosome numbers (Figure 2F). Next, to determine the fate of these cells we performed a TUNEL assay and observed that 8.5% of the Mad2 overexpressing cells underwent apoptotic cell death as compared to 0.3% in the non transgenic controls (Figure 2G) after 48 h of transgene activation. Thus, high levels of Mad2 overexpression delay the exit from mitosis but allow the generation of polyploid/aneuploid cells with very low viability.
Mad2 overexpression leads to chromosomal instability
In order to further assess the chromosomal instability induced by Mad2 overexpression, we performed karyotype analyses of metaphase spreads generated from early passage primary MEFs overexpressing moderate (tTA) or high (rtTA) levels of Mad2 for short durations. In these cells, upon transgene activation for 24 hours, we observed both aneuploid (2n ± x) and tetraploid cells with accompanying aneuploidy (4n ± x) (Figure 3A). Comparable albeit slightly lower rates of aneuploidy were observed in the rtTA MEFs despite higher levels of Mad2 perhaps due to some cell death as described above. Indeed in all subsequent analyses of CIN in which Mad2 is overexpresed for short durations, comparable effects are observed in tTA and rtTA systems and we do not distinguish between the two. Primary wild type MEFs, under normal culture conditions, spontaneously become tetraploid. However, we found that the percentage of binucleated cells was significantly higher in the Mad2 overexpressing MEFs (75%) than in the non transgenic controls (7.7%) (representative example in Figure 3C). In addition, overexpression of Mad2 led to a significant increase in the number of chromosomal breaks and fragments, end-to-end fusions (dicentric and acentric chromosomes) as well as chromatid breaks and gaps as compared to wild type (Figure 3B and D and Supplemental Figure 1) or uninduced populations (data not shown). MEFs that overexpressed high levels of Mad2 also showed evidence of heterochromatin separation at the centromeres, possibly as a result of prolonged microtubule tension generated during the metaphase arrest (see Supplemental Figure 1B).
Figure 3.
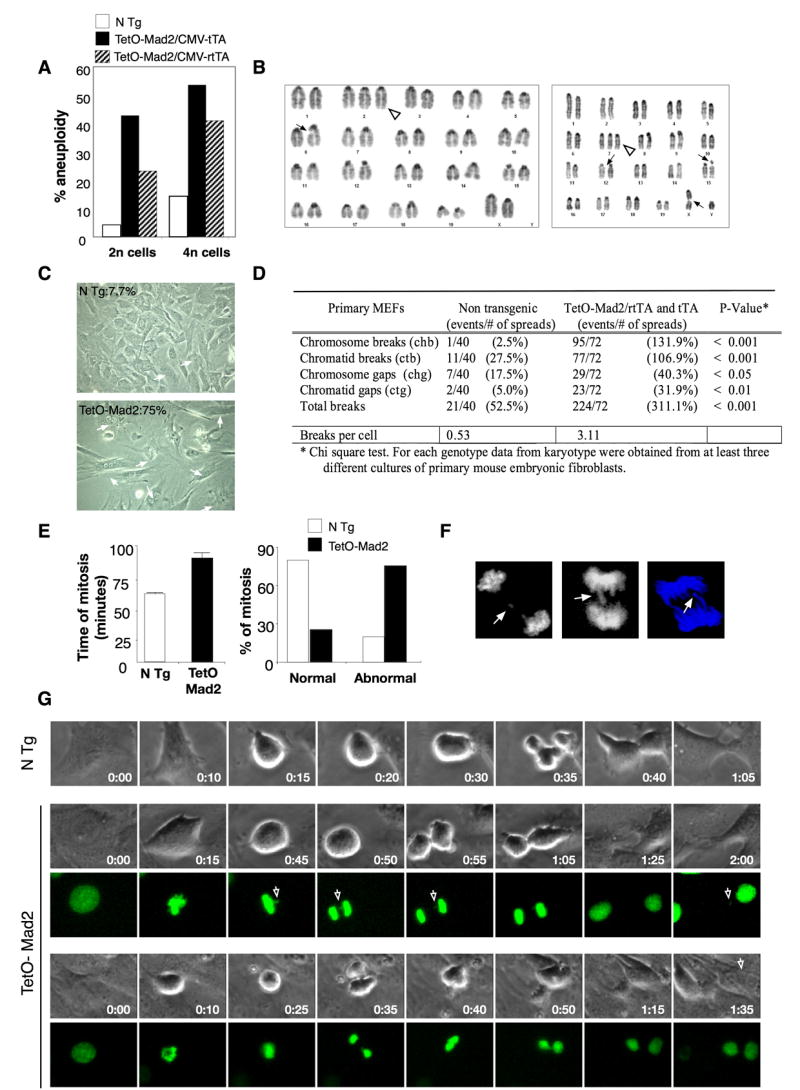
Overexpression of Mad2 leads to chromosomal instability. (A) Percentage of aneuploidy in the 2n and 4n population in TetO-Mad2 overexpressing MEFs in the Tet-On and Tet-Off systems. (B) Karyotype of a Tet-On cell with an extra chromosome (2) (white triangle) and a chromatid break (6) (ctb) (left panel), and karyotype of another cell with an extra chromosome 7 (white triangle), a ctb (X), ctb (15) and chromatid gap (ctg) (12) (right panel). (C) Representative picture of Tet-On P2 MEFs in culture showing binucleated cells (white arrows). (D) Number of chromosomal breaks on primary MEFs. (E, right panel) Time of mitosis of non transgenic (n = 98) and TetO-Mad2 (n = 90) MEFs was followed by time-lapse microscopy. Mean time of total mitosis is shown. (E, left panel) Percentage of cells with normal or abnormal mitosis (binucleated cells, furrow regression, chromosome bridges and mitotic catastrophe) as assessed by time-lapse microscopy. (F) Evidence of lagging chromosomes (top panel) and chromosome bridges (bottom 2 panels). (G) Time-lapse micrography of non transgenic and TetO-Mad2 MEFs. (Upper) N Tg cell entering mitosis at T=0 min and completing cytokinesis by 1 hour. (Middle) Representative cell overexpressing Mad2 with a chromosome bridge (white arrow) stays longer in mitosis and exits at 1hr 25 min with a missegregated chromosome (arrow). (Lower) Example of a cell with a chromosome bridge that suffers furrow regression and exits mitosis as a binucleated cell.
To further examine the abnormal mitoses observed by karyotype analysis, we used live cell imaging of MEFs infected with a retrovirus expressing histone 2B (H2B)-GFP. Cells were monitored by phase and fluorescent microscopy and mitotic timing scored setting time T=0 min at the point when nuclear envelope breakdown (NBD) was observed (as judged by loss of nuclear integrity and chromosome condensation). Whereas non-transgenic or uninduced MEFs underwent a normal and rapid mitosis (59 ± 1.6 min), cells overexpressing Mad2 (in both backgrounds, tTA and rtTA), took an unusually long time to finish the process (89 ± 5 min), displaying marked difficulties in completing cytokinesis and frequent defects in chromosome segregation, as previously reported (Hernando et al., 2004). The frequency of abnormal mitoses was increased in cells with elevated levels of Mad2 compared to wild type (Figure 3E) or uninduced populations (Supplemental Figure 1C). We observed cells in which lagging chromosomes and/or chromosome bridges gave way to two presumably aneuploid daughters and others in which furrow regression took place giving rise to a binucleate cell, confirming our findings of both aneuploid and tetraploid cells by karyotype analysis (Figure 3G). We interpret the images of lagging chromosomes and chromosome bridges (Figures 3F and G) as an attempt by the cell to bypass the mitotic block imposed by Mad2 overexpression prior to complete dissolution of the cohesins holding sister chromatids together. Overall, these data indicate that Mad2 overexpression can acutely produce genomic instability, a hallmark of human cancer.
Mad2 overexpression leads to a wide spectrum of tumors
Mad2 overexpression has been found in a wide spectrum of human tumors but whether such overexpression can causally initiate tumorigenesis is unexplored. We followed a cohort of 40 CMV-tTA mice overexpressing Mad2 in order to detect spontaneous tumor initiation. Fifty percent of Mad2 overexpressing mice were dead by 75 weeks as compared to no deaths in their non transgenic littermates (p value <0.001) (Figure 4F). Necropsy analysis of these mice that spontaneously died between 45 and 85 weeks showed a wide spectrum of tumors, including hepatoma and hepatocellular carcinoma, lung adenomas, fibrosarcomas and lymphomas (Table 1 and Figure 4A–D). Other non-tumor lesions observed included fallopian tube dysplasia (Figure 4E), testicular atrophy, hepatocellular regeneration, hepatomegaly, and splenomegaly due to extramedullary hematopoiesis (not shown). Most tumors observed developed with latencies greater than 12 months. Only in the case of an endometrial fibrosarcoma (by histological examination), an intestinal tumor and two lung tumors (by MRI) did we observe tumors arising before 12 months of transgene activation.
Figure 4.

Tumor susceptibility in TetO-Mad2/CMV-tTA mice. Micrography of H&E stainings of a lung adenoma (A); a metastatic lymphoma in the colon (B); an hepatocellular carcinoma (C); a fibrosarcoma in the skin (D) and a dysplasia of the fallopian tubes (E). Insets on A,B,C: macroscopic pictures of the corresponding tissues. Inset B, C lower panel: detail of abnormal mitoses; Blue bar 1cm. Red bar 80μm and black bar 100 μm. (F) Survival curve of TetO-Mad2/CMV-tTA mice where blue line represents TetO-Mad2/CMV-tTA mice (n=40) and green line are non transgenic littermates (n=15). (G) Chromosomal abnormalities detected by comparative genomic hybridization array analysis of TetO-Mad2/CMV-tTA normal livers and liver tumors compared to a wild type liver showing amplification and deletion of specific regions as well as a whole chromosome gain (X).
Table 1.
Tumor incidence in TetO-Mad2/CMV-tTA mice
| Tumor type | Incidence |
|---|---|
| Lung adenoma | 14 (35%) |
| Hepatoma | 9 (22.5%) |
| Hepatocellular carcinoma | 1 (2.5%) |
| Intestinal tumor | 5 (12.5%) |
| Lymphoma | 3 (7.5%) |
| Fibrosarcoma | 2 (5%) |
| Prostate tumor | 2 (5%) |
| Angiomyolipoma | 1 (2.5%) |
| Mammary adenocarcinoma | 1 (2.5%) |
| Testicular atrophy | 3 (7.5%) |
We also followed a cohort of 28 mice on the CMV-rtTA background maintained on normal diet or in food containing doxycycline. Fifty percent (9 out of 18) of the TetO-Mad2/CMV-rtTA mice maintained on a doxycycline diet developed tumors between 4 and 18 months of age while 0 out of 10 (0%) TetO-Mad2/CMV-rtTA mice fed a normal diet developed tumors at similar ages. The tumors found in the rtTA system were of the same histological origin and spectrum as the ones described for the tTA system (data not shown).
In light of the chromosomal alterations seen by karyotype analysis of MEFs, we performed comparative genomic hybridization in 3 liver tumors from Mad2 transgenic mice as well as in 3 liver samples that did not show obvious tumor development but overexpressed Mad2 (Figure 4G). In all cases of liver tissue analyzed (tumor and non tumor) we confirmed large chromosomal abnormalities such as whole chromosome gains and arm deletions and amplifications. Thus, Mad2 overexpression leads to extensive chromosome instability in vivo which may take place prior to overt transformation.
Mad2 overexpression is not required for tumor maintenance
One of the advantages of the tetracycline inducible system is the ability to temporally modulate the expression of the transgene. In order to test whether sustained Mad2 overexpression is required for tumor progression and maintenance, we monitored tumor growth in live bitransgenic mice by serial MRI scans. Twelve TetO-Mad2/CMV-tTA mice had significant masses evident by MRI after 13–16 months (Figure 5A). These mice were fed doxycycline and a second scan performed after 2 weeks. In all cases, the tumors persisted and continued to grow after Mad2 downregulation. We confirmed that the lesions observed in the MR images were in fact tumors by sacrificing the mice 13 weeks after the second MRI scan (maintained on doxycycline) followed by histological analyses. Turning off Mad2 has little effect on tumor progression as hepatomas harvested 15 weeks after addition of doxycycline to the feed were of the same size and histopathology as the untreated controls (Figure 5B). Ki67 staining also demonstrated that downregulation of the Mad2 transgene did not affect the proliferative index of the tumor (Figure 5B). QRT-PCR analysis showed that the levels of the transgene were indeed downregulated in the tumors (Figure 5C).
Figure 5.
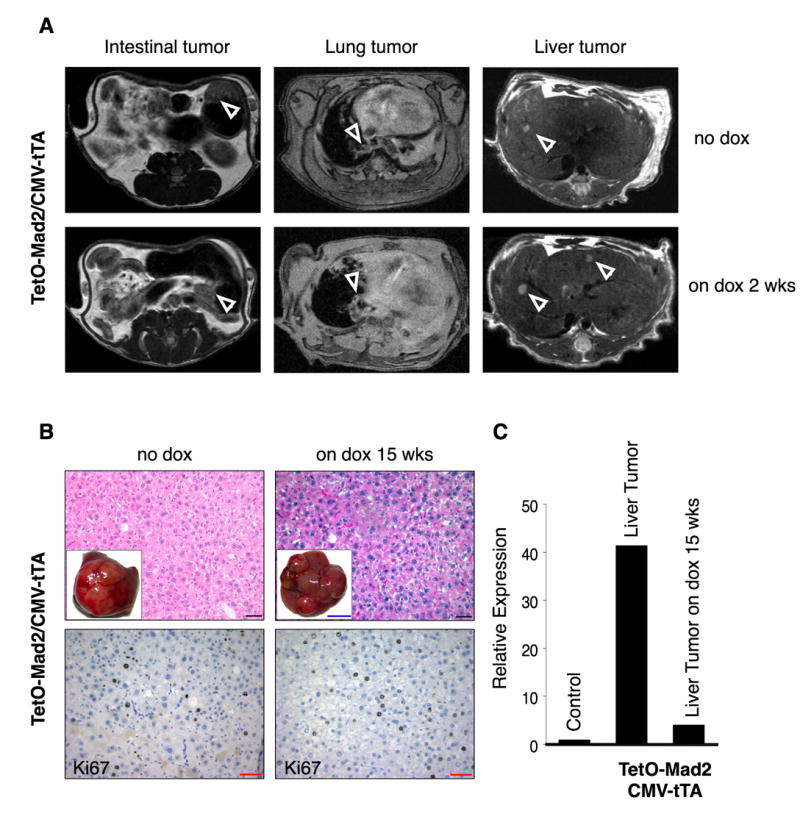
Overexpression of Mad2 is not required for tumor maintenance. (A) Axial MR images of the abdomen and the lungs of bitransgenic mice on normal diet (Mad2 on) and after 2 weeks on doxycycline food (Mad2 off) showing the presence of tumors (white arrows). (B) H&E (top) and Ki67 staining (bottom) of a hepatoma in a bitransgenic TetO-Mad2/CMV-tTA mouse untreated (Mad2 on) (left) and a mouse fed doxycycline for 15 weeks after confirming the presence of a tumor by MRI. (C) QRT-PCR confirming the downregulation of Mad2 in the tumor. Black bar 100μm, red bar 20μm and blue bar 1cm.
Similarly, tumors which developed in TetO-Mad2/CMV-rtTA mice on a doxycycline diet and then placed on a doxycycline free diet showed no evidence of tumor regression (Supplemental Figure 2). Importantly, in this case, the levels of Mad2 after doxycycline withdrawal were near background levels and are clearly not able to support tumorigenesis as described above.
Mad2 expression is significantly upregulated in a subset of human cancers
We decided to confirm the involvement of Mad2 deregulation in cancer initiation or progression by searching for aberrant Mad2 expression in human tumors. To this end, we used the ONCOMINE database that contains gene expression data compiled from multiple microarray analyses (http://www.oncomine.org) (Rhodes et al., 2004). Interestingly, the spectrum of human tumors in which Mad2 was found transcriptionally overexpressed largely overlapped with the cancer types found in the Mad2 transgenic mice. Thus, hepatocellular ((Chen et al., 2002) p=1.5 exp-17) and lung carcinomas ((Garber et al., 2001) p=1.9 exp-6), showed a significant upregulation of Mad2 as compared to the corresponding normal tissues. Moreover, analyzing data derived from a comparative multi-lymphoma study (Alizadeh et al., 2000), we noted that Mad2 was expressed at significantly higher levels in diffuse large B cell lymphomas (DLBCL) compared to chronic lymphocytic leukemia/small lymphocytic lymphoma or follicular lymphoma (FL) (p=2.9 exp-10).
As the microarray data described above suggested an involvement of Mad2 in DLBCL, we decided to validate this observation by performing an immunohistochemistry analysis of tissue microarrays (TMA) with a monoclonal antibody specific for Mad2. These TMAs contain tissue from 81 cases of DLBCL, 105 cases of FL (Hedvat et al., 2002)(Fig. 6A), 35 small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL) and 35 mantle cell lymphomas (MCL), as well as a variety of T cell lymphomas, lymphomas with plasmacytic differentiation and plasma cell myelomas. We observed that numerous DLBCL (58.7%) presented strong or moderate levels of Mad2 staining (Figure 6A). A survey of Mad2 expression in other subtypes (e.g. MCL, SLL/CLL, etc) did not show Mad2 upregulation in these other lymphomas with the exception of a subset of grade 3 Follicular lymphomas (FL) (Figure 6A), Burkitt’s lymphomas and T cell lymphoblastic lymphomas (Figure 6B).
Figure 6.
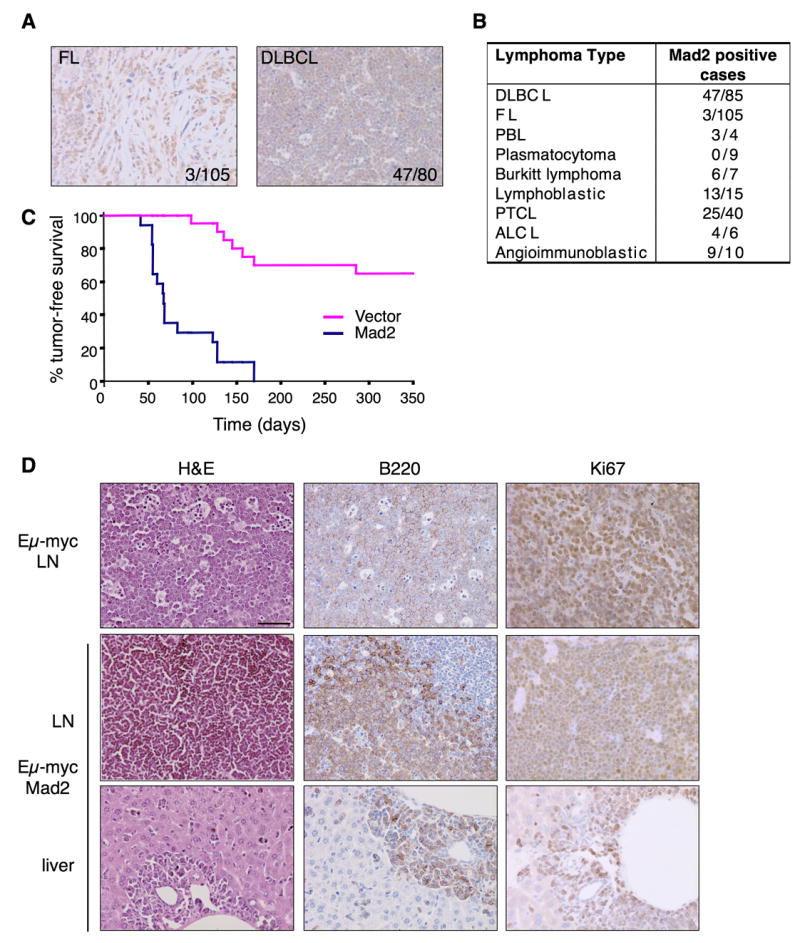
Mad2 overexpression, a common feature of certain human lymphomas, accelerates myc-driven lymphomagenesis. (A) Immunohistochemical evaluation of Mad2 expression in human FL and DLBCL. Two representative positive cases are shown. (B) Results of Mad2 immunohistochemistry analysis in multiple human lymphoma subtypes. (C) Tumor free survival curve of animals transplanted with Eμ-myc/vector vs. Eμ-myc/Mad2 HSCs showing statistically significant acceleration of tumor initiation mediated by Mad2 induction. (D) Histological and immunohistochemical evaluation of Eμ-myc and Eμ-myc/Mad2-derived lymphomas (LN) and liver metastases.
Mad2 overexpression accelerates lymphomagenesis in the Eμ-myc model
The fact that Mad2 is overexpressed in certain human B cell lymphomas prompted us to investigate whether Mad2 could be oncogenic in the B cell lineage. To do this, we took advantage of a well characterized system of lymphomagenesis involving c-myc, an oncogene that contributes to DLBCL and other lymphomas. Thus, we infected hematopoietic stem cells (HSCs) derived from Eμ-myc transgenic animals (Adams et al., 1985) with retroviruses containing the Mad2 cDNA co-expressing GFP and used them to reconstitute sublethally irradiated recipient mice. Adoptive transfer of Eμ-myc fetal liver cells from these animals gives rise to lymphomas in irradiated wild type recipients between 3–6 months of age (Schmitt et al., 2002). Mad2 overexpression accelerated Eμ-myc lymphomagenesis, with a marked reduction in tumor latency to 6–9 weeks (Figure 6C). Only 40% of mice reconstituted with fetal liver cells infected with control vector (MSCV) developed lymphomas by 400 days whereas 100% of mice reconstituted with Eμ-myc fetal livers infected with Mad2 retrovirus developed lymphoma (median tumor-free survival = 85 days) (p < 0.01; n=17 vs. n=10). Overall survival of mice was similarly affected. Importantly, all Eμ-myc/Mad2 lymphomas analyzed were positive for GFP expression, and overexpressed Mad2 by western blotting (not shown). Whole body imaging (data not shown) and pathological examination revealed that the Eμ-myc/Mad2 lymphomas were reminiscent of large cell lymphomas, highly aggressive and involved all major lymph node groups causing splenomegaly and thymic enlargement (data not shown).
Immunological analysis showed that all the Eμ-myc/Mad2 lymphomas were of B-cell origin (B220+), and had a similar proliferative index to that observed for Eμ-myc alone (Figure 6D). However, in contrast to Eμ-myc, the more accelerated Eμ-myc/Mad2 tumors tested displayed an immature B cell phenotype (B220+ IgM− ) (not shown). Tumor infiltration was commonly found in the liver (Figure 6D), lungs, and kidneys (not shown). Thus, Mad2 efficiently cooperated with Myc to produce aggressive lymphomas. The fact that TetO-Mad2/CMV-rtTA mice overexpress Mad2 in HSCs (Supplemental Figure 3) and developed lymphomas at low penetrance and only after 12 months latency argues that indeed the effects we are seeing are due to cooperation between Eμ-myc and Mad2 and not simply a consequence of Mad2 overexpression alone.
Mad2 overexpression leads to securin and cyclin B stabilization
Lymphocytes isolated from TetO-Mad2/CMV-rtTA mouse spleens were stimulated in vitro upon addition of ionomycine and PMA, and cells colleted at different time points. Doxycycline was also added to TetO-Mad2 lymphocytes in culture to stimulate the expression of the transgene. Mouse spleenocytes enter the cell cycle synchronously, allowing us to monitor cyclin B and securin levels as cells progress through S phase and mitosis. No significant differences in MPM2 kinetics were observed between non-transgenic and TetO-Mad2 cells (Figure 7A) suggesting an attempt by the Mad2 ooverexpressing cells to exit mitosis on schedule. Expression of securin and cyclinB was detected in both cell types from t=24h however protein degradation was remarkably delayed in Mad2 overexpressing cells as compared to non-transgenic (Figure 7B and C) lymphocytes. These results confirm in vivo previous data on Mad2-retrovirally transduced human fibroblasts and tumor cell lines (Hernando et al., 2004), and support the hypothesis that Mad2 overexpression causes a hyperactive spindle checkpoint, which could account for the mitotic defects and chromosomal instability underlying tumorigenesis in this model.
Figure 7.
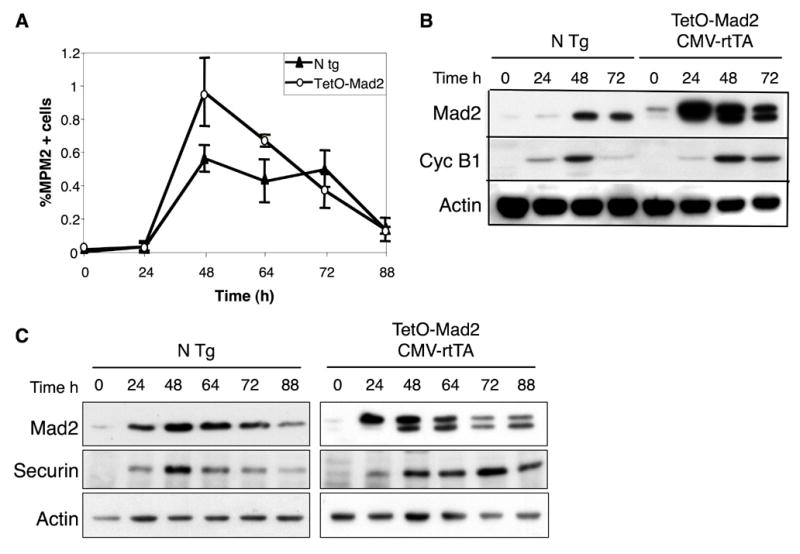
Mitotic progression of in vitro stimulated lymphocytes. (A) Percentage of lymphocytes positive for MPM2 as a marker of mitosis. (B) Western blot analysis of in vitro stimulated lymphocytes isolated from spleen of non transgenic mice and TetO-Mad2/CMV-rtTA mice in the presence of doxycycline showing stabilization of cyclin B1 in the Mad2 overexpressing cells as compared to the non transgenic cells (C) Western blot analysis of in vitro stimulated lymphocytes showing stabilization of securin in the TetO-Mad2/CMV-rtTA cells compared to the non transgenic control.
Discussion
Mad2 overexpression is common in human tumors (Alizadeh et al., 2000; Chen et al., 2002; Garber et al., 2001; Hernando et al., 2004; Li et al., 2003; Tanaka et al., 2001; van ‘t Veer et al., 2002) and has been shown to promote genomic instability in cell culture models (Hernando et al., 2004). Here we extend this analysis and show that Mad2 overexpression can initiate tumorigenesis and cooperate with other oncogenic stimuli. Consistent with a role for Mad2 in promoting genomic instability, Mad2-induced tumors have frequent genomic rearrangements, whole chromosome gains or losses and sustained Mad2 overexpression is not required for continued tumor growth.
Although we show that Mad2 overexpression can initiate tumorigenesis, activating mutations have not been reported in human cancers. However, studies suggest that Mad2 is under the control of E2F which is deregulated in many human cancers (Hernando et al., 2004). Thus, cells suffering mutations in the Rb pathway not only gain a proliferative advantage, but, as suggested in this study, can gain an instability that (again, as shown here) may contribute to tumorigenesis even if present only transiently. It should be noted that while the effect of Mad2 overexpression on tumor initiation and acceleration is likely to result from the observed chromosome instability, other unknown effects of Mad2 overexpression cannot be ruled out at this time.
Mad2 overexpression leads to a highly penetrant induction of a wide range of tumors in mice including lung adenomas, hepatomas and hepatocellular carcinomas, lymphomas and fibrosarcomas. Other cell types might also be susceptible to the effects of Mad2 overexpression but could have been masked by a variety of factors such as low expression levels from the CMV promoter or early lethality. This issue can now be addressed by the use of other tissue specific Mad2 alleles. As described, Mad2 overexpression was also observed to accelerate tumorigenesis in a well-established model of lymphoma driven by the expression of the myc oncogene in the B cell lineage. In addition, higher levels of Mad2 mRNA have been reported in DLBCL (Alizadeh et al., 2000) as compared to most other B cell lymphoma subtypes, confirmed by our expression analyses. Interestingly, DLBCL display a highly aggressive biological behavior and represent the most aberrant B cell lymphomas in terms of ploidy alterations. These data suggest that in addition to tumor initiation, Mad2 overexpression may play an important role in tumor progression and mortality. Indeed, as reported previously, Mad2 is a poor prognostic marker for neuroblastoma (Hernando et al., 2004) consistent with this hypothesis.
Clearly, in tumors the “penalty” for loss of a whole chromosome induced by Mad2 overexpression is balanced by growth advantages which likely result from LOH at tumor suppressor loci. Once a cell has acquired the CIN phenotype, theoretical considerations suggest that there is an optimal chromosome loss rate (between 10exp-2 and 10exp-3 per chromosome per generation) (Komarova and Wodarz, 2004) which will maximize the loss of tumor suppressor genes and expansion of transformed clones. Indeed, we and others have observed a threshold for cell viability in cell culture and animal models as high level of expression of Mad2 (this study), complete loss of separase in mice (Kumada et al., 2006; Wirth et al., 2006) and complete loss of mitotic checkpoint function all lead to a profound cell death and early embryonic lethality (Babu et al., 2003; Dobles et al., 2000; Kops et al., 2004; Michel et al., 2004; Wang et al., 2004). It is likely for this reason that no human tumors to date have been identified which have sustained a complete loss of Mad2 function, although partial loss of function has been observed (Percy et al., 2000; Wang et al., 2002; Wang et al., 2000). It will be of interest to see if separase heterozygous mice will develop tumors with prolonged latency. While high levels of Mad2 overexpression in the rtTA system caused cell death of MEFs in culture, it is likely that in tumors that develop in the rtTA mice there is selection for levels of Mad2 which allow cell viability but promote cellular transformation.
The simplest explanation for the chromosome instability observed in the Mad2 overexpressing mice is that the stabilization of securin and cyclin B, observed previously in primary IMR90 cells (Hernando et al., 2004) and now in Mad2 overexpressing lymphocytes (Figure 7), inhibits the activity of separase leading to non-disjunction events and to cytokinesis inhibition. This is consistent with the oncogenic role of securin (PTTG) overexpression (Pei and Melmed, 1997). Formal genetic demonstration of this hypothesis awaits the analysis of the Mad2 transgenics crossed with the securin knockout animals which is currently underway.
The cause of the observed interstitial deletions amplifications in the Mad2 overexpressing cells is unclear at this time. It is possible that when cohesiveness of sister chromatids is maintained during the exit from mitosis, chromosome breakage and rejoining events facilitate this type of chromosome instability. Indeed, our real time microscopy of Mad2 overexpressing cells shows evidence of chromatin trapped in extended cytoplasmic bridges during cytokinesis followed by a breakage event (see Supplemental movie 1). In addition, karyotype analysis of MEFs overexpressing Mad2 show clear evidence of chromosome breakage in addition to whole chromosome gains and losses.
The rate of acquisition of CIN in tumors must be comparable to spontaneous mutation rates in order to compete with mutational LOH at tumor suppressor loci and therefore play a role in tumor initiation. The acquisition of CIN may in fact be the second hit after a mutation at a TS locus since whole chromosome loss is not rate limiting in a CIN cell (see discussion in (Nowak et al., 2002)). Interestingly, since Mad2 overexpression induces both interstitial deletions and amplifications and whole chromosome loss it might induce both the initial loss of function event at tumor suppressor loci as well as LOH. This would serve to minimize the deleterious effects of whole chromosome loss. Alternatively, spontaneous mutation of tumor suppressor genes may be the event that is selected for in the clonal expansion of the initiating tumor cell. Further analysis of the tumors that arise in the Mad2 transgenic mice is required to address this question.
It has been suggested that lagging and bridging chromosomes in human cells in culture are insufficient to induce cleavage furrow regression and tetraploidization (Shi and King, 2005). Rather, these mislocalized chromatids must end up in the wrong daughter cell in order to induce tetraploidization and aneuploidy is generally acquired after the tetraploidization event. However, this notion is at odds with recently published studies in S. cerevisiae in which chromatin in the cleavage furrow induces an ipl dependent signaling cascade which results in furrow regression (Norden et al., 2006). Mad2 overexpression would be predicted to induce a high rate of non-disjunction events due to persistence of cohesion and tetraploidy prior to the appearance of an aneuploid cell. However, we observe aneuploidy early after Mad2 induction in murine cells with chromosome numbers in the 2N and 4N range. A similar result has been reported recently in CENP-E knockout MEFs (Weaver et al., 2006) This discrepancy is unlikely to be a murine specific effect as has been suggested (Shi and King, 2006) since a similar result has been observed in Mad2 overexpressing human cells (Hernando et al., 2004) and in human cells which show high rates of non-disjunction due to the loss of one copy of Mad2 (Michel et al., 2001). We conclude that in several different settings aneuploidy can be established independently of tetraploidization.
Turning off Mad2 transgene expression in established tumors has little effect on tumor progression, at least in the case of the hepatomas examined. This is in contrast to the oncogene-dependence observed in other systems (for review see (Jonkers and Berns, 2004)). We presume that in the case of Mad2, the lack of dependence is a reflection of the early induction of chromosome instability by Mad2 which would persist after Mad2 levels are normalized. This hit-and-run effect of Mad2 overexpression may lead to an underestimation of the fraction of human tumors which have experienced Mad2 overexpression or overexpression of other mitotic checkpoint components during the early phases of the oncogenic process. In sum then, our results suggest that deregulation of mitotic checkpoint pathways by Rb inactivation or other mechanisms may be an early and transient event in the initiation and evolution of a wide variety of common cancers.
Experimental procedures
Generation of Mad2 inducible Mice
The pTRE vector from Clontech, containing the tetracycline operator and the SV40 polyadenylation sequence, was linearized with EcoRI and BamHI. The murine Mad2 cDNA was amplified with specific primers containing the HA epitope tag and the corresponding restriction enzymes and ligated into the pTRE vector. Restriction digests and sequencing were used to identify clones in which the Mad2 cDNA had inserted into the correct orientation.
Animal husbandry and genotyping
TetO-Mad2 transgenic mice, CMV-tTA and CMV-rtTA mice were kept in a pathogen-free housing under guidelines approved by the MSKCC Institutional Animal Care and Use Committee and Research Animal Resource Center. Eμ-myc mice (Adams et al., 1985) and CMV-tTA mice (Furth et al., 1994) have been previously described. CMV-rtTA mice contain an interstitial deletion on chromosome 5 and will be described elsewhere. Doxycycline was administered by feeding mice with doxycycline-impregnated food pellets (625ppm; Harlan-Teklad). Tail DNA was isolated using Qiaprep Tail DNeasy isolation kit (QIAGEN) according to the manufacturer’s protocol. TetO-Mad2 transgenic mice were genotyped using the following primers: Mad2F: 5′-CCATCCACGCTGTTTTGACCTC-3′; Mad2R: 5′-GGCTTTCTGGGA CTTTTCTCTACG-3′ and CMV-rtTA mice: rtTAF: 5′- GTGAAGTGGGTCCGCGTA CAG-3′ and rtTAR: 5′- GTACTCGTCAATTCCAAGGGCATCG-3′.
Preparation of MEFs and lymphocytes and tissue culture
Mouse embryonic fibroblasts (MEFs) were isolated from E13.5 embryos and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM glutamine, 1% penicillin/streptomycin, 10% tetracycline free fetal bovine serum (FBS) and 1 μg/ml of doxycycline when indicated. For proliferation assays, 1x105 cells were plated on 6-well plates in duplicate as described previously (Sotillo et al., 2001). Primary lymphocytes were isolated from the spleen of 6-month-old mice, cultured in RPMI + 10% FBS and stimulated with PMA and ionomycine (Sigma) in the presence or absence of doxycycline and cell cycle profiles were analyzed by cytometry.
Retroviral-mediated gene transfer and lymphoma generation
Eμ-myc HSCs derived from fetal livers at embryonic day 13–15 were transduced with retroviruses expressing Mad2 or the MSCV vector alone and used to reconstitute the hematopoietic compartment of lethally irradiated C57BL/6 mice (Schmitt et al., 2002; Schmitt et al., 2000). Mice were observed for lymphoma onset with periodic palpation of peripheral lymph nodes, overall morbidity and by whole body fluorescence imaging. After the appearance of well-palpable lymphomas, tumors were harvested and either fixed for histological evaluation or rendered as single-cell suspensions, analyzed or stored frozen in 10% DMSO.
Magnetic resonance imaging
Individual mice were subjected to MRI assessment for detection of tumors. In brief, mice were anesthetized with 2% isofluorane and images were obtained on a Bruker 4.7T 40cm bore magnet with a commercial 7-cm inner diameter birdcage coil in the Animal Imaging MRCore Facility at MSKCC. Low-resolution axial scout images were obtained initially, followed by a high-spatial-resolution T2-weighted axial images (repetition interval (TR)=3,800ms, effective echo time (TE)=35ms, eight echoes per phase encoding step, spatial resolution=1.0mm slice thickness x 112μmX112μm in plane resolution, and four repetitions of data acquisition for 8–9 min of imaging time).
FACS, Karyotyping, FISH, and Live Cell Imaging
For FACS analysis, trypsinized cells were washed in PBS, fixed in 70% ethanol and stained with propidium iodide (50μg/ml). 104 cells were analyzed by using a FACScalibur (Becton Dickinson). Apoptotic cells were labeled by fluorescent TUNEL assay (In Sity Cell Death Detection Kit, Roche) and quantified by FACS. For karyotyping, cells were incubated in medium containing Colcemid (0.05 μg/ml) for 40 minutes and harvested by standard cytogenetic procedures. Metaphase spreads were stained with DAPI (0.08%) in 2xSSC. For FISH analysis we made probes using pairs of BAC clones near the centromeres for each chromosome. Additional details are listed in the Supplemental Material. Mitotic index was quantified by measuring MPM2 expression (anti-MPM2, Upstate Biotechnology) versus DNA content (PI) by FACS. For live cell imaging, primary MEFs were infected twice with a retrovirus expressing H2B-GFP (Yamamoto et al., 2004) and were cultivated in a glass-bottom culture (Delta TPG) dish. Imaging was performed as previously described (Michel et al., 2004).
Array CGH analysis
Genomic DNA extracted from normal and tumor livers from TetO-Mad2/CMV-tTA and CMV-rtTA mice was subjected to comparative genomic hybridization array analysis at the MSKCC Genomics Core Lab. For each mouse, genomic DNA extracted from the liver of a wild type littermate was used as a reference and hybridized into mouse CGH Agilent arrays (44A version). Results were analyzed using a special normalization method correcting for the GC content of the probes (adapted from (Tonon et al., 2005)).
RNA and protein analysis
RNA was isolated using the RNeasy kit (Qiagen, Valencia, CA). RNA was treated with DNaseI (Ambion) to eliminate any contaminating DNA. RT-PCR reactions were performed with SuperScript III (Invitrogen) according to the manufacturer’s instructions. For quantitative RT-PCR, reactions were performed using the ABI7900 Sequence Detection System (Applied Biosystems). Primer sequences and amplification conditions and protein expression are described in the Supplemental Material.
Tissue microarrays of human lymphomas
We analyzed the Oncomine database for expression of Mad2 on different set of microarray data comparing normal vs. cancer samples and established a p<0.05 as cut-off limit. Several tissue microarrays (TMA) comprising 168 cases of FL (Hedvat et al., 2002), 81 cases of DLBCL, 35 small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL), 35 mantle cell lymphomas (MCL), 15 T cell lymphoblastic lymphomas, 10 angioimmunoblastic lymphomas, 40 peripheral T cell lymphoma (PTCL), 6 anaplastic large cell lymphoma (ALCL), 7 Burkitt’s lymphomas, 9 plasmatocytomas and 4 plasmablastic lymphomas were analyzed for Mad2 expression by immunohistochemistry analysis. Patient samples were obtained through institutionally approved protocols.
Histopathology
For immunohistochemistry analysis, representative sections were deparaffinized, rehydrated in graded alcohols, and processed using the avidin-biotin immunoperoxidase method. Sections were subjected to antigen retrieval by microwave oven treatment using standard procedures. Diaminobenzidine was used as the chromogen and hematoxylin to counterstain nuclei. The antibodies used for inmunohistochemistry are listed in the Supplemental Material. TMAs were scored (by JTF and EH) evaluating % of positivity of tumor cells and intensity of nuclear staining.
Supplementary Material
Acknowledgments
We thank H. Varmus and F. Cong for the CMV-rtTA mice and members of the Varmus laboratory for discussions. C. Le and M. Lupu for help with the MRI analysis; M. Leversha for karyotype analysis; D. Domingo for FACs analysis; A. Viale for assistance with CGH analysis; Y. Chin, S. Curelariu, M.E. Dudas, E. Charytonowicz, M. Asher and I. Linkov for technical assistance; M.T Hemann and J. Friedman for assistance in HSC transplants; S. Kogan for immunophenotypic characterization of Eμ-myc/Mad2 tumors; the Wahl laboratory for the H2B-GFP retrovirus; and members of the Benezra laboratory, especially J. Schvartzman for discussions and critical reading of the manuscript. RS is a postdoctoral fellow from the Charles H. Revson Foundation and received support from La Fundacion Caja Madrid-CNIO. CC-C, SWL and RB are supported by grants from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, van Deursen JM. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol. 2003;160:341–353. doi: 10.1083/jcb.200211048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 2004;23:2016–2027. doi: 10.1038/sj.onc.1207374. [DOI] [PubMed] [Google Scholar]
- Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, Lai KM, Ji J, Dudoit S, Ng IO, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–1939. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciosk R, Zachariae W, Michaelis C, Shevchenko A, Mann M, Nasmyth K. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93:1067–1076. doi: 10.1016/s0092-8674(00)81211-8. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, Mahmood R, Yang YM, Xu M, Rao CV. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–445. doi: 10.1158/0008-5472.can-03-3119. [DOI] [PubMed] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, Musacchio A. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol. 2005;15:214–225. doi: 10.1016/j.cub.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–645. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- Ewald D, Li M, Efrat S, Auer G, Wall RJ, Furth PA, Hennighausen L. Time-sensitive reversal of hyperplasia in transgenic mice expressing SV40 T antigen. Science. 1996;273:1384–1386. doi: 10.1126/science.273.5280.1384. [DOI] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation [In Process Citation] Genes Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth PA, St Onge L, Boger H, Gruss P, Gossen M, Kistner A, Bujard H, Hennighausen L. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc Natl Acad Sci U S A. 1994;91:9302–9306. doi: 10.1073/pnas.91.20.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habu T, Kim SH, Weinstein J, Matsumoto T. Identification of a MAD2-binding protein, CMT2, and its role in mitosis. Embo J. 2002;21:6419–6428. doi: 10.1093/emboj/cdf659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedvat CV, Hegde A, Chaganti RS, Chen B, Qin J, Filippa DA, Nimer SD, Teruya-Feldstein J. Application of tissue microarray technology to the study of non-Hodgkin’s and Hodgkin’s lymphoma. Hum Pathol. 2002;33:968–974. doi: 10.1053/hupa.2002.127438. [DOI] [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Jonkers J, Berns A. Oncogene addiction: sometimes a temporary slavery. Cancer Cell. 2004;6:535–538. doi: 10.1016/j.ccr.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Komarova NL, Wodarz D. The optimal rate of chromosome loss for the inactivation of tumor suppressor genes in cancer. Proc Natl Acad Sci U S A. 2004;101:7017–7021. doi: 10.1073/pnas.0401943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004;101:8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Kumada K, Yao R, Kawaguchi T, Karasawa M, Hoshikawa Y, Ichikawa K, Sugitani Y, Imoto I, Inazawa J, Sugawara M, et al. The selective continued linkage of centromeres from mitosis to interphase in the absence of mammalian separase. J Cell Biol. 2006;172:835–846. doi: 10.1083/jcb.200511126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GQ, Li H, Zhang HF. Mad2 and p53 expression profiles in colorectal cancer and its clinical significance. World J Gastroenterol. 2003;9:1972–1975. doi: 10.3748/wjg.v9.i9.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gorbea C, Mahaffey D, Rechsteiner M, Benezra R. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci U S A. 1997;94:12431–12436. doi: 10.1073/pnas.94.23.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci U S A. 2004;101:4459–4464. doi: 10.1073/pnas.0306069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–359. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih Ie M, Vogelstein B, Lengauer C. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci U S A. 2002;99:16226–16231. doi: 10.1073/pnas.202617399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG) Mol Endocrinol. 1997;11:433–441. doi: 10.1210/mend.11.4.9911. [DOI] [PubMed] [Google Scholar]
- Percy MJ, Myrie KA, Neeley CK, Azim JN, Ethier SP, Petty EM. Expression and mutational analyses of the human MAD2L1 gene in breast cancer cells. Genes Chromosomes Cancer. 2000;29:356–362. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1044>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Rao CV, Yang YM, Swamy MV, Liu T, Fang Y, Mahmood R, Jhanwar-Uniyal M, Dai W. Colonic tumorigenesis in BubR1+/-ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 2005;102:4365–4370. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell. 2002;1:289–298. doi: 10.1016/s1535-6108(02)00047-8. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med. 2000;6:1029–1035. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]
- Shi Q, King RW. Cell biology: Nondisjunction, aneuploidy and tetraploidy (Reply) Nature. 2006;442:E10. doi: 10.1038/nature05139. [DOI] [PubMed] [Google Scholar]
- Sotillo R, Dubus P, Martin J, de la Cueva E, Ortega S, Malumbres M, Barbacid M. Wide spectrum of tumors in knock-in mice carrying a Cdk4 protein insensitive to INK4 inhibitors. Embo J. 2001;20:6637–6647. doi: 10.1093/emboj/20.23.6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann O, Zou H, Gerber SA, Gygi SP, Kirschner MW. Dual inhibition of sister chromatid separation at metaphase. Cell. 2001;107:715–726. doi: 10.1016/s0092-8674(01)00603-1. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nishioka J, Kato K, Nakamura A, Mouri T, Miki C, Kusunoki M, Nobori T. Mitotic checkpoint protein hsMAD2 as a marker predicting liver metastasis of human gastric cancers. Jpn J Cancer Res. 2001;92:952–958. doi: 10.1111/j.1349-7006.2001.tb01186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonon G, Wong KK, Maulik G, Brennan C, Feng B, Zhang Y, Khatry DB, Protopopov A, You MJ, Aguirre AJ, et al. High-resolution genomic profiles of human lung cancer. Proc Natl Acad Sci U S A. 2005;102:9625–9630. doi: 10.1073/pnas.0504126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC- dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu T, Fang Y, Xie S, Huang X, Mahmood R, Ramaswamy G, Sakamoto KM, Darzynkiewicz Z, Xu M, Dai W. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 2004;103:1278–1285. doi: 10.1182/blood-2003-06-2158. [DOI] [PubMed] [Google Scholar]
- Wang X, Jin DY, Ng RW, Feng H, Wong YC, Cheung AL, Tsao SW. Significance of MAD2 expression to mitotic checkpoint control in ovarian cancer cells. Cancer Res. 2002;62:1662–1668. [PubMed] [Google Scholar]
- Wang X, Jin DY, Wong YC, Cheung AL, Chun AC, Lo AK, Liu Y, Tsao SW. Correlation of defective mitotic checkpoint with aberrantly reduced expression of MAD2 protein in nasopharyngeal carcinoma cells. Carcinogenesis. 2000;21:2293–2297. doi: 10.1093/carcin/21.12.2293. [DOI] [PubMed] [Google Scholar]
- Wasch R, Cross FR. APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature. 2002;418:556–562. doi: 10.1038/nature00856. [DOI] [PubMed] [Google Scholar]
- Wassmann K, Benezra R. Mitotic checkpoints: from yeast to cancer. Curr Opin Genet Dev. 2001;11:83–90. doi: 10.1016/s0959-437x(00)00161-1. [DOI] [PubMed] [Google Scholar]
- Wassmann K, Liberal V, Benezra R. Mad2 phosphorylation regulates its association with Mad1 and the APC/C. Embo J. 2003;22:797–806. doi: 10.1093/emboj/cdg071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Cleveland DW. Cell biology: nondisjunction, aneuploidy and tetraploidy. Nature. 2006;442:E9–10. doi: 10.1038/nature05139. discussion E10. [DOI] [PubMed] [Google Scholar]
- Wirth KG, Wutz G, Kudo NR, Desdouets C, Zetterberg A, Taghybeeglu S, Seznec J, Ducos GM, Ricci R, Firnberg N, et al. Separase: a universal trigger for sister chromatid disjunction but not chromosome cycle progression. J Cell Biol. 2006;172:847–860. doi: 10.1083/jcb.200506119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G, Luo X, Habu T, Rizo J, Matsumoto T, Yu H. Conformation-specific binding of p31(comet) antagonizes the function of Mad2 in the spindle checkpoint. Embo J. 2004;23:3133–3143. doi: 10.1038/sj.emboj.7600322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Jiang P, Yang M, Xu M, Yamauchi K, Tsuchiya H, Tomita K, Wahl GM, Moossa AR, Hoffman RM. Cellular dynamics visualized in live cells in vitro and in vivo by differential dual-color nuclear-cytoplasmic fluorescent-protein expression. Cancer Res. 2004;64:4251–4256. doi: 10.1158/0008-5472.CAN-04-0643. [DOI] [PubMed] [Google Scholar]
- Yamamoto TM, Iwabuchi M, Ohsumi K, Kishimoto T. APC/C-Cdc20-mediated degradation of cyclin B participates in CSF arrest in unfertilized Xenopus eggs. Dev Biol. 2005;279:345–355. doi: 10.1016/j.ydbio.2004.12.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.