

Abstract
Indian hedgehog (Ihh) is essential for chondrocyte and osteoblast proliferation/differentiation during prenatal endochondral bone formation. The early lethality of various Ihh-ablated mutant mice, however, prevented further analysis of its role in postnatal bone growth and development. In this study, we describe the generation and characterization of a mouse model in which the Ihh gene was successfully ablated from postnatal chondrocytes in a temporal/spatial-specific manner; postnatal deletion of Ihh resulted in loss of columnar structure, premature vascular invasion, and formation of ectopic hypertrophic chondrocytes in the growth plate. Furthermore, destruction of the articular surface in long bones and premature fusion of growth plates of various endochondral bones was evident, resulting in dwarfism in mutant mice. More importantly, these mutant mice exhibited continuous loss of trabecular bone over time, which was accompanied by reduced Wnt signaling in the osteoblastic cells. These results demonstrate, for the first time, that postnatal chondrocyte-derived Ihh is essential for maintaining the growth plate and articular surface and is required for sustaining trabecular bone and skeletal growth.
Keywords: cre/loxP, endochondral bone, tamoxifen-inducible, cartilage, osteoblast
Bones develop through two different processes: intramembranous ossification and endochondral ossification (1). Some of the skeletal elements derive directly from mesenchymal cells (intramembranous ossification), but the majority of the skeleton forms by endochondral ossification, a process that involves a cartilage intermediate; cells aggregate and then differentiate into chondrocytes and start to deposit a cartilage-specific extracellular matrix rich in collagen type II. Once these cells stop proliferating, they become hypertrophic, deposit collagen type X-rich matrix protein that becomes calcified, and eventually undergo apoptosis. Perichondrial cells surrounding the cartilage differentiate into osteoblasts and form a bone collar (1–4). The chondrocyte and osteoblast differentiation pathways are interrelated during endochondral bone formation. Transcription factors of the Sox- and Runt-domain families and signaling molecules, such as Indian hedgehog (Ihh), parathyroid hormone-related peptide (PTHrP), and FGFs, play essential roles in regulating complex endochondral bone formation (5).
Ihh expression, in mouse, has been detected in various soft tissues, in prehypertrophic chondrocytes (6), and in osteoblasts (7, 8). In rats, Ihh has been detected in tibial growth plates at 4 and 12 weeks of age (9); similarly, IHH expression has been detected in growth plates, with the highest expression during the early stages of puberty in humans (10). These observations clearly suggest a role of Ihh in postnatal bone formation; however, the effects of Ihh produced by postnatal chondrocytes on bone growth and osteoblast maturation are not clear and represent an intense area of research.
Hedgehog signals are transduced through smoothened (Smo), a putative G protein-coupled seven-transmembrane domain protein (11). In the absence of hedgehog proteins, Smo is repressed by the Ihh target gene, patched (Ptch), another cell surface receptor for hedgehog. The in vivo physiological role of Ihh has been determined mostly by knockout studies (12). The majority of Ihh-null embryos die during early development, when Ihh expression is detected in the visceral endoderm. Ihh-null mice display abnormal chondrocyte proliferation and maturation, with no mature osteoblasts. In contrary, misexpression of Ihh in the chicken limb inhibits chondrocyte differentiation. Earlier studies have suggested PTHrP-dependent suppression of chondrocyte hypertrophy by Ihh (13, 14). Of relevance, studies have also documented PTHrP-independent effects of Ihh on chondrocytes (15, 16). Chondrocyte specific Smo conditional knockout mice showed reduced proliferation of chondrocytes in the presence of PTHrP expression. Furthermore, overexpression of either Ihh or a constitutively active Smo allele specifically in cartilage resulted in increased activity of the Ihh signaling pathway to promote chondrocyte proliferation (15). Together, these observations suggest that Ihh has the ability to act directly on chondrocytes to induce their proliferation, independent of PTHrP activity.
Studies have also suggested an important role of Ihh in osteoblast differentiation. Chimeric mouse studies of the growth plate proposed that Ihh is secreted by prehypertrophic and hypertrophic chondrocytes and is responsible for signaling to the periarticular surface and for induction of bone collar formation in the adjacent perichondrium (17). Furthermore, conditional knockout mice lacking Smo from perichondrial cells demonstrated that Ihh signaling is required for the formation of a normal bone collar and the development of a primary spongiosa (18). Wnt signaling, downstream of Ihh, also was suggested to regulate osteoblast differentiation during embryonic skeletogenesis (19). Removal of canonical Wnt signaling by conditional deletion of the β-catenin gene from early osteoblast progenitors results in failure of terminal differentiation to osteocalcin-positive osteoblasts.
Recently, we have generated Ihh-null mice, in which Ihh was selectively ablated from chondrocytes; the phenotype of these mice resembled conventional Ihh-null mice at birth and provides the evidence of the essential role of chondrocyte-derived Ihh in prenatal endochondral bone formation (20). However, due to early lethality of these mutants, the role of Ihh in postnatal bone growth and remodeling could not be studied (20).
To resolve this issue, we have generated tamoxifen-inducible conditional col2α1-Cre ER*; floxed Ihh-knockout mice. These mutants provided the in vivo tool to assess the role of Ihh in the postnatal growth plate after the endochondral bone formation process has already been initiated. Our results demonstrate that Ihh expression in the postnatal chondrocytes is essential for maintenance of the growth plate and the articular surface and for sustaining a primary spongiosa, possibly through activation of Wnt signaling, and eventual bone growth after birth.
Results
Efficiency and Specificity of Cre-loxP Mediated in Vivo Gene Ablation.
Transgenic mice in which Cre recombinase fused to a mutated estrogen receptor protein was driven by the col2α1 promoter were generated as reported earlier (21) and are described in Materials and Methods and shown schematically in Fig. 1A. In these mice, Cre recombinase activity is controlled by the injection of tamoxifen in a time-specific manner. To test Cre specificity and efficiency, we crossed col2α1-Cre ER* animals to Rosa26; Cre indicator mice (22) which express LacZ upon Cre activation. We i.p. injected a single dose of either 0.2 mg of 4-hydroxy tamoxifen (tamoxifen) or vehicle alone at postnatal day (P)0 into col2α1-Cre ER*; Rosa26 reporter animals and performed LacZ staining at P1, P4, P7, and P14 (Fig. 1 B–G). By P1, we could observe weak LacZ staining in the appendicular skeleton (Fig. 1B). Significant LacZ expression was obvious at P4 in the growth plate and articular cartilage of the long bones. Because the size of the growth plate diminishes with age and a secondary ossification center is formed, LacZ expression was confined accordingly and only seen in a restricted area within the epiphysis after P4. No staining could be found in vehicle-treated (corn oil-injected) controls. We confirmed the specific LacZ staining on sections of tibiae at P7 and P14 (Fig. 1 C and D). Furthermore, at P4 LacZ expression also was observed in cartilaginous tissues, such as ribs, sternum (Fig. 1E), paws (Fig. 1F), nasal region, and in the secondary cartilage that forms in the lateral region of the interparietal bone (Fig. 1G), corresponding to the areas where col2α1 is normally expressed. As evident by the LacZ staining in Fig. 1 C and D, the col2α1 promoter used to drive Cre recombinase in these experiments was very specific.
Fig. 1.

Specificity of Col2α1-Cre ER* using floxed ROSA reporter mice. (A) Col2α1-Cre ER* construct. (B–L) LacZ staining of Col2α1-Cre ER*; Rosa26cre mice. (B–G) Col2α1-Cre ER*; Rosa26cre mice were injected with tamoxifen (Tam) or vehicle (Veh) at P0 followed by LacZ staining at P1, P4, P7, and P14. (B) LacZ staining of hindlimbs. (C and D) Sections of proximal tibiae at P7 (C) and P14 (D). (E–G) Ribs and sternum (E), paws (F), and skull (G) at P4. (H–L) LacZ staining of tibiae at P15, P18, P21, and P28 (H) and of ribs (I), sternum (J), paws (K), and tail (L) at P18 after injection of tamoxifen at P14.
We extended our studies by injecting tamoxifen at a later stage, such as P14. For this purpose col2α1-Cre ER*; Rosa26 reporter animals were injected i.p. with a single dose of either 2 mg of tamoxifen or vehicle, followed by examination of LacZ expression at P15, P18, P21, and P28 (Fig. 1 H–L). Interestingly, we could detect strong LacZ-expressing cells at all these later time points, indicating the presence of cells in which the ROSA gene was activated at P14. The finite LacZ expression in the cartilaginous areas is suggestive of the specificity of the staining (Fig. 1H) and as demonstrated in various cartilaginous tissues at P18 (Fig. 1 I–L). No staining could be detected in vehicle-treated controls. In the current study, we used these col2α1-Cre ER* transgenic mice to delete the Ihh gene from chondrocytes after birth.
Generation and Characterization of col2α1-Cre ER*; Ihh d/d Animals.
To generate col2α1-Cre ER*; Ihh fl/fl animals, we bred Col2α1-Cre ER* transgenic mice into the floxed Ihh animal background (Ihh fl/fl). Col2α1-Cre ER*; Ihh fl/fl mice were born with the expected rate of Mendelian inheritance and did not show any abnormal phenotype. We then injected col2α1-Cre ER*; Ihh fl/fl and Ihh fl/fl control mice with a single dose of either 0.2 mg of tamoxifen or vehicle alone at P0. No changes in body weight, size, or length of the mutant animals were evident until P7 after injection; however, 1 week later, at P14, col2α1-Cre ER*; Ihh d/d mice were significantly smaller in size measured from nose to tail (24%; P < 0.01) than control littermates (Fig. 2A), with a significant reduction in the length of tail, trunk, and limbs. Interestingly, x-rays from hindlimbs of tamoxifen-injected col2α1-Cre ER*; Ihh d/d animals showed complete loss of the growth plates in both the proximal and distal ends of the mutant long bones when compared with Ihh fl/fl control animals (Fig. 2B, arrows), indicating premature closure of the growth plates. Although these mutants were able to live beyond our study period, their size and body weight did not increase or recover after P14, as demonstrated at 8 weeks of age (Fig. 2 A and C; 45% smaller, P < 0.0001).
Fig. 2.

Phenotype of Col2α1-Cre ER*; Ihh d/d mice at P14 (A and B) and 8 weeks (A and C). (A) At P14, mutant mice were 24% smaller in size than control littermates (∗, P < 0.01); at 8 weeks, the difference in size increased to 45% (∗∗∗, P < 0.0001; n = 7, 5, 8, 5, 7, and 3 in each group, respectively from left to right). (B) X-ray analysis of tibiae at P14. Arrows depict the translucency of the growth plates in the control (Left) and the fusion of the growth plate in the mutant proximal and distal tibia (Right). (C) Macroscopic image of 8-week-old littermates. Mutant (right) did not increase body size after P14 (see also A).
Impaired Mineralization and Shape of Endochondral Formed Bones.
At P7, a slight increase of Alizarin Red S staining was evident in the epiphyseal cartilage of the mutant tibia, suggesting minor changes in the mineralization pattern due to absence of Ihh (data not shown). To confirm this observation we performed histological analyses and detected von Kossa staining in the center of the growth plate of the long bones at P7 (Fig. 3F). At P14, Alizarin Red S staining of various skeletal elements, such as ribs, paws, knee joint, and tail vertebrae showed enhanced mineralization (Fig. 3 A–D). The chondro–osseus junction of the ribs was enlarged in mutant mice when compared with the one in the control mice (Fig. 3A, arrow). The growth plates of phalanges, tibiae, and tail vertebrae had completely disappeared, and mutant vertebrae were significantly smaller in size (Fig. 3 D and E). Histological examination of the vertebrae at P14 also demonstrated the loss of mutant growth plates (Fig. 3E). Furthermore, a closer analysis of the vertebrae showed that in contrast to the normal intervertebral disks, in which the nucleus pulposus was surrounded by a well formed annulus fibrosus, the discs of col2α1-Cre ER*; Ihh d/d animals exhibited enlarged nuclei pulposi with no annulus fibrosus on the articular surface of the vertebrae (Fig. 3E, arrow).
Fig. 3.

Mineralization of various skeletal elements. (A–D) Mineralization pattern of ribs (A, arrow), paws (B), tibia (C), and tail vertebrae (D) of controls (Co) and mutants (M) by Alizarin red/Alcian blue at P14. Arrows in B–D depict the loss of the growth plates. (E) Mutant vertebrae showed loss of annulus fibrosus (arrow) surrounding the significantly enlarged nucleus pulposus (NP). (F–I) von Kossa staining of tibiae of Ihh fl/fl (Upper) and col2α1-Cre ER*; Ihh d/d (Lower) at P7 (F), P14 (G) (complete loss of growth plate is evident), and 8 weeks (H and I) (note loss of trabecular bone in H). Arrows in I point to the decreased thickness of the endosteal seam along the diaphysis in the mutant tibia.
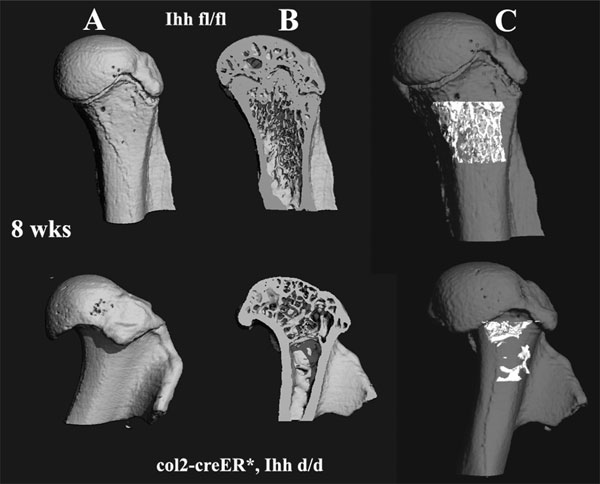
An abnormal process that started with initial ectopic mineralization of the mutant tibial growth plate at P7 (Fig. 3F) eventually led to a complete loss of the growth plate in mutant bones at P14; such changes resulted in an abnormal shape of the tibia with complete loss of the rounded articular surface at the distal end (Fig. 3G). These data clearly suggest that chondrocyte-derived Ihh is involved in regulating the postnatal mineralization process in the growth plate possibly by influencing chondrocyte differentiation. Interestingly, after closure of the growth plate we noticed a significant loss of trabeculae in the metaphyseal region as demonstrated at 8 weeks of age, and their articular surfaces were severely disrupted (Fig. 3H). Microcomputed tomographic analysis confirmed less trabecular bone in the mutant mice, suggesting reduced bone mineral density in the metaphyseal region at 8 weeks [supporting information (SI) Fig. 6]. Moreover, we found a significant diminution of the thickness of the endosteal seam along the diaphysis, indicating a substantial decrease in osteoblast formation (Fig. 3I, arrows). These data indicate that loss of Ihh signaling from the growth plate not only altered chondrocyte differentiation but also affected osteoblast development in bone. This observation provides in vivo evidence that Ihh signals from postnatal chondrocytes to osteoblasts and that this signal is required to maintain trabecular bone.
Chondrocyte-Derived Ihh Is Required for Maintenance of Normal Growth Plate After Birth.
Morphological analysis of the proximal growth plate of col2α1-Cre ER*; Ihh d/d animals at P7 demonstrated an absolute loss of columnar structure of chondrocytes in the tibia when compared with controls (SI Fig. 7A), where normal proliferating columns of cartilaginous cells are evident. The mutant growth plate was primarily composed of hypertrophic chondrocytes that express collagen type X but not collagen type II indicating an abnormally advanced maturation stage for these cells (SI Fig. 7 B and C). The expression pattern of the PTH/PTHrP receptor also was changed, with positive signals found in cells surrounding the area of ectopically differentiated hypertrophic chondrocytes in mutants (SI Fig. 7D). Furthermore, the expression of PTHrP was observed in the adjacent regions to articular and prospective secondary ossification center in controls. Alternatively, PTHrP expression was observed around ectopic hypertrophic chondrocytes in mutants (SI Fig. 7E). These results suggest that postnatal deletion of Ihh from chondrocytes affects the regular distribution of PTHrP and its receptor expression, and such abnormal regulation of the PTHrP system might alter chondrocyte differentiation.
Examination of Endochondral Bone Formation in the Col2α1-Cre ER*; Ihh d/d Tibia.
During bone development, chondrocytes proliferate and differentiate and hypertrophic chondrocytes are replaced by bone, a process that begins with blood vessel invasion. We and others (15, 20) have shown that Ihh is required for chondrocyte proliferation. To examine the mechanism causing the loss of the growth plate in tamoxifen-injected mutant mice, we determined the rate of chondrocyte proliferation and vascular invasion in the growth plate. The number of proliferative chondrocytes decreased dramatically in mutants when compared with controls, as determined by proliferating cell nuclear antigen (PCNA)-stained cell populations (Fig. 4A). Moreover, premature blood vessel invasion was detected in the middle of growth plates in mutants at P7 (Fig. 4B). These results suggest that decreased proliferation and premature differentiation of mutant chondrocytes, along with abnormal vascular invasion, lead to advanced replacement of mineralized cartilage by bone. PTHrP protein (Fig. 4C) was detected in the same cell population as PCNA, which also expressed PTH/PTHrP receptors. The possibility that the loss of Ihh signal could affect cell death of chondrocytes was ruled out, because no obvious changes in apoptosis were found by TUNEL staining between control and mutant chondrocytes (data not shown). Similarly, no significant differences in the number of tartrate-resistant acidic phosphatase-positive multinucleated osteoclasts were found in the mutant bones when compared with the ones of control littermates (data not shown).
Fig. 4.

Analyses of endochondral bone formation in tibiae at P7. (A) Proliferation was analyzed by PCNA staining and was significantly decreased in col2α1-Cre ER*; Ihh d/d. Lower represents magnifications of the boxed areas in Upper. (B) Atypical invasion of blood vessels in the middle of growth plate (arrows) was detected by PECAM-1 (CD31) staining. (C) Altered expression pattern of PTHrP protein in the mutant proximal tibia. (D) Protein levels for β-catenin were significantly decreased in bone collar (arrowheads) and primary spongiosa (boxed area) of col2α-Cre ER*; Ihh d/d animals.
We then studied whether specific pathways that regulate osteoblast maturation could be affected by the loss of Ihh expression from postnatal chondrocytes. Recently, β-catenin, a Wnt signaling target, was shown to be required for terminal differentiation of osteoblasts from its progenitors at the chondro–osseus junction adjacent to the growth plates (19). We examined whether loss of Ihh expression from postnatal chondrocytes could influence Wnt signaling to affect osteoblast maturation; immunohistochemical analyses showed a significant reduction in β-catenin expression in the bone collar and primary spongiosa of col2α-Cre ER*; Ihh d/d mice, suggesting that postnatal chondrocyte-derived Ihh is required for osteoblast maturation through activation of the Wnt signaling pathway and is thereby partly regulating their functionality (Fig. 4D).
Analysis of Osteoblast Differentiation in the Col2α1-Cre ER*; Ihh d/d Tibia.
We have found that the severity of the abnormal phenotype in Col2α1-Cre ER*; Ihh d/d mutants is dependent on the degree of Ihh deletion. Notably efficient deletion of the Ihh gene (Fig. 5A) from chondrocytes in the postnatal growth plate leads to a complete loss of expression of its receptor, Ptch, not only in the surrounding cartilaginous cells, but more importantly in the primary spongiosa (Fig. 5B, arrow). These data together with the loss of β-catenin expression (Fig. 4D) indicate that postnatal chondrocyte-derived Ihh is involved in a tight regulation and communication between chondrocytes and osteoblasts during postnatal bone growth and development.
Fig. 5.

Analyses of osteoblast maturation at P7. (A–D) In situ hybridization using riboprobes for Ihh (A), Ptch (B), collagen type I (Col I) (C), and osteocalcin (OC) (D). The arrow in B points to the complete loss of Ptch expression in the mutant primary spongiosa. Boxed area in C depicts collagen type I expression in the bone collar, which is absent in the mutant. (E) Bar graph showing mRNA expression of Dkk1 and OPG in the primary spongiosa of Ihh fl/fl and col2α1-Cre ER*; Ihh d/d mice at P7. Data are expressed as fold over controls. The expression of both Dkk1 and OPG was significantly reduced in mutants (P < 0.05; n = 3 measured in triplicates).
By P7, a decrease in the expression of the early osteoblast marker, collagen type I, was detected in the bone collar adjacent to the growth plate (Fig. 5C). Moreover, a significant reduction in osteocalcin expression, a marker of terminally differentiated osteoblasts, was evident in the mutant bone (Fig. 5D). These results demonstrate that Ihh expressed by chondrocytes in the postnatal growth plate is required for normal osteoblast differentiation and, therefore, maintenance of endochondral bone. To further study the effects of Ihh deletion from postnatal chondrocytes on osteoblasts, we isolated RNA from the primary spongiosa of the tibia of Ihh fl/fl and col2α1-Cre ER*; Ihh d/d animals and performed quantitative real-time PCR for Dickkopf1 (Dkk1) and Osteoprotegerin (OPG), two downstream target molecules of the Wnt signaling pathway. In accordance with less β-catenin expression, a >50% reduction in expression of Dkk1 and OPG was evident in the mutant bones (P < 0.05), indicating that Ihh expressed by postnatal chondrocytes is required for osteoblast differentiation through activation of Wnt signaling (Fig. 5E).
Discussion
In the current study, we have demonstrated an essential role for chondrocyte-derived Ihh in maintaining postnatal growth plate structures and trabecular bones by generating tamoxifen-inducible conditional Ihh knockout mice. Our results suggest that Ihh expression in postnatal chondrocytes is required to prevent ectopic hypertrophy and to maintain a normal structure of the growth plate. Earlier studies have shown that Ihh is required for columnar chondrocyte differentiation from precursor cells independent of PTHrP (16). Our present study reemphasizes the fact that Ihh is essential to induce columnar chondrocyte differentiation to maintain a growth plate, even after birth, despite the presence of PTHrP protein in the remaining chondrocytes in the growth plate at P7 in mutant mice (Fig. 4C).
In the present study, Ihh is found to be essential for the maintenance of the postnatal growth plates of numerous skeletal sites, including long bones, vertebrae, and digits (Fig. 3). Time-course experiments also demonstrated that the deletion of Ihh from col2α1-expressing cells at P0 resulted in the premature vascularization in the middle of the tibial growth plate at P7 (Fig. 4B). Moreover, the formation of a secondary ossification center could not be observed, presumably because of the premature loss of the growth plate. Furthermore, intervertebral disks of mutant vertebrae showed an enlarged nucleus pulposus with no annulus fibrosus on the articular surface of the vertebrae (Fig. 3E). Although Ihh is not directly expressed in the intervertebral disks, it appears to be a key molecular signal in the growth and differentiation of cells within the vertebral endplate (23), suggesting an important role for Ihh in preventing disk degeneration. These findings demonstrate that chondrocyte-derived Ihh is required to maintain a normal growth plate and to form a secondary ossification center and is required for the longitudinal growth of the endochondral skeleton in postnatal life.
The results also support the notion that Ihh plays a key role in endochondral bone formation after birth and that other factors cannot compensate for this loss. For example, FGF-receptor 3 (FGFR3) is known to be an upstream negative regulator of the hedgehog signaling pathway in the growth plate (24). Mice with a constitutively activating mutation in FGFR3 show a phenotype that resembles the phenotype of our mutants, which is distinguished by their dwarfism and premature fusion of growth plates in the long bones and in vertebrae (25). However, we could not detect any premature apoptosis of chondrocytes in the growth plates of Ihh mutant mice at P7 (data not shown), as is found in the FGFR3 mutants. The loss of the growth plate in Col2α1-Cre ER*; Ihh d/d mutants could be explained by the advanced replacement of the abnormally ectopic, mineralized hypertrophic chondrocytes by bone, accompanied by enhanced angiogenesis and matrix degradation, leading to the a significant decrease in the number of proliferative chondrocytes.
Studies have suggested that Ihh, expressed by chondrocytes and osteoblasts, can directly affect bone formation (18); because osteoblast differentiation in those mutants was disturbed, the question regarding the origin and effect of Ihh on osteoblast maturation remained unknown. The results of our present study, in which the Ihh gene was deleted selectively from chondrocytes at a time when mature osteoblasts already existed and endochondral bone has already formed, clearly demonstrate that postnatal chondrocyte-derived Ihh is essential for the maintenance of a growth plate and, more importantly, that Ihh produced by osteoblasts is not sufficient to compensate for this loss. It is worth mentioning that we did not observe any significant differences in osteoclast number in control and mutant mice, as determined by tartrate-resistant acidic phosphatase staining (data not shown). These data would indicate that the loss of endochondral bone in our newly generated mutant mice is due to decreased osteoblastic activities and not to an increased osteoclast number. The loss of trabecular bone could be explained either by the failure to generate enough chondrocytes and, therefore, insufficient production of extracellular matrix needed to support osteoblast development or by the lack of direct Ihh signaling from chondrocytes to osteoblasts to maintain bone remodeling. This latter hypothesis is supported by the fact that osteoblasts located in the adjacent bone collar and primary spongiosa that do not express Ptch, the receptor for Ihh (Fig. 5B), also express less collagen type I and osteocalcin (OC) (Fig. 5 C and D); more importantly, these cells exhibit significantly reduced expression of β-catenin protein (Fig. 4D). The reduced expression of these cellular markers is suggestive of impaired differentiation of osteoblasts, and the likely effect is decreased bone formation. Moreover, the expression of molecular Wnt signaling targets, such as Dkk1 and OPG (Fig. 5E) was significantly reduced (P < 0.05) in the primary spongiosa, which would indicate that the regulation of osteoblast differentiation by Ihh is mediated through Wnt signaling in postnatal life. Taken together, these data suggest that osteoblast maturation requires an Ihh signal from chondrocytes and that loss of such a signal results in a failure of osteoblast differentiation, even in the postnatal period. Furthermore, the significant reduction of the number of osteoblasts located in the endosteal seam of the diaphysis also supports such possibilities. A failure in osteoblast maturation could explain the loss of trabecular bone, as shown by microcomputed tomographic analysis at 8 weeks (SI Fig. 6). The results of the time course experiments, together with the decreased numbers of mature osteoblasts, are very supportive of such possibilities.
In summary, the data presented in this study clearly show the importance of Ihh signaling from postnatal chondrocytes to maintain a growth plate, articular surface, and adjacent endochondral bone formation. Furthermore, this work demonstrates the necessity for a functioning communication between cartilage and bone, even in postnatal life, to maintain postnatal growth plate structures and for preserving trabecular bones. These findings regarding the regulation of postnatal bone growth by Ihh signaling may open an avenue to treat skeletal diseases, such as achondroplasia, osteoarthritis, disk degeneration and osteoporosis, by manipulating either Ihh expression or its signaling components.
Materials and Methods
Generation of col2α1-Cre ER*; Ihh fl/fl Animals.
Col2α1-Cre ER* transgenic mice were interbred with Ihh fl/fl animals to obtain Ihh fl/fl and col2α1-Cre ER*; Ihh fl/fl offspring. Animals were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were studied according to Institutional Animal Care and Use Committee approved protocols.
Immunohistochemistry.
Expression of PCNA, CD31, PTHrP, and β-catenin was examined by using anti-PCNA antibody (Zymed; Invitrogen, Carlsbad, CA), MEC 13.3 rat monoclonal anti-mouse PECAM-1 antibody (Pharmingen, Franklin Lakes, NJ), goat polyclonal anti mouse PTHrP (E-19) antibody (Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit polyclonal anti human β-catenin (H-102) antibody (Santa Cruz Biotechnology). Sections were incubated at 4°C overnight with primary antibodies, processed further with a commercially available staining kit, and visualized with DAB staining.
Quantitative PCR.
At P7, 60-μm frozen sections were prepared from the tibiae of tamoxifen-injected (P0) mice. Primary spongiosa was dissected and collected in lysis buffer (Agilent Technologies, Santa Clara, CA) and 2-mercaptoethanol (Sigma–Aldrich, St. Louis, MO). Total RNA was extracted (Agilent Technologies), and processed further for real-time PCR with the following specific primers: 5′-GTGGTACGACCAGAGGCA TAC-3′ (β-actin forward), 5′-AAGGCCAACCGTGAAAAGAT-3′ (β-actin reverse), 5′-CTTGCGCTGA AGATGAGGAGT-3′ (Dkk1 forward), 5′-GAGGGCATGCATATT CCATTT-3′ (Dkk1 reverse), 5′-CAGTTTCTGG GTCATAATGCAA- 3′ (OPG forward), and 5′-ATGAACAAGTGGCTGTGCTG-3′ (OPG reverse). The relative expression of mutants was normalized to that of controls by using the calculations described earlier (26).
Statistical Analysis.
Statistically significant differences between groups were evaluated by Mann–Whitney's U test. A P value of <0.05 was considered to be statistically significant. Analyses were performed by using Excel (Microsoft, Redmond, WA) and Prism 3.0 (GraphPad, San Diego, CA).
Supplementary Material
Acknowledgments
We thank Drs. H. M. Kronenberg, B. Olsen, M. Noda, M. Whitman, J. Stern, N. Fukai, D. Medici, T. Kobayashi, W. P. Kuo, and K. Tsuji for helpful suggestions and D. Creel for microcomputed tomography analyses. This work was supported by a grant from the Volkswagen Foundation, by the Harvard School of Dental Medicine, and by National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant AR050560 (to B.L.).
Abbreviations
- PCNA
proliferating cell nuclear antigen
- Pn
postnatal day n
- PTHrP
parathyroid hormone-related peptide.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0608449104/DC1.
References
- 1.Horton W. Growth Genet Horm. 1990;6:1–3. [Google Scholar]
- 2.Gilbert S. Developmental Biology. Sunderland, MA: Sinauer; 1997. [Google Scholar]
- 3.Hall B. Am Sci. 1988;76:174–181. [Google Scholar]
- 4.Erlebacher A, Filvaroff E, Gitelman S, Derynck R. Cell. 1995;80:371–378. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- 5.de Crombrugghe B, Lefebvre V, Nakashima K. Curr Opin Cell Biol. 2001;13:721–727. doi: 10.1016/s0955-0674(00)00276-3. [DOI] [PubMed] [Google Scholar]
- 6.Bitgood MJ, McMahon AP. Dev Biol. 1995;172:126–138. doi: 10.1006/dbio.1995.0010. [DOI] [PubMed] [Google Scholar]
- 7.Murakami S, Nifuji A, Noda M. Endocrinology. 1997;138:1972–1978. doi: 10.1210/endo.138.5.5140. [DOI] [PubMed] [Google Scholar]
- 8.Murakami S, Noda M. Calcif Tissue Int. 2000;66:272–276. doi: 10.1007/pl00005843. [DOI] [PubMed] [Google Scholar]
- 9.van der Eerden BC, Karperien M, Gevers EF, Lowik CW, Wit JM. J Bone Miner Res. 2000;15:1045–1055. doi: 10.1359/jbmr.2000.15.6.1045. [DOI] [PubMed] [Google Scholar]
- 10.Kindblom JM, Nilsson O, Hurme T, Ohlsson C, Savendahl L. J Endocrinol. 2002;174:R1–6. doi: 10.1677/joe.0.174r001. [DOI] [PubMed] [Google Scholar]
- 11.Alcedo J, Noll M. Biol Chem. 1997;378:583–590. doi: 10.1515/bchm.1997.378.7.583. [DOI] [PubMed] [Google Scholar]
- 12.St-Jacques B, Hammerschmidt M, McMahon AP. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Science. 1996;273:613–621. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 14.Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LHK, Ho C, Mulligan RC, et al. Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 15.Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Development (Cambridge, UK) 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi T, Soegiarto DW, Yang Y, Lanske B, Schipani E, McMahon AP, Kronenberg HM. J Clin Invest. 2005;115:1734–1742. doi: 10.1172/JCI24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung UI, Schipani E, McMahon AP, Kronenberg HM. J Clin Invest. 2001;107:295–304. doi: 10.1172/JCI11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. Development (Cambridge, UK) 2004;131:1309–1318. doi: 10.1242/dev.01006. [DOI] [PubMed] [Google Scholar]
- 19.Rodda SJ, McMahon AP. Development (Cambridge, UK) 2006;133:3231–3244. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 20.Razzaque MS, Soegiarto DW, Chang D, Long F, Lanske B. J Pathol. 2005;207:453–461. doi: 10.1002/path.1870. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura E, Nguyen MT, Mackem S. Dev Dyn. 2006;07:58. doi: 10.1002/dvdy.20892. [DOI] [PubMed] [Google Scholar]
- 22.Soriano P. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 23.DiPaola CP, Farmer JC, Manova K, Niswander LA. J Orthop Res. 2005;23:1112–1119. doi: 10.1016/j.orthres.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Naski MC, Colvin JS, Coffin JD, Ornitz DM. Development (Cambridge, UK) 1998;125:4977–4988. doi: 10.1242/dev.125.24.4977. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Spatz MK, Kannan K, Hayk H, Avivi A, Gorivodsky M, Pines M, Yayon A, Lonai P, Givol D. Proc Natl Acad Sci USA. 1999;96:4455–4460. doi: 10.1073/pnas.96.8.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}